

Summary
Este artigo tem como objetivo fornecer a metodologia para transgênese lentiviral em embriões de ratos usando múltiplas injeções de uma suspensão do vírus no espaço de perivitelline zigogote. Ratos fêmeas que são acasalados com uma cepa masculina fértil com uma cor de pele dominante diferente é usado para gerar mães adotivas pseudográvidas.
Abstract
Modelos animais transgênicos são fundamentalmente importantes para a pesquisa biomédica moderna. A incorporação de genes estrangeiros em embriões de camundongos ou ratos primitivos é uma ferramenta inestimável para a análise da função genética em organismos vivos. O método de transgênese padrão é baseado na microinjeção de fragmentos de DNA estranho susceptante em um pronúcleo de um oócito fertilizado. Esta técnica é amplamente utilizada em camundongos, mas permanece relativamente ineficiente e tecnicamente exigente em outras espécies animais. O transgene também pode ser introduzido em embriões em estágio unicelular através de infecção lentiviral, fornecendo uma alternativa eficaz às injeções pronuclears padrão, especialmente em espécies ou cepas com uma estrutura de embriões mais desafiadora. Nesta abordagem, uma suspensão que contém vetores lentivirais é injetada no espaço perivitelline de um embrião de rato fertilizado, que é tecnicamente menos exigente e tem uma taxa de sucesso maior. Os vetores lentivirais mostraram incorporar eficientemente o transgene no genoma para determinar a geração de linhas transgênicas estáveis. Apesar de algumas limitações (por exemplo, requisitos de Biossegurança Nível 2, limites de tamanho do fragmento de DNA), a transgênese lentiviral é um método de transgênese rápida e eficiente. Além disso, o uso de ratos fêmeas que são acasalados com uma cepa masculina fértil com uma cor de pele dominante diferente é apresentado como uma alternativa para gerar mães adotivas pseudográvidas.
Introduction
Por muitos anos, roedores de laboratório, como ratos e camundongos, têm sido usados para modelar condições fisiológicas e patológicas humanas. A pesquisa em animais levou a descobertas que eram inatingíveis por qualquer outro meio. Inicialmente, estudos genéticos se concentraram na análise de distúrbios e fenótipos de ocorrência espontânea que são considerados para imitar de perto a condição humana1. O desenvolvimento de métodos de engenharia genética permitiu a introdução ou exclusão de genes específicos para obter um fenótipo desejado. Portanto, a geração de animais transgênicos é reconhecida como uma técnica fundamental na pesquisa moderna que permite estudos da função genética em organismos vivos.
A tecnologia animal transgênica tornou-se possível através de uma combinação de realizações em embriologia experimental e biologia molecular. Na década de 1960, o embriologista polonês A. K. Tarkowski publicou o primeiro trabalho sobre manipulação de embriões de camundongos durante os estágios iniciais do desenvolvimento2. Além disso, biólogos moleculares desenvolveram técnicas para gerar vetores de DNA (ou seja, portadores) para a introdução entre outros de DNA estranho no genoma do animal. Esses vetores permitem a propagação de genes selecionados e sua modificação apropriada, dependendo do tipo de pesquisa que é realizada. O termo "animal transgênico" foi introduzido por Gordon e Ruddle3.
A primeira espécie amplamente aceita que foi usada em neurobiologia, fisiologia, farmacologia, toxicologia e muitos outros campos das ciências biológicas e médicas foi o rato norueguês Rattus norvegicus4. No entanto, devido à dificuldade em manipular embriões de ratos, o rato musculus da casa tornou-se a espécie animal dominante na pesquisa genética5. Outra razão para a primazia do camundongo em tal pesquisa foi a disponibilidade de tecnologia de células-tronco embrionárias para gerar animais eliminados para esta espécie. A técnica mais utilizada de transgênese (2-10% dos descendentes transgênicos em relação a todos os animais nascidos) é a microinjeção de fragmentos de DNA em um pronúcleo de um oócito fertilizado. Em 1990, essa abordagem, que foi introduzida pela primeira vez em camundongos, foi adaptada para ratos6,7. A transgênese de ratos por injeção pronuclear é caracterizada pela menor eficiência8 em relação aos camundongos, o que está estritamente relacionado com a presença de plasma elástico emembranaspronuclear9 . Embora a sobrevivência dos embriões após a manipulação seja 40-50% menor do que em camundongos, essa técnica é considerada um padrão na geração de ratos geneticamente modificados10. Abordagens alternativas que podem garantir a incorporação transgênica eficiente e maiores taxas de sobrevivência de zigotes injetados têm sido investigadas.
O principal determinante da expressão transgênica estável e transmissão à prole é sua integração ao genoma celular hospedeiro. Os lentivírus (LVs) têm a característica distinta de serem capazes de infectar células divisórias e não divisórias. Seu uso como ferramenta para a incorporação de genes heterólogos em embriões provou ser altamente eficiente11, e os indivíduos transgênicos são caracterizados pela expressão estável do fragmento de DNA incorporado. A eficácia dos vetores lentivirais foi confirmada para a modificação genética de camundongos12,13, ratos12,14, e outras espécies11. Neste método, a suspensão lv é injetada sob a zona pellucida do embrião na fase de dois pronucleis. Esta técnica essencialmente garante 100% de sobrevivência dos embriões porque o oolema permanece inalterado. A produção de suspensões lv de alta qualidade e relativamente altamente concentradas são fatores cruciais. No entanto, concentrações mais baixas de suspensões de LV podem ser superadas por injeções repetidas11, o que aumenta a quantidade de partículas virais na superfície do ovo, sem afetar a integração da membrana. Embriões que são submetidos a injeções repetidas no espaço perivitelline desenvolvem-se ainda mais, e descendentes transgênicos podem transmitir o transgene através da germinação. A eficiência da geração transgênica de ratos por transgênese lentiviral pode ser de até 80%12.
