

Summary
Questo articolo ha lo scopo di fornire la metodologia per la transgenesi lentivirale negli embrioni di ratto utilizzando iniezioni multiple di una sospensione di un virus nello spazio perivitelina dello zigote. I ratti femmina che vengono accoppiati con un ceppo maschile fertile con un diverso colore di pelliccia dominante viene utilizzato per generare madri adottive pseudoincinte.
Abstract
I modelli animali transgenici sono fondamentalmente importanti per la ricerca biomedica moderna. L'incorporazione di geni estranei negli embrioni di topo o ratto precoce è uno strumento inestimabile per l'analisi delle funzioni geniche negli organismi viventi. Il metodo di transgenesi standard si basa sulla microiniettatura di frammenti di DNA estraneo in un pronucle di un ovocita fecondato. Questa tecnica è ampiamente utilizzata nei topi, ma rimane relativamente inefficiente e tecnicamente impegnativa in altre specie animali. Il transgene può anche essere introdotto negli embrioni a una cellula attraverso l'infezione lentivirale, fornendo un'alternativa efficace alle iniezioni pronucleari standard, specialmente nelle specie o nei ceppi con una struttura embrionale più impegnativa. In questo approccio, una sospensione che contiene vettori lentivirali viene iniettata nello spazio perivitelina di un embrione di ratto fecondato, che è tecnicamente meno esigente e ha un tasso di successo più elevato. È stato dimostrato che i vettori lentivirali incorporano in modo efficiente il transgene nel genoma per determinare la generazione di linee transgeniche stabili. Nonostante alcune limitazioni (ad esempio, requisiti di livello di biosicurezza 2, limiti di dimensione del frammento di DNA), la transgenesi lentivirale è un metodo di transgenesi rapida ed efficiente. Inoltre, l'uso di ratti femminili che vengono accoppiati con un ceppo maschile fertile con un diverso colore di pelliccia dominante è presentato come un'alternativa per generare madri adottive pseudoincinte.
Introduction
Per molti anni, i roditori di laboratorio, come ratti e topi, sono stati utilizzati per modellare le condizioni fisiologiche e patologiche umane. La ricerca sugli animali ha portato a scoperte irraggiungibili con qualsiasi altro mezzo. Inizialmente, gli studi genetici si sono concentrati sull'analisi di disturbi e fenotipi che si verificano spontaneamente che sono considerati imitare da vicino la condizione umana1. Lo sviluppo di metodi di ingegneria genetica ha permesso l'introduzione o la cancellazione di geni specifici per ottenere un fenotipo desiderato. Pertanto, la generazione di animali transgenici è riconosciuta come una tecnica fondamentale nella ricerca moderna che consente studi sulla funzione genica negli organismi viventi.
La tecnologia animale transgenica è diventata possibile attraverso una combinazione di risultati nell'embriologia sperimentale e nella biologia molecolare. Negli anni sessanta, l'embriologo polacco A. K. Tarkowski pubblicò il primo lavoro sulla manipolazione degli embrioni di topo durante le prime fasi dello sviluppo2. Inoltre, i biologi molecolari hanno sviluppato tecniche per generare vettori di DNA (cioè portatori) per l'introduzione tra l'altro del DNA estraneo nel genoma dell'animale. Questi vettori consentono la propagazione di geni selezionati e la loro modifica appropriata, a seconda del tipo di ricerca che viene condotta. Il termine "animale transgenico" è stato introdotto da Gordon e Ruddle3.
La prima specie ampiamente accettata che è stata utilizzata in neurobiologia, fisiologia, farmacologia, tossicologia, e molti altri campi delle scienze biologiche e mediche è stato il ratto norvegese, Rattus norvegicus4. Tuttavia, a causa della difficoltà di manipolare gli embrioni di ratto, il topolino mus musculus è diventato la specie animale dominante nella ricerca genetica5. Un altro motivo per il primato del topo in tale ricerca è stata la disponibilità di una tecnologia di cellule staminali embrionali per generare animali knockout per questa specie. La tecnica di transgenesi più comunemente utilizzata (2-10% della prole transgenica rispetto a tutti gli animali nati) è la microiniezione di frammenti di DNA in un pronucle di un ovocita fecondato. Nel 1990, questo approccio, introdotto per la prima volta nei topi, è stato adattato per i ratti6,7. La transgenesi del ratto per iniezione pronucleare è caratterizzata da una minore efficienza8 rispetto ai topi, che è strettamente correlata alla presenza di plasma elastico e membrane pronucleari9. Anche se la sopravvivenza degli embrioni dopo la manipolazione è inferiore del 40-50% rispetto ai topi, questa tecnica è considerata uno standard nella generazione di ratti geneticamente modificati10. Sono stati studiati approcci alternativi in grado di garantire un'efficace corporazione di transgeni e tassi di sopravvivenza più elevati di zigoti iniettati.
Il fattore determinante dell'espressione transgene stabile e della trasmissione alla progenie è la sua integrazione nel genoma della cellula ospite. I lentivirus (LV) hanno la caratteristica distintiva di essere in grado di infettare sia le cellule divisorie che non dividendo. Il loro uso come strumento per l'incorporazione di geni eterologhi in embrioni si è dimostrato altamente efficiente11, e gli individui transgenici sono caratterizzati da espressione stabile del frammento di DNA incorporato. L'efficacia dei vettori lentivirali è stata confermata per la modificazione genetica dei topi12,,13,ratti12,14e altre specie11. In questo metodo, la sospensione LV viene iniettata sotto la zona pellucida dell'embrione nello stadio di due pronuclei. Questa tecnica garantisce essenzialmente la sopravvivenza al 100% degli embrioni perché l'oolemma rimane inalterato. La produzione di sospensioni LV di alta qualità e relativamente altamente concentrate sono fattori cruciali. Tuttavia, concentrazioni più basse di sospensioni LV possono essere superate da ripetute iniezioni11, che aumenta la quantità di particelle virali sulla superficie dell'uovo senza influenzare l'integrazione della membrana. Gli embrioni sottoposti a iniezioni ripetute nello spazio perivitelina si sviluppano ulteriormente, e la prole transgenica può trasmettere il transgene attraverso la germinale. L'efficienza della generazione di ratti transgenici mediante transgenesi lentivirale può essere fino all'80%12.