Aqui, descrevemos a produção de lentivírus recombinante derivado do HIV-1 que foi pseudodigitado com proteína envelope de estomatite vesicular (VSV) G. O uso do pseudotipo VSV de segunda geração determina a ampla infectividade das partículas virais e permite a produção de vetores altamente estáveis que podem ser concentrados por ultracentrifugação e criopreservação. Após a verificação do título, os vetores estão prontos para serem usados como um veículo para a entrega de transgênicos em zigotes de rato Albino Wistar. Após uma série de injeções, os embriões podem ser cultivados durante a noite e transferidos no estágio de duas células para mães adotivas. Neste ponto, uma das duas abordagens alternativas pode ser considerada. O procedimento padrão utiliza fêmeas pseudográvidas como receptores de embriões. No entanto, quando a taxa de gravidez é baixa após o acasalamento com machos vasectomizados, os embriões podem ser implantados em fêmeas grávidas wistar/sprague-dawley (SD) que são acasaladas com ratos machos férteis com uma cor de pele escura (por exemplo, ratos da Noruega Marrom [BN)." A cor da pele permite a distinção da prole da gravidez natural de descendentes que se originam dos embriões manipulados transferidos.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
A produção e aplicação de vetores virais estava de acordo com as diretrizes do Nível 2 de Biossegurança e foi aprovada pelo Ministério do Meio Ambiente polonês. Todos os procedimentos experimentais em animais descritos abaixo foram aprovados pelo Comitê Deética Local. Os animais foram alojados em gaiolas ventiladas individualmente a uma temperatura estável (21-23 °C) e umidade (50-60%) com acesso a ad libitum à água e alimentos sob um ciclo de luz/escuridão de 12 h/12 h.
1. Produção de vetores lentivirais
- Transfecção de células HEK 293T
NOTA: O protocolo aqui apresentado é projetado para a transfecção de vinte pratos de cultura Ø10 cm que produz aproximadamente 200 mL de sobrenadante vetorial bruto.- Células DE Cultura HEK 293T no meio DMEM que é suplementada com soro bovino fetal (10%, v/v) em uma incubadora de CO2 umidificada a 37 °C. Para a transfecção, prepare vinte placas de 10 cm de diâmetro e sementes 1,5-2 x 106 HEK 293T células por prato.
- Quando a confluência atinge ~70%, transfectas as células que utilizam reagente de polietilenoina (PEI), pH 7.0, a uma razão de 3 μg de PEI por 1 μg de DNA.
- Prepare a mistura de transfecção para cinco pratos (prepare o número de repetições de acordo com o número total de pratos). A 1 mL do Meio Águia Modificada de Dulbecco (DMEM; sem soro), adicione a mistura de três plasmídeos para que atinjam uma quantidade final de 25 μg de plasmídeo VSVg, 50 μg de delta R8.2 e 50 μg de plasmídeo de codificação.
- Pipeta para cima e para baixo, e adicione 125 μL de PEI a uma concentração de 3 μg/μL. Incubar em temperatura ambiente por 15 min, invertendo o tubo três vezes durante a incubação. Adicione 200 μL da mistura de transfecção por placa. Em seguida, incubar as placas em uma incubadora de CO2 umidificada a 37 °C.
- Concentração de vetores lentivirais
- Quarenta e oito horas após a transfecção, colher o meio que contém partículas lv. Use tubos cônicos de 50 mL.
NOTA: Ao usar um plasmídeo com uma etiqueta fluorescente, as células podem ser visualizadas neste momento para verificar a eficiência da transfecção. Uma nova porção do meio DMEM pode ser adicionada, e as células podem ser incubadas por mais 24 horas. O rendimento de LV é comparável quando coletado nos pontos de tempo de 48 e 72 h após a transfecção. - Centrifugar o meio a 3.000 xg por 5 min e temperatura ambiente para remover células separadas.
- Filtre o sobrenadante (0,45 μm) e despeje-o em novos tubos.
NOTA: Esta etapa pode ser omitida. - Adicione DNase I (livre de RNase, 1 μg/mL) e MgCl2 (1 mM), e incubar em banho-maria a 37 °C por 15 min.
- Transfira os tubos de polietileno médio para descartáveis e a ultracentrífuga em um rotor oscilante a 115.000 x g e 4 °C por 1,5 h.
- Após o centrifugação, drene suavemente as paredes dos tubos dos resíduos médios.
- Mergulhe a pelota com solução salina tampoada por fosfato estéril (PBS; 70-80 μL por tubo).
- Incubar por 2 h a 4-8 °C.
- Resuspenda os vetores virais na PBS por pipetting suave.
ATENÇÃO: Evite espumar. - Transfira para um tubo de centrífuga de 1,5 mL e centrífuga a 7.000 x g e 4 °C por 30 s. Transfira o sobrenadante para um novo tubo. Repita este passo até que nenhuma pelota de detritos celulares esteja visível.
- Alíquota e congele a -80 °C. Evite recongelar a alíquota lv.
- Quarenta e oito horas após a transfecção, colher o meio que contém partículas lv. Use tubos cônicos de 50 mL.
- Determinação do título de vírus usando reação quantitativa da cadeia de polimerase
NOTA: A titulação dos vetores virais é realizada utilizando PCR quantitativo (qPCR). Este método baseia-se na amplificação de um fragmento de DNA de 84 bp de comprimento de dupla retinência dentro da longa região de repetição terminal do genoma viral15.- Prepare a curva padrão fazendo diluições seriais do plasmídeo codificador de LV: 1:500, 1:1.000, 1:5.000, 1:10.000, 1:100.000 e 1:1.000.000. Determine o número de cópias do plasmídeo usado para a curva padrão. Use a seguinte fórmula: número de cópias/μL = (concentração [g/μL] x 6,02 x 1023 [número/mol]) / (660 [g/mol] x tamanho plasmídeo [bp]), onde 6,02 x 1023 número/mol é o número de Avogadro, e 660 g/mol é o peso da bp.
NOTA: Podem ser utilizadas calculadoras de números de cópia on-line. - Preparar diluições da suspensão lentiviral: 1:100, 1:500 e 1:1.000.
- Prepare a mistura de reação (volumes por poço): 10 μL de qPCR Mastermix, 1 μL de 10 μM Forward primer, 1 μL de 10 μM de primer reverso e 7 μL de H2O. Pipeette a mistura nos poços de 96-well placas.