Qui, descriviamo la produzione di lentivirus ricombinante derivante da HIV-1 che è stato pseudotipato con proteina di involucro G del virus della stomatite vescicolare (VSV). L'uso dello pseudotipo VSV del sistema di imballaggio di seconda generazione determina l'ampia infettività delle particelle virali e permette la produzione di vettori altamente stabili che possono essere concentrati dall'ultracentrifugazione e dalla crioconservazione. Dopo la verifica del tibieto, i vettori sono pronti per essere utilizzati come veicolo per la consegna transgene in zigoti ratti albino Wistar. Dopo una serie di iniezioni, gli embrioni possono essere coltivati durante la notte e trasferiti allo stadio a due cellule per le madri affidatarie. A questo punto, è possibile considerare uno dei due approcci alternativi. La procedura standard utilizza femmine pseudoincinte come embrioni. Tuttavia, quando il tasso di gravidanza è basso dopo l'accoppiamento con maschi vasectomizzati, gli embrioni possono essere impiantati in femmine incinte Wistar/Sprague-Dawley (SD) che sono accoppiate con ratti maschi fertili con un colore di pelliccia scura (ad esempio, ratti Brown Norway [BN]). Il colore della pelliccia permette la distinzione della prole dalla gravidanza naturale dalla prole che ha origine dagli embrioni manipolati trasferiti.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
La produzione e l'applicazione di vettori virali erano conformi alle linee guida del livello di biosicurezza 2 ed è stata approvata dal Ministero dell'Ambiente polacco. Tutte le procedure sperimentali sugli animali descritte di seguito sono state approvate dal Comitato Etico Locale. Gli animali sono stati alloggiati in gabbie ventilate individualmente a una temperatura stabile (21-23 gradi centigradi) e umidità (50-60%) con accesso ad libitum all'acqua e al cibo in base a un ciclo di luce/buio di 12 h/12 h.
1. Produzione vettoriale Lentivirale
- Trasfezione di cellule HEK 293T
NOTA: Il protocollo qui presentato è progettato per la trasfezione di venti piatti di coltura di 10 cm che produce circa 200 mL di supernatante vettoriale grezzo.- Coltura HEK 293T cellule in mezzo DMEM che è integrato con siero bovino fetale (10%, v/v) in un incubatore di CO2 umidificato a 37 gradi centigradi. Per la trasfezione, preparare venti piastre di 10 cm di diametro, e semi 1.5–2 x 106 celle HEK 293T per piatto.
- Quando la confluenza raggiunge il 70%, trasfectare le cellule utilizzando il reagente di polietilenimina (PEI), pH 7,0, con un rapporto di 3 g di PEI per 1 g di DNA.
- Preparare la miscela di trasfezione per cinque piatti (preparare il numero di ripetizioni in base al numero totale di piatti). A 1 mL di Dulbecco's Modified Eagle Medium (DMEM; senza siero), aggiungete la miscela di tre plasmidi in modo che raggiungano una quantità finale di 25 g di plasmide VSVg, 50 g di delta R8.2 e 50 g di plasmide di codifica.
- Pipetta su e giù, e aggiungere 125 l of PEI ad una concentrazione di 3 g/l. Incubare a temperatura ambiente per 15 minuti, invertendo il tubo tre volte durante l'incubazione. Aggiungere 200 l della miscela di trasfezione per piastra. Successivamente, incubare le piastre in un'incubatrice di CO2 umidificata a 37 gradi centigradi.
- Concentrazione di vettori lentivirali
- Quarantotto ore dopo la trasfezione, raccogliere il mezzo che contiene particelle DiV. Utilizzare tubi conici da 50 ml.
NOTA: Quando si utilizza un plasmide con un tag fluorescente, le celle possono essere visualizzate a questo punto per verificare l'efficienza della trasfezione. È possibile aggiungere una nuova porzione di supporto DMEM e le cellule possono essere incubate per ulteriori 24 ore. Il rendimento Di LV è paragonabile se raccolto a4 e 72 h punti di tempo dopo la trasfezione. - Centrifugare il mezzo a 3.000 x g per 5 min e temperatura ambiente per rimuovere le cellule staccate.
- Filtrare il super-natante (0,45 m) e versarlo in nuovi tubi.
NOTA: questo passaggio può essere omesso. - Aggiungere DNase I (senza RNase, 1 g/mL) e MgCl2 (1 mM) e incubare in un bagno d'acqua a 37 gradi centigradi per 15 min.
- Trasferire il mezzo ai tubi monouso in polietilene, e ultracentrifuga in un rotore oscillante a 115.000 x g e 4 s per 1,5 h.
- Dopo la centrifuga, scolare delicatamente le pareti dei tubi dai residui medi.
- Immergere il pellet con una salina sterile con buffer fosfato (PBS; 70-80 l per tubo).
- Incubare per 2 ore a 4-8 gradi centigradi.
- Risospendere i vettori virali in PBS con un pipettaggio delicato.
AVVISO: Evitare la schiuma. - Trasferire su un tubo centrifuga di 1,5 ml e centrifugare a 7.000 x g e 4 gradi centigradi per 30 s. Trasferire il supernatante in un nuovo tubo. Ripetere questo passaggio fino a quando non è visibile alcun pellet di detriti cellulari.
- Aliquota e congelare a -80 gradi centigradi. Evitare di ricongelare l'aliquota LV.
- Quarantotto ore dopo la trasfezione, raccogliere il mezzo che contiene particelle DiV. Utilizzare tubi conici da 50 ml.
- Determinazione del titro del virus utilizzando la reazione a catena quantitativa della polimerasi
NOTA: la titolazione dei vettori virali viene eseguita utilizzando la PCR quantitativa (qPCR). Questo metodo si basa sull'amplificazione di un frammento di DNA lungo 84 bp a doppio filamento all'interno della regione di ripetizione del terminale lungo del genoma virale15.- Preparare la curva standard effettuando diluizioni seriali del plasmide codifica LV: 1:500, 1:1,000, 1:5.000, 1:10.000, 1:100.000 e 1:1.000.000. Determinare il numero di copie del plasmide utilizzato per la curva standard. Utilizzare la seguente formula: numero di copie/zL (concentrazione [g/l] x 6,02 x 1023 [numero/mol]) / (660 [g/mol] x plasmid size [bp]), dove 6,02 x 1023 numero/mol è il numero di Avogadro e 660 g/mol è il peso bp.
NOTA: è possibile utilizzare calcolatori di numeri di copia online. - Preparare le diluizioni della sospensione lentivirale: 1:100, 1:500 e 1:1,000.
- Preparare la miscela di reazione (volumi per pozzo): 10 l di qPCR Mastermix, 1 : L di 10 M primer avanti, 1 L di 10M inverso e 7 -L di H2O. Pipette la miscela nei pozzetti di piastre di 96 pozzetti.
NOTA: Primer in avanti: 5'-AGCTTGCCTTGAGTGCTTCA. Primer inverso: 5'-TGACTAAAAGGGGCTGGGGGGGGA. - Aggiungere in triplice sospensione standard di diluizione e lentivirale di 1 L di ciascuna diluizione standard e sospensione lentivirale.
- Eseguire il qPCR secondo i seguenti parametri: 50 s per 2 min, 96 s per 5 min, e 35 cicli di 96 s per 20 s, 60 C per 40 s e 70 s per 1 min, seguito da fase di curva di fusione: 95 s per 1 min e 60 C a 30 s.
- Analizzare i risultati confrontando il numero di molecole ricevute per ogni diluizione con la curva standard. Determinare la concentrazione di molecole vettoriali come la media di tre repliche per ogni diluizione.
NOTA: La quantificazione presentata dà la concentrazione fisica delle particelle virali. Non deve essere trattato come un titro funzionale.
- Preparare la curva standard effettuando diluizioni seriali del plasmide codifica LV: 1:500, 1:1,000, 1:5.000, 1:10.000, 1:100.000 e 1:1.000.000. Determinare il numero di copie del plasmide utilizzato per la curva standard. Utilizzare la seguente formula: numero di copie/zL (concentrazione [g/l] x 6,02 x 1023 [numero/mol]) / (660 [g/mol] x plasmid size [bp]), dove 6,02 x 1023 numero/mol è il numero di Avogadro e 660 g/mol è il peso bp.
2. Generazione di ratti transgenici
- Superovulation e raccolta di embrioni fecondati
- Amministrare gonadotropine.
NOTA: Per aumentare il numero di embrioni raccolti (circa 30 per femmina), utilizzare femmine Wistar immature di 5 settimane per la stimolazione ormonale.- Il primo giorno (12:00-13:00), iniettare intraperitonealmente la gonadotropina del siero della cavalla incinta (PMSG; 25 IU per femmina). Preparare 1 mL aliquote di soluzione di lavoro ad una concentrazione di 125 IU/mL sciogliendo polvere ormonale in 0.9% NaCl. Conservare a -20 gradi centigradi per un massimo di 1 mese o -80 gradi centigradi per un massimo di 6 mesi.
- Il giorno 3 (12 PM-1PM), iniettare intraperitonealmente gonadotrofina colionica umana (hCG; 30 IU per femmina). Preparare 1 mL aliquote di soluzione di lavoro (150 IU/mL) sciogliendo polvere ormonale in 0.9% NaCl. Conservare a -20 gradi centigradi per un massimo di 1 mese o -80 gradi centigradi per un massimo di 6 mesi.
- Dopo l'amministrazione hCG, mate femmine 1:1 con maschi sessualmente fertili (3-10 mesi).
- La mattina successiva (giorno 4 alle 8-10), controllare le femmine per la presenza di un tappo vaginale. Controllare l'apertura vaginale per la presenza di un tappo di accoppiamento whitish, che per la migliore visualizzazione deve essere controllato la mattina presto dopo la notte di accoppiamento. Per la raccolta degli embrioni, utilizzare solo femmine con un tappo visibile.
- Raccogliere gli embrioni alle 10:00. Sacrificare gli animali per accisare gli ovidotti e raccogliere gli ovidotti in un piatto con mezzo M2 preriscaldato.
- Trasferire gli ovidotti in un piatto da 35 mm che contiene un mezzo M2 preriscaldato con ialuronidasi da testicoli bovini ad una concentrazione di 0,5 mg/mL.
- Aprire le pareti dell'ovulo utilizzando pinze sottili sotto uno stereomicroscopio e premere l'ampulla (cioè, la parte gonfia dell'ovuldotto che contiene embrioni fecondati che sono circondati da cellule cumuliche) fino a quando gli embrioni vengono liberati.
NOTA: L'ialuronidasi digerisce enzymaticamente le cellule cumuli, rilasciando embrioni.
AVVISO: l'esposizione prolungata all'ialuronidasi è deleteria per gli embrioni; pertanto, questo passaggio non dovrebbe durare più di 5 min. - Per facilitare il rilascio di embrioni da cellule cumuli, pipette delicatamente su e giù utilizzando una pipetta di trasferimento di vetro che è collegato a un tubo aspiratore a bocca.
- Per produrre la pipetta di trasferimento, tirare una pasteurtta di vetro sopra una fiamma per produrre una punta dritta di 5-10 cm. Rompere la pipetta lasciando una punta di 4 cm.
- Lavare gli embrioni un paio di volte in M2 mezzo per rimuovere ialuronidasi e detriti cellulari. Trasferire gli embrioni in un piatto di 60 mm che contiene (50 dollari l) gocce di mezzo M16 pre-equilibrato, coperto da paraffina liquida o olio minerale, in un incubatore umido di 37 gradi centigradi con un'atmosfera di CO2 del 5%.
- Amministrare gonadotropine.
- Microiniezione di vettori lentivirali a embrioni monocellulare sotto la zona pellucida
NOTA: Utilizzare embrioni a uno stadio con due pronuclei visibili per microiniezione (Figura 1).- Scongelare l'aliquota LV a temperatura ambiente e centrifugare a 10.000 x g e RT per 2 min a pellet eventuali detriti cellulari rimanenti.
- Configurazione microiniezione
- Preparare pipette di vetro (borosilicate vetro capillare) utilizzando una microforgia. Tirare il vetro capillare su una fiamma per produrre una punta di 5-10 cm. Rompere la pipetta lasciando una punta di 4 cm. Il diametro esterno dovrebbe essere di 80-120 m.
NOTA: Assicurarsi che la punta della pipetta sia perfettamente dritta e liscia. - Assemblare la pipetta tirata in una microforgiera con la punta di fronte al filamento riscaldante. Riscaldare il filamento molto vicino alla punta della pipetta e lasciarlo ridurre ad un diametro di 15 m (circa il 20% delle dimensioni dell'embrione). Posizionare la pipetta perpendicolarmente al filamento riscaldante, 2-3 mm dalla punta della pipetta, e iniziare a riscaldarsi. Il vetro si ammorbidisce. Riscaldare fino a raggiungere un angolo di 15 gradi.