NOTA: Primer dianteiro: 5'-AGCTTGCCTTGAGTGCTTCa. Primer reverso: 5'-TGACTAAAAGGGTCTGAGGGA. - Adicione 1 μL de cada diluição padrão e suspensão lentiviral em triplicado.
- Execute o qPCR de acordo com os seguintes parâmetros: 50 °C para 2 min, 96 °C para 5 min e 35 ciclos de 96 °C para 20 s, 60 °C para 40 s e 70 °C para 1 min, seguido de estágio de curva de derretimento: 95 °C para 1 min e 60 °C a 30 s.
- Analise os resultados comparando o número de moléculas recebidas para cada diluição à curva padrão. Determine a concentração de moléculas vetores como a média de três réplicas para cada diluição.
NOTA: A quantificação apresentada proporciona a concentração física das partículas virais. Não deve ser tratado como um título funcional.
- Prepare a curva padrão fazendo diluições seriais do plasmídeo codificador de LV: 1:500, 1:1.000, 1:5.000, 1:10.000, 1:100.000 e 1:1.000.000. Determine o número de cópias do plasmídeo usado para a curva padrão. Use a seguinte fórmula: número de cópias/μL = (concentração [g/μL] x 6,02 x 1023 [número/mol]) / (660 [g/mol] x tamanho plasmídeo [bp]), onde 6,02 x 1023 número/mol é o número de Avogadro, e 660 g/mol é o peso da bp.
2. Geração de ratos transgênicos
- Superovulação e coleta de embriões fertilizados
- Administre gonadotropinas.
NOTA: Para aumentar o número de embriões coletados (aproximadamente 30 por fêmea), use fêmeas Wistar imaturas de 5 semanas de idade para estimulação hormonal.- No dia 1 (12:00-1PM), injete intraperitoneicamente o soro de égua grávida gonadotropina (PMSG; 25 UI por fêmea). Preparar alíquotas de 1 mL de solução de trabalho em uma concentração de 125 UI/mL dissolvendo o pó hormonal em 0,9% naCl. Armazene a -20 °C por até 1 mês ou -80 °C por até 6 meses.
- No dia 3 (12 PM-1 PM), injete intraperitoneicamente gonadotrophina coriônica humana (hCG; 30 UI por fêmea). Preparar alíquotas de 1 mL de solução de trabalho (150 UI/mL) dissolvendo o pó hormonal em 0,9% de NaCl. Armazene a -20 °C por até 1 mês ou -80 °C por até 6 meses.
- Após a administração do HCG, mate fêmeas 1:1 com machos sexualmente férteis (3-10 meses de idade).
- Na manhã seguinte (dia 4 às 8-10 am), verifique as fêmeas para a presença de um plug vaginal. Verifique a abertura vaginal para a presença de um plugue de acasalamento esbranquiçado, que para melhor visualização deve ser verificado no início da manhã após a noite de acasalamento. Para a coleta de embriões, utilize apenas fêmeas com um plugue visível.
- Coletar embriões às 10 da manhã. Sacrifique os animais para extirpar os ovidutos, e colete os ovidutos em um prato com meio M2 pré-aquecido.
- Transfira os ovidutos para um prato de 35 mm que contenha meio M2 pré-aquecido com hialuronidase de testículos bovinos a uma concentração de 0,5 mg/mL.
- Abra as paredes do oviduto usando pinças finas sob um microscópio enomicroscópio e pressione a ampulla (ou seja, a parte inchada do oviduto que contém embriões fertilizados que estão cercados por células cumulus) até que os embriões sejam liberados.
NOTA: A hialuronidase digere enzimáticamente as células cumulus, liberando embriões.
ATENÇÃO: A exposição prolongada à hialuronidase é deletério aos embriões; portanto, este passo não deve durar mais do que 5 min. - Para facilitar a liberação de embriões de células cumulus, tubos suavemente para cima e para baixo usando uma pipeta de transferência de vidro que é conectada a um tubo aspirador operado pela boca.
- Para produzir a pipeta de transferência, puxe uma pipeta pasteur de vidro sobre uma chama para produzir uma ponta reta ~5-10 cm. Quebre a pipeta deixando uma ponta de ~4 cm.
- Lave os embriões algumas vezes em meio M2 para remover os detritos hialuronidase e celular. Transfira os embriões para um prato de 60 mm que contenha (~50 μL) gotas de meio M16 pré-equilibrado, coberto por parafina líquida ou óleo mineral, em uma incubadora de 37 °C umidificada com uma atmosfera de 5% de CO2.
- Administre gonadotropinas.
- Microinjeção de vetores lentivirais em embrião em estágio unicelular sob a zona pellucida
NOTA: Use embriões em estágio unicelular com duas pronucéis visíveis para microinjeção (Figura 1).- Descongele a alíquota LV à temperatura ambiente e centrífuga a 10.000 x g e RT por 2 min para pellet quaisquer detritos celulares restantes.
- Configuração de microinjeção
- Prepare as pipetas de retenção de vidro (capilar de vidro borossilicato) usando uma microforge. Puxe o vidro capilar sobre uma chama para produzir uma ponta de 5 a 10 cm. Quebre a pipeta deixando uma ponta de ~4 cm. O diâmetro externo deve ser de ~80-120 μm.
NOTA: Certifique-se de que a ponta da pipeta é perfeitamente reta e lisa. - Monte a pipeta puxada em uma microforge com a ponta na frente do filamento de aquecimento. Aqueça o filamento muito perto da ponta da pipeta e deixe-o encolher para um diâmetro de ~15 μm (aproximadamente 20% do tamanho do embrião). Posicione a pipeta perpendicularmente ao filamento de aquecimento, a 2-3 mm da ponta da pipeta, e comece a aquecer. O vidro vai amolecer. Aqueça até atingir um ângulo de 15°.
- Prepare capilares de vidro borosilicato de microinjeção com um filamento usando um puxador de pipeta. Insira o capilar na câmara de puxar. Execute um teste de rampa (pela primeira vez para vidro novo e todas as vezes depois de trocar o filamento). Defina o Calor para o valor de rampa -10, Puxe para 100, Velocity para 150 e Time para 100.