- Preparare i capillari di vetro di microiniezione con un filamento utilizzando un tiratore pipetta. Inserire il capillare nella camera di trazione. Eseguire un test di rampa (per la prima volta per il vetro nuovo e ogni volta dopo aver cambiato il filamento). Impostare il valore Calore -10, Tira su 100, Velocità su 150 e Tempo su 100.
NOTA: Modificare i parametri per ottenere un'iniezione ottimale capillare. - Sotto un cappuccio a flusso laminare biosicurezza, caricare circa 2 ll della soluzione virale nella pipetta di microiniezione con una punta di microloader.
- Preparare un piatto di microiniezione (coperchio da 60 mm piatto Petri) con una goccia di 100 -L di M2 medio (nel mezzo), coperto da paraffina liquida o olio minerale.
- Montare la pipetta di tenuta e microiniezione capillare che viene caricato con soluzione virale a un micromanipolatore e microiniezione piatto sotto un microscopio invertito.
- Preparare pipette di vetro (borosilicate vetro capillare) utilizzando una microforgia. Tirare il vetro capillare su una fiamma per produrre una punta di 5-10 cm. Rompere la pipetta lasciando una punta di 4 cm. Il diametro esterno dovrebbe essere di 80-120 m.
- Eseguire la microiniezione.
- Trasferire 15-20 embrioni a uno stadio a una goccia di M2 sulla parabola di microiniezione. Tenere l'embrione usando una pipetta di possesso.
- Utilizzando l'ingrandimento 400x, iniettare la soluzione LV sotto la zona pellucida allo spazio perivitelline utilizzando il capillare di vetro collegato ad un iniettore automatico. Tieni il capillare sotto la zona pellucida per un momento.
NOTA: Utilizzando una leggera pressione positiva, la soluzione virale fluirà continuamente fuori dall'iniezione capillare, ma il volume delle sospensioni che viene consegnato non può essere controllato. - Utilizzando una pipetta fine, riportare gli embrioni al piatto di coltura nell'incubatrice a 37 gradi centigradi in un'atmosfera di CO2 del 5%. Il numero di iniezioni di uno zigote può variare e può essere adattato in base alla concentrazione vettoriale virale.
NOTA: gli embrioni iniettati possono essere trasferiti alle madri affidatarie allo stadio a una cellula o incubate O/N nel mezzo M16 prima di essere trasferiti allo stadio a due cellule. Si dovrebbe evitare una coltura prolungata in vitro degli embrioni di ratto.
- Trasferimento di embrioni iniettati alle madri affidate
- Preparare le madri adottive accoppiando femmine SD sessualmente mature con maschi fertili di BN o con maschi SD vasectomizzati (la procedura vasectomia è descritta nella sezione 3 di seguito) il giorno 3 (per il trasferimento di embrioni in fase a una cellula) o il giorno 4 (per il trasferimento di embrioni nella fase a due cellule).
NOTA: Per il trasferimento di ovidotti, utilizzare 0,5 giorni dopo il coitum (dpc) femmine. - La mattina successiva, controllare le femmine SD per un tappo vaginale, e utilizzare solo quelli con una spina visibile.
- Eseguire il trasferimento dell'embrione.
NOTA: Condurre la procedura chirurgica con strumenti sterili sotto uno stereomicroscopio. Prima del giorno dell'intervento, forbici autoclave, pinze sottili, porta aghi e portabissi.- Anestesizzare una femmina con somministrazione i.p. di ketamina (50 mg/kg) e soluzione di medetomidina (0,5 mg/kg). Testare i riflessi per confermare l'anestesia prima di iniziare la procedura chirurgica.
- Iniettare sottocutaneamente l'animale con acido tolefenamico (2 mg/kg), maabbaororato (1 mg/kg) e enrofloksacin (5-10 mg/kg) rispettivamente per prevenire infiammazioni, dolore e infezione.
- Applicare la lubrificazione dell'unguento oftalmico su entrambi gli occhi per evitare l'essiccazione corneale. Rasare la pelliccia dalla parte posteriore e sterilizzare la pelle con scrub chirurgico seguito dal 70% di alcol utilizzando pastiglie sterili non adere. Lasciare asciugare la pelle.
- Iniettare l'animale sottocutaneo con 100 -L di 0,25% bupivacaina (anestetico locale) nel sito di incisione. Trasferire l'animale in una posizione prona su una superficie pulita su un pad di riscaldamento sotto l'obiettivo di un microscopio chirurgico. Coprire il ratto con un drappo sterile con un piccolo foro tagliato sulla parte bassa della schiena.
- Eseguire un'incisione cutanea di circa 2 cm, parallela alla colonna vertebrale lombare.
- Utilizzando forbici affilate, fare un taglio nella parete addominale. Prendi un cuscinetto di grasso ovarico con pinze, e tira fuori l'ovaio e l'ovidotto e mettili su garza che è bagnato con 0.9% NaCl.
- Aspirare mezzo M2, tre bolle d'aria, e gli embrioni nel trasferimento capillare. Numero totale raccomandato di embrioni da trasferire (unilaterali o bilaterali): femmina incinta (15-16 embrioni), femmina pseudoincinta (30 embrioni).
- Fare una piccola incisione nell'ovulo (tra l'infundibulum e l'ampulla) utilizzando microforbici e inserire la pipetta di trasferimento nell'ovidotto.
- Espellere delicatamente gli embrioni e le bolle d'aria dalla pipetta all'ovidotto. Con pinze smussate, posizionare il tratto riproduttivo nella cavità addominale.
- Suturare la parete addominale con suture assorbenti dell'acido poliglicolico e chiudere l'incisione della pelle con clip di ferita. A seconda del numero di embrioni disponibili, ripetere questa procedura per l'altro ovidotto.
- Iniettare l'animale intraperitonenalmente con atipamezolo (0,5 mg/kg) per invertire l'effetto dell'anestesia.
- Trasferire l'animale in una gabbia pulita e tenerlo su una piastra riscaldante per recuperare completamente dall'anestesia. La consegna nei ratti avviene dopo 21 giorni.
NOTA: Quando i ratti BN maschi vengono utilizzati per l'accoppiamento, solo i cuccioli bianchi sono potenzialmente transgenici; cuccioli marroni sono da gravidanza naturale. - Raccogliere frammenti di tessuto (preferibilmente dall'orecchio) al genotipo cuccioli di 3 settimane.
- Preparare le madri adottive accoppiando femmine SD sessualmente mature con maschi fertili di BN o con maschi SD vasectomizzati (la procedura vasectomia è descritta nella sezione 3 di seguito) il giorno 3 (per il trasferimento di embrioni in fase a una cellula) o il giorno 4 (per il trasferimento di embrioni nella fase a due cellule).
3. Vasectomia
NOTA: Prima del giorno dell'intervento, forbici autoclave, pinze sottili e portaa ago.
- Anestesizzare un ratto SD maschio di 5 settimane con soluzione di somministrazione i.p. di ketamina (50 mg/kg) e medetomidina (0,5 mg/kg). Testare i riflessi per confermare l'anestesia prima di iniziare la procedura chirurgica.
- Somministrare l'acido tolfenamico (2 mg/kg), mailintoolo tartrate (1 mg/kg) e enrofloksacin (5-10 mg/kg) sottocutaneamente per prevenire l'infiammazione, dolore, e infezione, rispettivamente.
- Applicare la lubrificazione dell'unguento oftalmico su entrambi gli occhi per evitare l'essiccazione corneale. Posizionare la supina del ratto su una superficie pulita su un pad di riscaldamento e sterilizzare la pelle sui testicoli con scrub chirurgico seguito dal 70% di alcol utilizzando pastiglie sterili non aderiscono. Lasciare asciugare la pelle. Coprire il ratto con un drappo sterile con un piccolo foro tagliato sopra i testicoli. Premere delicatamente l'addome per esporre i testicoli nel sacco scrotale.
- Utilizzando forbici chirurgiche, fare un'incisione di 0,5 cm al centro del sacco scrotale. Individuare la parete della linea mediana (linea biancastra) tra i testicoli.
- Fare un'incisione di 5 mm nella membrana del testicolo vicino al lato sinistro della parete mediana.
- Spingere con attenzione i testicoli a sinistra e individuare vas deferens (tra il teste e la linea mediana) come condotto bianco con un singolo vaso sanguigno.
- Tirare delicatamente i vas deferens fuori dal sacco scrotale utilizzando le pinze di un orologiaio. Tenere il vas deferens con una coppia di pinze, e tagliarlo con forbici sottili (o cauterizzare con punte rosso-caldo di una seconda coppia di pinze). Rimuovere un frammento di 1 cm del condotto.
NOTA: Se viene eseguita la cauterizzazione, tenere la punta della seconda coppia di pinze nella fiamma. - Ripetere la procedura di cui sopra per gli altri testiri. Suturare la pelle con suture assorbenti dell'acido poliglicolico e iniettare all'animale intraperitonelmente atipamezolo (0,5 mg/kg).
- Mettere il ratto in una gabbia pulita su una piastra riscaldante fino a quando l'animale non si riprende dall'anestesia.
NOTA: I maschi possono essere utilizzati negli accoppiamenti di prova dopo un periodo di recupero di 2 settimane. Dopo la sterilità è confermata, possono essere utilizzati per l'induzione pseudo-gravidanza.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Utilizzando il protocollo qui descritto, sono stati prodotti vettori lentivirali che trasportavano il costrutto Syn-TDP-43-eGFP (titro lv fisico 3,4 x 108/L) e quindi potrebbero essere utilizzati per iniezioni di sottozonali embrionali a una cella. Solo gli embrioni con due pronuclei visibili sono stati sottoposti alla procedura. Il numero di iniezioni di sospensioni virali è stato determinato sperimentalmente. L'elevata efficienza dell'impianto e la mancanza simultanea di prole transgenica sono stati considerati indicatori di un numero insufficiente di particelle virali per una trasduzione di successo. In questo caso, il numero di iniezioni è stato aumentato. La singola somministrazione di LV portò alla nascita di 20 ratti di generazione F0, nessuno dei quali era transgenico. Un aumento del numero di iniezioni di un ordine di grandezza non ha provocato la nascita di ratti, ma il 100% degli embrioni sviluppati fino allo stadio a due cellule. Negli esperimenti successivi, il numero di iniezioni è stato aumentato di uno rispetto al valore per il quale è stata ottenuta la prole. Per la variante di due iniezioni sono nati otto ratti, tre dei quali sono stati confermati per trasportare il transgene (riassunto nella tabella 1). Uno dei fondatori non ha trasferito il transgene alla prole. Il numero di embrioni che sono stati iniettati e trasferiti in ogni variante sperimentale era 48 nelle varianti LV x1 e LV x2 e 45 in LV x10. Per ogni configurazione sperimentale sono state utilizzate tre femmine affidatarie. L'approccio scelto ha permesso la generazione di linee di ratto transgenico stabili che esprimevano la proteina di fusione TDP-43-eGFP sotto il controllo del promotore neuronale Synapsin-1 in tutto il sistema nervoso centrale (Figura 2A,B)14. La transgenesi basata sul lentivirus ha portato all'inserimento di una singola copia del transgene, come dimostrato da qPCR (Figura 2C).
Nella configurazione sperimentale descritta in precedenza, il tasso di sopravvivenza degli embrioni iniettati era del 95%. Risultati simili sono stati ottenuti quando lo stesso metodo è stato utilizzato per altri vettori lentivirali, come riepilogato nella tabella 2. La percentuale di embrioni sopravvissuti alle iniezioni pronucleari è stata significativamente inferiore (29-45%). La Tabella 2 riassume i risultati rappresentativi dell'efficienza di impianto degli zigoti manipolati, considerando il trasferimento di femmine pseudoincinte e incinte. L'uso di embrioni non manipolati insieme a embrioni iniettati è stato precedentemente riportato16. I nostri risultati complessivi suggeriscono che i ratti femmine incinte possono essere utilizzati come madri adottivi con efficienza comparabile. Abbiamo ottenuto una percentuale simile di impianto di embrioni estranei in ratti in gravidanza e pseudopregnantmente (media generale per diverse configurazioni sperimentali: 15% contro 16%). Tuttavia, il tasso di impianto era più alto quando gli embrioni subivano una manipolazione più sottile, il che significa un'iniezione subzonale (10% contro 21%). In particolare, i dati numerici analizzati per singoli cicli di microiniezione hanno indicato che l'efficacia dell'impianto dipendeva dal numero di iniezioni di un embrione(tabella 1, ultima colonna) e dipendeva indirettamente dal carico virale.
| Vettore | numero di iniezioni/embrioni | numero di embrioni iniettati | numero di cuccioli | numero di madri affidatarie | numero di fondatori transgenici | Efficienza di impianto per ogni variante |
| Syn-TDP-43WTLV | 1 | 48 | 20 | 3 | 0 | 42% |
| 10 | 45 | 0 | 3 | 0 | 0% | |
| 2 | 48 | 8 | 3 | 3 | 17% |
Tabella 1: Riepilogo del numero di iniezioni subzonali di zigoti convettori lentivirali Syn-TDP-43WT.