NOTA: Modifique os parâmetros para obter um capilar de injeção ideal. - Sob uma coifa de fluxo laminar biossafety, carregue aproximadamente 2 μL da solução viral na pipeta de microinjeção com uma ponta de microcarregador.
- Prepare uma placa de microinjeção (tampa de placa de Petri de 60 mm) com uma gota de 100 μL de médio M2 (no meio), coberta por parafina líquida ou óleo mineral.
- Monte a pipeta de retenção e o capilar de microinjeção que é carregado com solução viral para um micromanipulador e microinjeção prato sob um microscópio invertido.
- Prepare as pipetas de retenção de vidro (capilar de vidro borossilicato) usando uma microforge. Puxe o vidro capilar sobre uma chama para produzir uma ponta de 5 a 10 cm. Quebre a pipeta deixando uma ponta de ~4 cm. O diâmetro externo deve ser de ~80-120 μm.
- Faça a microinjeção.
- Transfira 15-20 embriões de estágio unicelular para a queda de M2 na antena de microinjeção. Segure o embrião usando uma pipeta de retenção.
- Utilizando a ampliação de 400x, injete a solução LV sob a zona pellucida para o espaço perivitelline usando o capilar de vidro que é conectado a um injetor automático. Segure o capilar sob a zona pellucida por um momento.
NOTA: Usando pressão positiva suave, a solução viral fluirá continuamente para fora da capilar de injeção, mas o volume da suspensão que é entregue não pode ser controlado. - Usando uma pipeta fina, devolva os embriões ao prato de cultura na incubadora a 37 °C em uma atmosfera de 5% de CO2. O número de injeções de um zigoto pode variar e pode ser adaptado com base na concentração de vetores virais.
NOTA: Os embriões injetados podem ser transferidos para mães adotivas em estágio unicelular ou O/N incubados em meio M16 antes de serem transferidos no estágio de duas células. A cultura in vitro prolongada dos embriões de ratos deve ser evitada.
- Transferência de embriões injetados para mães adotivas
- Preparar mães adotivas acasalando fêmeas sd sexualmente maduras com machos fértil sd ou com machos sd vasectomizados (o procedimento de vasectomia é descrito na seção 3 abaixo) no dia 3 (para transferência de embriões em estágio de uma célula) ou dia 4 (para transferência de embriões no estágio de duas células).
NOTA: Para a transferência de ovidutos, use 0,5 dias pós-coito (dpc) femininos. - Na manhã seguinte, verifique as fêmeas da SD para um plugvaginal, e use apenas aquelas com um plugue visível.
- Realizar transferência de embriões.
NOTA: Conduzir o procedimento cirúrgico com instrumentos estéreis sob um microscópio estereomicroscópio. Antes do dia da cirurgia, tesoura autoclave, fórceps finos, porta-agulhas e suporte de bisturi.- Anestesiar uma fêmea com i.p. administração de cetamina (50 mg/kg) e medetomidina (0,5 mg/kg) solução. Teste para reflexos para confirmar a anestesia antes de iniciar o procedimento cirúrgico.
- Injete o animal subcutâneamente com ácido tolefenâmico (2 mg/kg), tarrato de butorphanol (1 mg/kg) e enrofloksacina (5-10 mg/kg) para prevenir inflamação, dor e infecção, respectivamente.
- Aplique lubrificação de pomada oftálmica em ambos os olhos para evitar a secagem da córnea. Raspe a pele das costas e esterilize a pele com esfoliante cirúrgico seguido de 70% de álcool usando almofadas não aderentes estéreis. Deixe a pele secar.
- Injete o animal subcutâneamente com 100 μL de bupivacaína (anestésico local) no local da incisão. Transfira o animal em posição propensa a uma superfície limpa em uma almofada de aquecimento sob o objetivo de um microscópio cirúrgico. Cubra o rato com uma cortina estéril com um pequeno orifício cortado na parte inferior das costas.
- Realize uma incisão cutânea de aproximadamente 2 cm, paralela à coluna vertebral lombar.
- Usando uma tesoura afiada, faça um corte na parede abdominal. Pegue uma almofada de gordura ovariana usando fórceps, retire o ovário e o oviduto e coloque-os em gaze que é molhada com 0,9% de NaCl.
- Aspirar m2 médio, três bolhas de ar, e os embriões para a transferência capilar. Número total recomendado de embriões a serem transferidos (unilaterais ou bilaterais): fêmea grávida (≤ 15-16 embriões), fêmea pseudográvida (≤ 30 embriões).
- Faça uma pequena incisão no oviduto (entre o infíbulo e a ampulla) usando microtesoura, e insira a pipeta de transferência no oviduto.
- Expulse suavemente embriões e bolhas de ar da pipeta para o oviduto. Com fórceps contundentes, coloque o trato reprodutivo de volta na cavidade abdominal.
- Suturar a parede abdominal com suturas absorvíveis de ácido poliglicólico e fechar a incisão da pele com clipes de ferida. Dependendo do número de embriões disponíveis, repita este procedimento para o outro oviduto.
- Injete o animal intraperitonealmente com atipamezol (0,5 mg/kg) para reverter o efeito da anestesia.
- Transfira o animal para uma gaiola limpa e mantenha-o em uma placa de aquecimento para se recuperar totalmente da anestesia. A entrega em ratos ocorre após ~21 dias.
NOTA: Quando os ratos BN machos são usados para acasalamento, apenas os filhotes brancos são potencialmente transgênicos; filhotes marrons são de gravidez natural. - Coletar fragmentos de tecido (de preferência da orelha) para filhotes de 3 semanas de idade.
- Preparar mães adotivas acasalando fêmeas sd sexualmente maduras com machos fértil sd ou com machos sd vasectomizados (o procedimento de vasectomia é descrito na seção 3 abaixo) no dia 3 (para transferência de embriões em estágio de uma célula) ou dia 4 (para transferência de embriões no estágio de duas células).
3. Vasectomia
NOTA: Antes do dia da cirurgia, tesoura autoclave, fórceps finos e porta agulha.
- Anestesiar um rato SD macho de 5 semanas de idade com administração i.p. de cetamina (50 mg/kg) e medetomidina (0,5 mg/kg) solução. Teste para reflexos para confirmar a anestesia antes de iniciar o procedimento cirúrgico.
- Administrar ácido tolfenamico (2 mg/kg), tartar butorphanol (1 mg/kg) e enrofloksacina (5-10 mg/kg) subcutâneamente para prevenir inflamação, dor e infecção, respectivamente.
- Aplique lubrificação de pomada oftálmica em ambos os olhos para evitar a secagem da córnea. Coloque o supino de rato sobre uma superfície limpa em uma almofada de aquecimento, e esterilize a pele nos testículos com esfoliante cirúrgico seguido de 70% de álcool usando almofadas não aderentes estéreis. Deixe a pele secar. Cubra o rato com uma cortina estéril com um pequeno buraco cortado sobre os testículos. Pressione suavemente o abdômen para expor os testículos no saco escrotal.
- Usando uma tesoura cirúrgica, faça uma incisão de ~0,5 cm no meio do saco escrotal. Localize a parede da linha média (linha esbranquiçada) entre os testículos.
- Faça uma incisão de 5 mm na membrana de testículo perto do lado esquerdo da parede da linha média.
- Empurre cuidadosamente o testículo para a esquerda e localize vas deferens (entre o testículo e a linha média) como um ducto branco com um único vaso sanguíneo.
- Puxe suavemente os vasos deferentes do saco escrotal usando os fórceps de um relojoeiro. Segure os vasos deferentes com um par de fórceps, e corte-o com uma tesoura fina (ou cauterize com pontas vermelhas de um segundo par de fórceps). Remova um fragmento de ~1 cm do duto.
NOTA: Se a cauterização for realizada, segure a ponta do segundo par de fórceps na chama. - Repita o procedimento acima para os outros testículos. Suturar a pele com suturas absorvíveis de ácido poliglicólico e injetar o animal intraperitonealmente com atipamezol (0,5 mg/kg).
- Coloque o rato em uma gaiola limpa em uma placa de aquecimento até que o animal se recupere da anestesia.
NOTA: Os machos podem ser usados nos acasalamentos de teste após um período de recuperação de ~2 semanas. Depois que a esterilidade for confirmada, eles podem ser usados para indução de pseudogravidez.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Utilizando-se o protocolo descrito aqui, foram produzidos vetores lentivirais que carregavam o construto Syn-TDP-43-eGFP (título de LV físico = 3,4 x 108/μL) e, em seguida, poderiam ser utilizados para injeções subzonais de embriões em estágio de célula. Apenas embriões com duas pronucéis visíveis foram submetidos ao procedimento. O número de injeções de suspensões virais foi determinado experimentalmente. Alta eficiência de implantação e falta simultânea de descendentes transgênicos foram considerados indicadores de um número insuficiente de partículas virais para transdução bem sucedida. Neste caso, o número de injeções foi aumentado. A administração única de LV resultou no nascimento de 20 ratos da geração F0, nenhum dos quais eram transgênicos. Um aumento no número de injeções por uma ordem de magnitude não resultou no nascimento de ratos, mas 100% dos embriões desenvolvidos para o estágio de duas células. Em experimentos subsequentes, o número de injeções foi aumentado em uma em comparação com o valor para o qual os filhotes foram obtidos. Para a variante de duas injeções, nasceram oito ratos, três dos quais foram confirmados para transportar o transgene (resumido na Tabela 1). Um dos fundadores não transferiu o transgene para a prole. O número de embriões que foram injetados e transferidos em cada variante experimental foi de 48 nas variantes LV x1 e LV x2 e 45 em LV x10. Três fêmeas adotivas foram usadas para cada configuração experimental. A abordagem escolhida permitiu a geração de linhas de ratos transgênicos estáveis que expressavam a proteína de fusão TDP-43-eGFP sob controle do promotor neuronal Sinapsin-1 em todo o sistema nervoso central (Figura 2A,B)14. A transgênese baseada em lentivírus resultou em uma única inserção de cópia do transgene, como demonstrado pela qPCR (Figura 2C).
Na configuração experimental descrita acima, a taxa de sobrevivência dos embriões injetados foi de 95%. Resultados semelhantes foram obtidos quando o mesmo método foi utilizado para outros vetores lentivirais resumidos na Tabela 2. O percentual de embriões que sobreviveram às injeções pronucleares foi significativamente menor (29-45%). A Tabela 2 resume os resultados representativos da eficiência de implantação de zigotos manipulados, considerando a transferência de pseudogestantes versus gestantes. O uso de embriões não manipulados juntamente com embriões injetados foi previamente relatado16. Nossos resultados sugerem que ratos fêmeas grávidas podem ser usadas como mães adotivas com eficiência comparável. Obtivemos uma porcentagem semelhante de implantação de embriões estrangeiros em ratos gestantes e pseudogestantes (média geral para várias configurações experimentais: 15% vs. 16%). No entanto, a taxa de implantação foi maior quando os embriões foram submetidos a manipulação mais sutil, o que significa uma injeção subzonal (10% vs. 21%). Notavelmente, os dados numéricos analisados para rodadas individuais de microinjeção indicaram que a eficácia da implantação dependia do número de injeções de um embrião(Tabela 1,última coluna) e dependia indiretamente da carga viral.
| Vetor | número de injeções/embrião | número de embriões injetados | número de filhotes | número de mães adotivas | número de fundadores transgênicos | Eficiência de implantação para cada variante |
| Syn-TDP-43WTLV | 1 | 48 | 20 | 3 | 0 | 42% |
| 10 | 45 | 0 | 3 | 0 | 0% | |
| 2 | 48 | 8 | 3 | 3 | 17% |
Tabela 1: Resumo do número de injeções subzonais de zigotos comvetores lentivirais Syn-TDP-43WT.