| Metodo | Vettore | Titro / Concentrazione | Numero di embrioni iniettati | Embrioni sopravvissuti | Tasso di sopravvivenza | Numero di madri affidatarie | Numero di cuccioli | Efficienza dell'impianto | Gravidanza (P) /Pseudogravidanza (PP) |
| Pni | TTYH1-Thy1-EGFP | 1 ng/L | 1083 | 424 | 39% | 16 | 54 | 13% | Pp |
| Pni | H3mCherry | 0,5-2 ng/L | 2229 | 647 | 29% | 29 | 67 | 10% | Pp |
| Pni | Syn-TDP-43-A315T | 2 ng/L | 1256 | 562 | 45% | 31 | 42 | 7% | Pp |
| Lv | Syn-TDP-43-A315T | 8,7 x 108 | 115 | 106 | 92% | 7 | 18 | 17% | P |
| Lv | Syn-TDP-43 WT | 3,4 x 108 | 152 | 141 | 93% | 9 | 28 | 20% | P |
| Lv | LVH3mcherry | 1,3 x 107 | 504 | 450 | 89% | 13 | 115 | 26% | Pp |
Tabella 2: Tasso di sopravvivenza dell'embrione e dell'efficienza dell'impianto, a seconda del metodo di iniezione utilizzato e della gravidanza rispetto all'induzione pseudo-gravidanza. PNI, iniezione pronucleare; LV, iniezione subzonale vettoriale vettoriale di lentivirale.

Figura 1: Fotografia microscopica dell'embrione di ratto a una cellula preparato per l'iniezione di vettore lentivirale subzonale. L'embrione è stato immobilizzato con una pipetta. Sono visibili due pronuclei che contenevano materiale genetico materno e paterno e il corpo polare. Barra della scala: 20 m. Fare clic qui per visualizzare una versione più grande di questa figura.
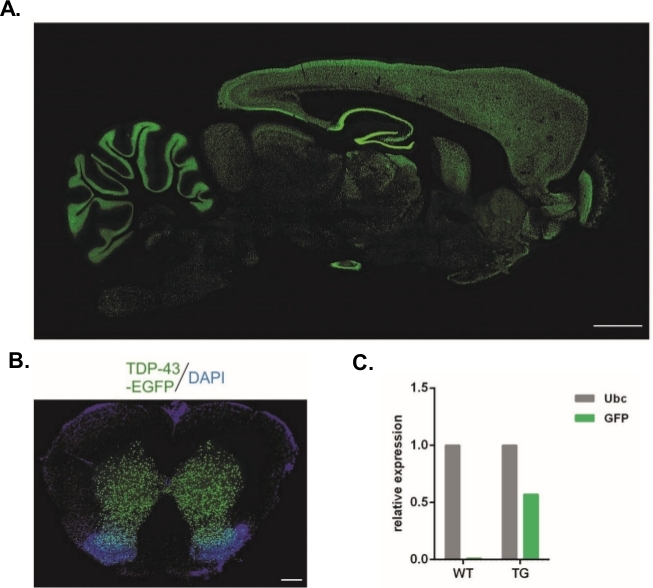
Figura 2: Generazione di linee di ratto transgenico stabili che hanno espresso la proteina di fusione TDP-43-eGFP sotto il controllo del promotore neuronale Synapsin-1 in tutto il sistema nervoso centrale. (A) Sinapsina-1 (Syn)-driven hTDP-43-eGFP modello di espressione in una sezione sagittale del cervello di ratto transgenico. La barra della scala 3 mm.(B) La sezione coronale del midollo spinale di un ratto transgenico in cui la fluorescenza eGFP, contrastata con DAPI, era limitata alla materia grigia del midollo spinale. Barra della scala: 250 m. (C) Espressione relativa della trascrizione transgenica GFP rispetto alla trascrizione di riferimento dell'ubiquitina C. n - 2 caratteri selvatici. n - 2 transgenico. La cifra è stata modificata a partire da14. Fare clic qui per visualizzare una versione più grande di questa figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
I progressi nelle tecnologie transgeniche hanno reso i modelli di roditori uno strumento inestimabile nella ricerca biomedica. Essi offrono l'opportunità di studiare le relazioni genotipo-fenotipo in vivo. Qui, presentiamo un'alternativa ampiamente disponibile per la transgenesi convenzionale mediante iniezioni pronucleari. L'uso della trasduzione genica lentivirale bypassa la necessità di microiniezioni impegnative perché i vettori virali possono essere iniettati sotto la zona pellucida. Questo approccio non influisce sull'integrità degli embrioni, che garantisce essenzialmente un tasso di sopravvivenza del 100% per gli zigoti iniettati. Il transgene che viene incorporato per mezzo di vettori lentivirali è stabilmente integrato nel genoma ospite, consentendo l'espressione a lungo termine e la trasmissione germinale. Inoltre, presentiamo due tecniche alternative per il trasferimento di embrioni modificati per le madri affidatarie. Una tecnica utilizza il trasferimento di embrioni a femmine pseudoincinte che sono precedentemente preparate accoppiandosi con maschi sterili vasectomizzati. L'altra tecnica si basa sull'uso di femmine naturalmente incinte che sono accoppiate con maschi fertili ma con un colore di pelliccia diverso (cioè, ratti BN). Questo corso di gravidanza più fisiologico permette il corretto sviluppo di embrioni che subiscono difficili modifiche genetiche16.
I primi tentativi riusciti di generare ratti transgenici sono stati segnalati nel 19907. Tuttavia, a causa delle difficoltà nella transgenesi dei ratti17, negli ultimi decenni 9 è stato generato un numero relativamente piccolo di linee di ratto transgenico.9 Diverse differenze principali sono osservate tra la transgenesi di topi e ratti utilizzando microiniezioni. Per i ratti, le linee principalmente di razza (ad esempio, Wistar e SD) sono utilizzate per la transgenesi. Per i topi, i ricercatori utilizzano principalmente la razza F1 di ceppi inbred a causa della loro maggiore fertilità, una migliore risposta alla superovulazione ormonale e uno sviluppo relativamente facile degli embrioni in vitro dallo stadio unicellulare alle blastocisti18. L'induzione della superovulazione nei ratti è molto meno efficiente rispetto ai topi che utilizzano la stimolazione ormonale standard PMSG/hCG. Per questo motivo, sono stati fatti tentativi di sviluppare protocolli alternativi per amministrare questi ormoni in ratti che utilizzano l'infusione continua di FSH invece di una singola somministrazione PMSG19. Tuttavia, superovulazione che è causata da PMSG/hCG o FSH/hCG ha dimostrato di avere efficienza comparabile20. A nostro parere, il fattore più critico che influenza l'efficacia della superovulazione è l'età delle femmine selezionate. Tuttavia, i parametri esatti devono essere testati per ogni ceppo di ratto, laboratorio, ecc.