| Método | Vetor | Titer/ Concentração | Número de embriões injetados | Embriões sobrevividos | Taxa de sobrevivência | Número de mães adotivas | Número de filhotes | Eficiência de implantação | Gravidez (P) /Pseudogravidez (PP) |
| Pni | TTYH1-Thy1-EGFP | 1 ng/μL | 1083 | 424 | 39% | 16 | 54 | 13% | Pp |
| Pni | H3mCherry | 0,5-2 ng/μL | 2229 | 647 | 29% | 29 | 67 | 10% | Pp |
| Pni | Syn-TDP-43-A315T | 2 ng/μL | 1256 | 562 | 45% | 31 | 42 | 7% | Pp |
| Lv | Syn-TDP-43-A315T | 8.7 x 108 | 115 | 106 | 92% | 7 | 18 | 17% | P |
| Lv | Syn-TDP-43 WT | 3.4 x 108 | 152 | 141 | 93% | 9 | 28 | 20% | P |
| Lv | LVH3mcherry | 1.3 x 107 | 504 | 450 | 89% | 13 | 115 | 26% | Pp |
Tabela 2: Taxa de sobrevivência do embrião e eficiência de implantação, dependendo do método de injeção utilizado e da gravidez versus indução pseudogravidez. PNI, injeção pronuclear; LV, injeção subzonal vetorial lentiviral.

Figura 1: Fotografia microscópica de embrião de rato em estágio de célula única que foi preparado para injeção de vetor lentiviral subzonal. O embrião foi imobilizado com uma pipeta de espera. Dois pronucleis que continham material genético materno e paterno e o corpo polar são visíveis. Barra de escala = 20 μm. Clique aqui para ver uma versão maior desta figura.
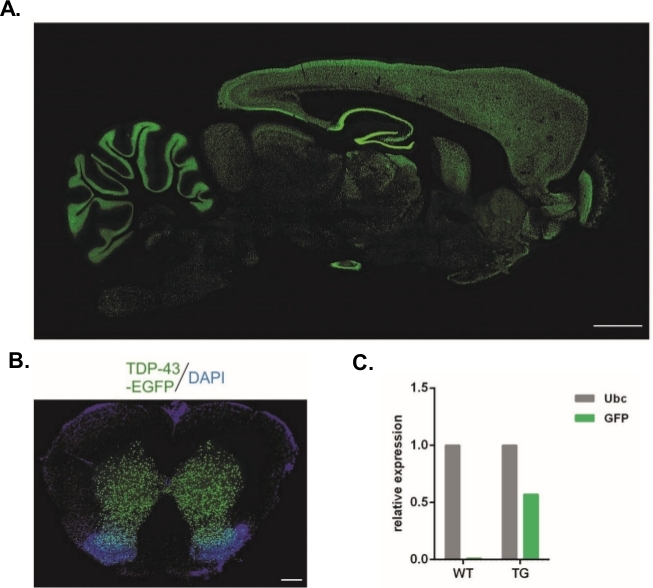
Figura 2: Geração de linhas de ratos transgênicos estáveis que expressaram a proteína de fusão TDP-43-eGFP sob controle do promotor neuronal Sinapsin-1 em todo o sistema nervoso central. (A) Padrão de expressão hTDP-43-eGFP orientado por sinapsina-1 em uma seção sagital do cérebro transgênico do rato. Barra de escala = 3 mm. (B) Seção coronal da medula espinhal de um rato transgênico onde a fluorescência eGFP, contrariada com DAPI, foi restrita à matéria cinzenta da medula espinhal. Barra de escala = 250 μm. (C) Expressão relativa da transcrição transgênica gfp em comparação com a transcrição de referência da ubiquitina C. n = 2 tipo selvagem. n = 2 transgênicos. A figura foi modificada a partir de14. Clique aqui para ver uma versão maior desta figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Os avanços nas tecnologias transgênicas tornaram os modelos de roedores uma ferramenta inestimável na pesquisa biomédica. Eles oferecem a oportunidade de estudar relações genótipo-fenótipo in vivo. Aqui, apresentamos uma alternativa amplamente disponível para transgênese convencional por injeções pronucleares. O uso da transdução do gene lentiviral ignora a necessidade de exigir microinjeções porque vetores virais podem ser injetados sob a zona pellucida. Essa abordagem não afeta a integridade do embrião, o que essencialmente garante uma taxa de sobrevivência de 100% para zigotes injetados. O transgene que é incorporado por meio de vetores lentivirais é integrado ao genoma hospedeiro, permitindo a expressão a longo prazo e a transmissão de germes. Além disso, apresentamos duas técnicas alternativas para transferência de embriões modificados para mães adotivas. Uma técnica utiliza a transferência de embriões para fêmeas pseudográvidas que são previamente preparadas por acasalamento com machos inférteis vasectomizados. A outra técnica baseia-se no uso de fêmeas naturalmente grávidas que são acasaladas com machos férteis, mas com uma cor de pele diferente (ou seja, ratos BN). Este curso mais fisiológico da gravidez permite o desenvolvimento adequado de embriões que sofrem modificações genéticas desafiadoras16.
As primeiras tentativas bem sucedidas de gerar ratos transgênicos foram relatadas em 19907. No entanto, devido às dificuldades na transgênese dos ratos17, um número relativamente pequeno de linhas transgênicas de ratos foram gerados nas últimas décadas9. Várias diferenças principais são observadas entre a transgênese do rato e do rato usando microinjeções. Para ratos, principalmente linhas de raça (por exemplo, Wistar e SD) são usadas para transgênese. Para camundongos, os pesquisadores usam principalmente o cruzamento de f1 de cepas de raça inbred devido à sua maior fertilidade, melhor resposta à superovulação hormonal e desenvolvimento relativamente fácil de embriões in vitro do estágio de uma célula para blastocistos18. A indução da superovulação em ratos é muito menos eficiente do que em camundongos que usam estimulação hormonal PMSG/hCG padrão. Por essa razão, foram feitas tentativas de desenvolver protocolos alternativos para administrar esses hormônios em ratos que utilizam infusão contínua de FSH em vez de uma única administração pmsg19. No entanto, a superovulação causada pelo PMSG/hCG ou FSH/hCG mostrou-se ter eficiência comparável20. Em nossa opinião, o fator mais crítico que afeta a eficácia da superovulação é a idade das fêmeas selecionadas. No entanto, os parâmetros exatos devem ser testados para cada cepa de rato, laboratório, etc.