La procedura per l'iniezione di una soluzione di DNA nel pronucleus di un embrione unicellulare è simile per entrambe le specie di roditori. Tuttavia, i pronuclei degli zigoti del ratto non hanno forme regolari come nei topi e tendono ad essere più difficili da definire nel citoplasma della cellula. Inoltre, la membrana cellulare dello zigote ratto e la membrana pronucleare sono più elastiche e viscose, complicando così l'inserimento di una micropipetta di vetro che viene caricata con la soluzione del DNA. Questi fattori portano a tassi di sopravvivenza degli ovuli di ratto più bassi dopo la microiniezione (31–65% contro 80% nei topi) e spiegano la minore efficienza transgenesi nei ratti9. Inoltre, l'intensa manipolazione meccanica dell'embrione può anche influenzare l'efficienza dell'impianto, che in molti laboratori, incluso il nostro, raggiunge un massimo del 10%. Questa resa relativamente bassa si osserva anche dopo l'impianto di un numero appropriato di embrioni21.
Un metodo che supera le difficoltà di cui sopra è l'infezione di embrioni unicellulari con retrovirus. I retrovirus contengono materiale genetico sotto forma di RNA, che al momento dell'ingresso nella cellula infetta viene trascritto nel DNA mediante trascrizione inversa del virus. Il DNA viene quindi trasportato attraverso i pori nucleari al nucleo cellulare, dove si integra nel genoma della cellula sotto forma di provirus. I vettori lentivirali sono stati utilizzati per generare topi transgenici e ratti12,14,22. Gli embrioni unicellulari che non hanno una zona pellucida possono essere incubati in una soluzione con un vettore lentivirale, oppure il vettore può essere iniettato sotto la zona pellucida nello spazio perivitelline. Il vantaggio principale di questo metodo è la sua efficienza estremamente elevata, raggiungendo oltre l'80% della prole transgenica. Dopo l'infezione con il vettore lentivirale, molte copie in siti diversi possono integrarsi nel genoma dello zigote, in contrasto con il metodo della transgenesi mediante microiniezione pronucleare, in cui un sito di integrazione è di solito osservato12. Nella prole del fondatore transgenico che viene fatta utilizzando vettori lentivirali, le singole copie del transgene sono segregate, che possono essere manifestate da diversi profili di espressione del transgene in ciascuna della progenie. Tuttavia, questo può aumentare la possibilità di ricevere un soggetto con il profilo di espressione desiderato che deriva dal transgene. Le restrizioni si applicano principalmente alla dimensione del transgene, che è limitata a circa 8 kb23.
Un'altra difficoltà nella transgenesi del ratto è la generazione di femmine che fungono da madri surrogate per embrioni geneticamente modificati. Nella procedura standard, le femmine sono incrociate con maschi vasectomizzati sterili per indurre la pseudogravidanza. Nei ratti, la tecnica di valutazione della pseudogravidanza è molto più difficile che nei topi, quindi la stimolazione con il gonadotropino che rilascia l'ormone agonista viene talvolta utilizzata pochi giorni prima dell'accoppiamento con i maschi. Per questi motivi, nel protocollo descritto forniamo due approcci alternativi per ottenere le madri affidate. L'efficienza complessiva di impianto degli zigoti manipolati quando vengono utilizzate femmine incinte o pseudoincinte è simile. Tuttavia, la presenza di embrioni naturali non manipolati insieme a quelli manipolati può migliorare il tasso di gravidanza16. Sebbene la differenza principale nel tasso di impianto sia la tecnica di manipolazione (cioè PNI contro LV, 10% contro 20%; vedi tabella 2), l'uso di femmine pseudoincinte come madri adottive può essere utile per alcuni esperimenti.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
L'autore (W.K.) ha i diritti sul brevetto, "Metodo di produzione di un animale transgenico", dall'ufficio brevetti della Repubblica di Polonia (n. P 355353; 21.03.2008).
Acknowledgments
Questo studio è stato sostenuto dal progetto ANIMOD nell'ambito del programma Team Tech Core Facility Plus della Fondazione per la scienza polacca, cofinanziato dall'Unione europea nell'ambito del Fondo europeo di sviluppo regionale per WK.