O procedimento para injetar solução de DNA no pronúcleo de um embrião unicelular é semelhante para ambas as espécies de roedores. No entanto, os pronucleis de zigotes de rato não têm formas regulares como em camundongos e tendem a ser mais difíceis de definir no citoplasma da célula. Além disso, a membrana celular de zigoto de rato e a membrana pronuclear são mais elásticas e viscosas, complicando assim a inserção de uma micropipeta de vidro carregada com solução de DNA. Esses fatores levam a menores taxas de sobrevivência de ovos de rato após a microinjeção (31-65% vs. 80% em camundongos) e explicam a menor eficiência transgênese em ratos9. Além disso, a manipulação intensiva e mecânica do embrião também pode afetar a eficiência de implantação, que em muitos laboratórios, incluindo o nosso, atinge um máximo de 10%. Esse rendimento relativamente baixo é observado mesmo após a implantação de um número adequado de embriões21.
Um método que supera as dificuldades acima mencionadas é a infecção de embriões unicelulares com retrovírus. Os retrovírus contêm material genético na forma de RNA, que ao entrar na célula infectada é transcrito no DNA por transcrição reversa do vírus. O DNA é então transportado através dos poros nucleares para o núcleo celular, onde se integra ao genoma da célula na forma de um provírus. Os vetores lentivirais têm sido utilizados para gerar camundongos transgênicos e ratos12,14,22. Embriões unicelulares que não possuem uma zona pellucida podem ser incubados em uma solução com um vetor lentiviral, ou o vetor pode ser injetado sob a zona pellucida no espaço perivitelline. A principal vantagem deste método é sua eficiência extremamente alta, atingindo mais de 80% dos descendentes transgênicos. Após a infecção com o vetor lentiviral, muitas cópias em diferentes locais podem se integrar ao genoma do zigote, em contraste com o método de transgênese por microinjeção pronuclear, na qual um local de integração é geralmente observado12. Na prole do fundador transgênico que é feita usando vetores lentivirais, são segregadas cópias individuais do transgênico, que podem se manifestar por diferentes perfis de expressão do transgênico em cada uma das prole. No entanto, isso pode aumentar a chance de receber um sujeito com o perfil de expressão desejado que é derivado do transgene. As restrições aplicam-se principalmente ao tamanho do transgene, que é limitado a aproximadamente 8 kb23.
Outra dificuldade na transgênese de ratos é a geração de fêmeas que servem como mães de aluguel para embriões geneticamente modificados. No procedimento padrão, as fêmeas são cruzadas com machos vasectomizados estéreis para induzir a pseudogravidez. Em ratos, a técnica de avaliação da pseudogravidez é muito mais difícil do que em camundongos, por isso a estimulação com a gonadotropina liberando agonista hormonal é às vezes usada alguns dias antes do acasalamento com os machos. Por essas razões, no protocolo descrito fornecemos duas abordagens alternativas para a obtenção de mães adotivas. A eficiência geral de implantação de zigotos manipulados quando mulheres grávidas ou pseudográvidas são usadas é semelhante. No entanto, a presença de embriões naturais e não manipulados em conjunto com os manipulados pode melhorar a taxa de gravidez16. Embora a principal diferença na taxa de implantação seja a técnica de manipulação (ou seja, PNI vs. LV, 10% vs. 20%; ver Tabela 2), o uso de mulheres pseudográvidas como mães adotivas pode ser benéfico para alguns experimentos.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
O autor (W.K.) tem direito à patente, "Método de produção de um animal transgênico", do escritório de patentes da República da Polônia (nº 355353; 21.03.2008).
Acknowledgments
Este estudo foi apoiado pelo projeto ANIMOD dentro do programa Team Tech Core Facility Plus da Fundação para a Ciência Polonesa, co-financiado pela União Europeia no âmbito do Fundo Europeu de Desenvolvimento Regional para a WK.
Materials
| Name | Company | Catalog Number | Comments |
| 7500 Real Time PCR System | Applied Biosystems | ||
| Aerrane (isoflurane) | Baxter | FDG9623 | |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma | A5177-5EA | |
| Atipam 5 mg/ml | Eurovet Animal Health BV | N/A | 0.5 mg/kg |
| Baytril 25 mg/ml (enrofloksacin) | Bayer | N/A | 5-10 mg/kg |
| Borosilicate glass capillaries with filament GC100TF-15 | Harvard Apparatus Limited | 30-0039 | injection capillary |
| Bupivacaine 25 mg/ml | Advanz Pharma | N/A | 0.25% in 0.9% NaCl |
| Butomidor 10 mg/ml (butorphanol tartrate) | Orion Pharma | N/A | 1 mg/kg |
| CELLSTAR Tissue Cell Culture Dish 35-mm | Greiner Bio-One | 627160 | |
| CELLSTAR Tissue Cell Culture Dish 60-mm | Greiner Bio-One | 628160 | |
| CellTram Oil | Eppendorf | 5176 000.025 | |
| Cepetor (Medetomidine) 1 mg/ml | cp-pharma | N/A | 0.5 mg/kg |
| Chorulon, Human Chorionic Gonadotrophin | Intervet | N/A | 150 IU/ ml ml 0.9% NaCl |
| DMEM low glucose | Sigma Aldrich | D6048 | |
| DNase, RNase-free | A&A Biotechnology | 1009-100 | |
| EmbryoMax Filtered Light Mineral Oil | Sigma | ES-005-C | |
| Envelope protein coding plasmid for lentiviral vectors (VSVg plasmid) | ADDGENE | 14888 | |
| FemtoJet | Eppendorf | 4i /5252 000.013 | |
| Fetal Bovine Serum | Sigma Aldrich | F9665-500ML | |
| Folligon, Pregnant Mare’s Serum Gonadotropin | Intervet | N/A | 125 IU/ml in .9% NaCl |