Materials
| Name | Company | Catalog Number | Comments |
| 7500 Real Time PCR System | Applied Biosystems | ||
| Aerrane (isoflurane) | Baxter | FDG9623 | |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma | A5177-5EA | |
| Atipam 5 mg/ml | Eurovet Animal Health BV | N/A | 0.5 mg/kg |
| Baytril 25 mg/ml (enrofloksacin) | Bayer | N/A | 5-10 mg/kg |
| Borosilicate glass capillaries with filament GC100TF-15 | Harvard Apparatus Limited | 30-0039 | injection capillary |
| Bupivacaine 25 mg/ml | Advanz Pharma | N/A | 0.25% in 0.9% NaCl |
| Butomidor 10 mg/ml (butorphanol tartrate) | Orion Pharma | N/A | 1 mg/kg |
| CELLSTAR Tissue Cell Culture Dish 35-mm | Greiner Bio-One | 627160 | |
| CELLSTAR Tissue Cell Culture Dish 60-mm | Greiner Bio-One | 628160 | |
| CellTram Oil | Eppendorf | 5176 000.025 | |
| Cepetor (Medetomidine) 1 mg/ml | cp-pharma | N/A | 0.5 mg/kg |
| Chorulon, Human Chorionic Gonadotrophin | Intervet | N/A | 150 IU/ ml ml 0.9% NaCl |
| DMEM low glucose | Sigma Aldrich | D6048 | |
| DNase, RNase-free | A&A Biotechnology | 1009-100 | |
| EmbryoMax Filtered Light Mineral Oil | Sigma | ES-005-C | |
| Envelope protein coding plasmid for lentiviral vectors (VSVg plasmid) | ADDGENE | 14888 | |
| FemtoJet | Eppendorf | 4i /5252 000.013 | |
| Fetal Bovine Serum | Sigma Aldrich | F9665-500ML | |
| Folligon, Pregnant Mare’s Serum Gonadotropin | Intervet | N/A | 125 IU/ml in .9% NaCl |
| HEK 293T cells | ATCC | ATCC CRL-3216 | |
| Hyaluronidase from Bovine Testis | Sigma | H4272-30MG | 0.5 mg/ml in M2 medium |
| Inverted Microscope | Zeiss | Axiovert 200 | |
| Ketamine 100mg/ml | Biowet Pulawy | N/A | 50 mg/kg |
| Liquid Paraffin | Merck Millipore | 8042-47-5 | |
| M16 medium EmbryoMax | Sigma | MR-016-D | |
| M2 medium | Sigma | M7167 | |
| Magnesium Chloride 1M | Sigma Aldrich | 63069-100ML | |
| Microforge | Narishige | MF-900 | |
| Mineral Oil | Sigma | M8410-500ML | |
| NaCl 0.9% | POLPHARMA OTC | N/A | sterile, 5ml ampules |
| Operation microscope | Inami Ophthalmic Instruments | Deca-21 | |
| Packaging system coding plasmid for lentiviral vectors (delta R8.2 plasmid) | ADDGENE | 12263 | |
| PEI reagent (Polyethylenimine, Mw ~ 25,000,), | Polysciences, Inc | 23966-1 | |
| Penicilin-streptomycin | Sigma Aldrich | P0781-100ML | |
| Phosphate Buffered Saline, pH 7.4, liquid, sterile-filtered, suitable for cell culture | Sigma Aldrich | 806552-500ML | |
| Puller | Sutter Instrument Co. | P-97 | |
| Reflex Clip Applier/Reflex Clips | World Precision Instruments | 500345/500346 | |
| Safil, polyglycolic acid, braided, coated, absorbable threads | B.Braun Surgical | 1048029 | |
| Stereomicroscope | Olympus | SZX16 | |
| Surgical Sewing Thread | B.Braun | C1048040 | |
| SYBR Green PCR Master Mix | Applied Biosystem | 4334973 | |
| Tolfedine 4% (tolfenamic acid) | Vetoquinol | N/A | 2 mg/kg |
| TransferMan NK2 | Eppendorf | N/A | |
| Trypsin EDTA solution | Sigma Aldrich | T3924-500ML | |
| Ultracentrifuge | Beckman Coulter | Optima L-100 XP | |
| VacuTip | Eppendorf | 5175108.000 | holders capillary |
| Vita-POS | Ursapharm | N/A | eye ointment |
| Warming Plate | Semic | N/A | |
| Watchmaker Forceps | VWR | 470018-868 |
References
- Lazar, J., Moreno, C., Jacob, H. J., Kwitek, A. E. Impact of genomics on research in the rat. Genome Research. 15 (12), 1717-1728 (2005).
- Tarkowski, A. K. Studies on mouse chimeras developed from eggs fused in vitro. National Cancer Institute Monographs. 11, 51-71 (1963).
- Gordon, J. W., Ruddle, F. H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science. 214 (4526), 1244-1246 (1981).
- Gill, T. J., Smith, G. J., Wissler, R. W., Kunz, H. W. The Rat as an Experimental Animal. Science. 245 (4915), 269-276 (1989).
- Aitman, T. J., et al. Progress and prospects in rat genetics: a community view. Nature Genetics. 40 (5), 516-522 (2008).
- Hammer, R. E., Maika, S. D., Richardson, J. A., Tang, J. P., Taurog, J. D. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 63 (5), 1099-1112 (1990).
- Mullins, J. J., Peters, J., Ganten, D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature. 344 (6266), 541-544 (1990).
- Menoret, S., Remy, S., Usal, C., Tesson, L., Anegon, I. Generation of Transgenic Rats by Microinjection of Short DNA Fragments. Rat Genomics: Methods and Protocols. 597, 81-92 (2010).
- Tesson, L., et al. Transgenic modifications of the rat genome. Transgenic Research. 14 (5), 531-546 (2005).
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: Technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Ritchie, W. A., Neil, C., King, T., Whitelaw, C. B. Transgenic embryos and mice produced from low titre lentiviral vectors. Transgenic Research. 16 (5), 661-664 (2007).
- Lois, C., Hong, E. J., Pease, S., Brown, E. J., Baltimore, D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 295 (5556), 868-872 (2002).
- Pfeifer, A., Ikawa, M., Dayn, Y., Verma, I. M. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proceedings of the National Academy of Sciences of the United States of America. 99 (4), 2140-2145 (2002).
- Koza, P., et al. Neuronal TDP-43 depletion affects activity-dependent plasticity. Neurobiology of Disease. 130, 104499 (2019).
- Scherr, M., Battmer, K., Blomer, U., Ganser, A., Grez, M. Quantitative determination of lentiviral vector particle numbers by real-time PCR. Biotechniques. 31 (3), 520 (2001).
- Canseco, R. S., et al. Gene transfer efficiency during gestation and the influence of co-transfer of non-manipulated embryos on production of transgenic mice. Transgenic Research. 3 (1), 20-25 (1994).
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Brinster, R. L., Chen, H. Y., Trumbauer, M. E., Yagle, M. K., Palmiter, R. D. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proceedings of the National Academy of Sciences of the United States of America. 82 (13), 4438-4442 (1985).
- Armstrong, D. T., Opavsky, M. A. Superovulation of immature rats by continuous infusion of follicle-stimulating hormone. Biology of Reproduction. 39 (3), 511-518 (1988).
- Popova, E., Krivokharchenko, A., Ganten, D., Bader, M. Comparison between PMSG- and FSH-induced superovulation for the generation of transgenic rats. Molecular Reproduction and Development. 63 (2), 177-182 (2002).
- Johnson, L. W., Moffatt, R. J., Bartol, F. F., Pinkert, C. A. Optimization of embryo transfer protocols for mice. Theriogenology. 46 (7), 1267-1276 (1996).
- van den Brandt, J., Wang, D., Kwon, S. H., Heinkelein, M., Reichardt, H. M. Lentivirally generated eGFP-transgenic rats allow efficient cell tracking in vivo. Genesis. 39 (2), 94-99 (2004).
- Remy, S., et al. The Use of Lentiviral Vectors to Obtain Transgenic Rats. Rat Genomics: Methods and Protocols. 597, 109-125 (2010).