| HEK 293T cells | ATCC | ATCC CRL-3216 | |
| Hyaluronidase from Bovine Testis | Sigma | H4272-30MG | 0.5 mg/ml in M2 medium |
| Inverted Microscope | Zeiss | Axiovert 200 | |
| Ketamine 100mg/ml | Biowet Pulawy | N/A | 50 mg/kg |
| Liquid Paraffin | Merck Millipore | 8042-47-5 | |
| M16 medium EmbryoMax | Sigma | MR-016-D | |
| M2 medium | Sigma | M7167 | |
| Magnesium Chloride 1M | Sigma Aldrich | 63069-100ML | |
| Microforge | Narishige | MF-900 | |
| Mineral Oil | Sigma | M8410-500ML | |
| NaCl 0.9% | POLPHARMA OTC | N/A | sterile, 5ml ampules |
| Operation microscope | Inami Ophthalmic Instruments | Deca-21 | |
| Packaging system coding plasmid for lentiviral vectors (delta R8.2 plasmid) | ADDGENE | 12263 | |
| PEI reagent (Polyethylenimine, Mw ~ 25,000,), | Polysciences, Inc | 23966-1 | |
| Penicilin-streptomycin | Sigma Aldrich | P0781-100ML | |
| Phosphate Buffered Saline, pH 7.4, liquid, sterile-filtered, suitable for cell culture | Sigma Aldrich | 806552-500ML | |
| Puller | Sutter Instrument Co. | P-97 | |
| Reflex Clip Applier/Reflex Clips | World Precision Instruments | 500345/500346 | |
| Safil, polyglycolic acid, braided, coated, absorbable threads | B.Braun Surgical | 1048029 | |
| Stereomicroscope | Olympus | SZX16 | |
| Surgical Sewing Thread | B.Braun | C1048040 | |
| SYBR Green PCR Master Mix | Applied Biosystem | 4334973 | |
| Tolfedine 4% (tolfenamic acid) | Vetoquinol | N/A | 2 mg/kg |
| TransferMan NK2 | Eppendorf | N/A | |
| Trypsin EDTA solution | Sigma Aldrich | T3924-500ML | |
| Ultracentrifuge | Beckman Coulter | Optima L-100 XP | |
| VacuTip | Eppendorf | 5175108.000 | holders capillary |
| Vita-POS | Ursapharm | N/A | eye ointment |
| Warming Plate | Semic | N/A | |
| Watchmaker Forceps | VWR | 470018-868 |
References
- Lazar, J., Moreno, C., Jacob, H. J., Kwitek, A. E. Impact of genomics on research in the rat. Genome Research. 15 (12), 1717-1728 (2005).
- Tarkowski, A. K. Studies on mouse chimeras developed from eggs fused in vitro. National Cancer Institute Monographs. 11, 51-71 (1963).
- Gordon, J. W., Ruddle, F. H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science. 214 (4526), 1244-1246 (1981).
- Gill, T. J., Smith, G. J., Wissler, R. W., Kunz, H. W. The Rat as an Experimental Animal. Science. 245 (4915), 269-276 (1989).
- Aitman, T. J., et al. Progress and prospects in rat genetics: a community view. Nature Genetics. 40 (5), 516-522 (2008).
- Hammer, R. E., Maika, S. D., Richardson, J. A., Tang, J. P., Taurog, J. D. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 63 (5), 1099-1112 (1990).
- Mullins, J. J., Peters, J., Ganten, D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature. 344 (6266), 541-544 (1990).
- Menoret, S., Remy, S., Usal, C., Tesson, L., Anegon, I. Generation of Transgenic Rats by Microinjection of Short DNA Fragments. Rat Genomics: Methods and Protocols. 597, 81-92 (2010).
- Tesson, L., et al. Transgenic modifications of the rat genome. Transgenic Research. 14 (5), 531-546 (2005).
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: Technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Ritchie, W. A., Neil, C., King, T., Whitelaw, C. B. Transgenic embryos and mice produced from low titre lentiviral vectors. Transgenic Research. 16 (5), 661-664 (2007).
- Lois, C., Hong, E. J., Pease, S., Brown, E. J., Baltimore, D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 295 (5556), 868-872 (2002).
- Pfeifer, A., Ikawa, M., Dayn, Y., Verma, I. M. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proceedings of the National Academy of Sciences of the United States of America. 99 (4), 2140-2145 (2002).
- Koza, P., et al. Neuronal TDP-43 depletion affects activity-dependent plasticity. Neurobiology of Disease. 130, 104499 (2019).
- Scherr, M., Battmer, K., Blomer, U., Ganser, A., Grez, M. Quantitative determination of lentiviral vector particle numbers by real-time PCR. Biotechniques. 31 (3), 520 (2001).
- Canseco, R. S., et al. Gene transfer efficiency during gestation and the influence of co-transfer of non-manipulated embryos on production of transgenic mice. Transgenic Research. 3 (1), 20-25 (1994).
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Brinster, R. L., Chen, H. Y., Trumbauer, M. E., Yagle, M. K., Palmiter, R. D. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proceedings of the National Academy of Sciences of the United States of America. 82 (13), 4438-4442 (1985).
- Armstrong, D. T., Opavsky, M. A. Superovulation of immature rats by continuous infusion of follicle-stimulating hormone. Biology of Reproduction. 39 (3), 511-518 (1988).
- Popova, E., Krivokharchenko, A., Ganten, D., Bader, M. Comparison between PMSG- and FSH-induced superovulation for the generation of transgenic rats. Molecular Reproduction and Development. 63 (2), 177-182 (2002).
- Johnson, L. W., Moffatt, R. J., Bartol, F. F., Pinkert, C. A. Optimization of embryo transfer protocols for mice. Theriogenology. 46 (7), 1267-1276 (1996).
- van den Brandt, J., Wang, D., Kwon, S. H., Heinkelein, M., Reichardt, H. M. Lentivirally generated eGFP-transgenic rats allow efficient cell tracking in vivo. Genesis. 39 (2), 94-99 (2004).
- Remy, S., et al. The Use of Lentiviral Vectors to Obtain Transgenic Rats. Rat Genomics: Methods and Protocols. 597, 109-125 (2010).
