

Summary
Ce protocole décrit une méthode non invasive pour identifier efficacement les cellules en phase S pour les études de microscopie en aval, telles que la mesure du recrutement des protéines de réparation de l’ADN par micro-irradiation laser.
Abstract
La réparation des dommages à l’ADN maintient l’intégrité génétique des cellules dans un environnement hautement réactif. Les cellules peuvent accumuler divers types de dommages à l’ADN dus à des sources endogènes et exogènes telles que les activités métaboliques ou le rayonnement UV. Sans réparation de l’ADN, le code génétique de la cellule devient compromis, sapant les structures et les fonctions des protéines et faisant potentiellement des maladies.
Comprendre la dynamique spatio-temporelle des différentes voies de réparation de l’ADN dans diverses phases du cycle cellulaire est crucial dans le domaine de la réparation des dommages à l’ADN. Les techniques actuelles de microscopie fluorescente fournissent d’excellents outils pour mesurer la cinétique de recrutement de différentes protéines de réparation après l’induction de dommages à l’ADN. La synthèse de l’ADN pendant la phase S du cycle cellulaire est un point particulier du destin cellulaire en ce qui concerne la réparation de l’ADN. Il fournit une fenêtre unique pour filtrer l’ensemble du génome à la recherche d’erreurs. Dans le même temps, les erreurs de synthèse de l’ADN constituent également une menace pour l’intégrité de l’ADN qui n’est pas rencontrée dans les cellules non divisées. Par conséquent, les processus de réparation de l’ADN diffèrent considérablement en phase S par rapport aux autres phases du cycle cellulaire, et ces différences sont mal comprises.
Le protocole suivant décrit la préparation de lignées cellulaires et la mesure de la dynamique des protéines de réparation de l’ADN en phase S sur des sites de dommages à l’ADN induits localement, à l’aide d’un microscope confocal à balayage laser équipé d’une ligne laser de 405 nm. Tagged PCNA (avec mPlum) est utilisé comme marqueur du cycle cellulaire combiné à une protéine réparatrice d’intérêt marquée AcGFP (c.-à-d. EXO1b) pour mesurer le recrutement des dommages à l’ADN en phase S.
Introduction
Plusieurs voies de réparation de l’ADN ont évolué pour traiter les différents types de lésions de l’ADN qui peuvent survenir dans les cellules, qui sont toutes fortement régulées dans l’espace et le temps. L’une des périodes les plus vulnérables du cycle cellulaire est la phase S, lorsque la synthèse de l’ADN se produit. Bien que la prolifération soit fondamentale pour la vie, elle constitue également un défi majeur. Les cellules doivent assurer une réplication fidèle de leur génome pour éviter que les mutations ne soient transmises aux générations futures. Par conséquent, la prolifération fournit un point d’intervention thérapeutique qui a été utilisé pour le développement d’approches thérapeutiques dans le domaine de l’oncologie.
Toutes les principales techniques utilisées pour étudier le recrutement des protéines au niveau des lésions de l’ADN ont leurs forces et leurs limites. La micro-irradiation a une meilleure résolution spatiale et temporelle1 que la plupart des méthodes alternatives comme l’imagerie immunofluorescente des foyers induits par les rayonnements ionisants (IRIF), l’immunoprécipitation de la chromatine (ChIP) ou le fractionnement biochimique. Cependant, la micro-irradiation instrait la robustesse des techniques susmentionnées qui permettent d’échantillonner un grand nombre de cellules en même temps.
Pour étudier la réparation de l’ADN en phase S, il faut être capable de distinguer les cellules en phase S dans une population de culture cellulaire asynchrone. Il existe de nombreuses méthodes bien connues pour résoudre ce problème, impliquant soit la synchronisation des cellules, soit la visualisation des différentes phases du cycle cellulaire. Cependant, les deux approches introduisent des défis importants et des artefacts possibles. Les méthodes de synchronisation chimique largement utilisées pour enrichir les cellules au début de la phase S (par exemple, le double bloc de thymidine, l’aphidicoline et le traitement à l’hydroxyurée) permettent d’obtenir une synchronisation par l’induction d’un stress de réplication et éventuellement de dommages à l’ADN lui-même. Cela limite l’utilisation de ces méthodes pour étudier les processus de réparation de l’ADN en phase S2. La synchronisation par famine et libération sérique n’est applicable qu’à un nombre limité de lignées cellulaires, excluant en grande partie les lignées cellulaires cancéreuses qui dépendent moins des facteurs de croissance pour la progression du cycle cellulaire que les lignées cellulaires non transformées. Le système FUCCI (Fluorescence Ubiquitin Cell Cycle Indicator) est un outil particulièrement utile pour étudier le cycle cellulaire, mais il présente une limitation fondamentale lors de la différenciation entre les phases du cycle cellulaire S et G23.
Ici, il est montré que l’utilisation de PCNA marqué par fluorescence comme marqueur non invasif pour la phase S limite les inconvénients des méthodes chimiques de synchronisation du cycle cellulaire, tout en permettant plus de spécificité et de flexibilité que le système FUCCI. En tant que marqueur unique, non seulement le PCNA peut mettre en évidence les cellules en phase S dans une population asynchrone, mais il peut également montrer la progression exacte des cellules dans la phase S (c’est-à-dire au début, au milieu ou à la fin de la phase S)4. De faibles niveaux d’expression de PCNA exogène marqué garantissent une interférence minimale avec la progression du cycle cellulaire et les processus de réparation de l’ADN. Il est important de se rendre compte que le PCNA sert également de contrôle interne pour l’induction correcte des dommages à l’ADN, car il est impliqué dans la réparation de plusieurs lésions de l’ADN et est recruté pour les sites de dommages à l’ADN induits localement1,4.
Les expériences présentées ici montrent comment mesurer la dynamique de recrutement d’EXO1b en phase S et comment celle-ci est affectée par l’inhibiteur bien établi de PARP, l’olaparib. L’activité de la nucléase EXO1b est pertinente pour un large éventail de voies de réparation de l’ADN, y compris la réparation des discordances (MMR), la réparation de l’excision nucléotidique (NER) et la réparation de la rupture double brin (DSB). En phase S, EXO1b joue un rôle majeur dans la recombinaison homologue (HR) par la formation de 3'ssDNA surplombs lors de la résection de l’ADN5. EXO1b a également été impliqué dans la réplication de l’ADN avec des rôles dans l’activation du point de contrôle pour redémarrer les fourches d’ADN bloquées ainsi que dans l’élimination de l’amorce et la maturation des fragments d’Okazaki au niveau du brin en retard lors du déplacement du brin dans la réplication5. Le recrutement d’EXO1b sur les sites d’ADN endommagés est régulé par l’interaction directe avec le poly (ADP-ribose) (PAR)6,7. En raison des nombreuses implications spécifiques au cycle cellulaire d’EXO1b, c’est un excellent choix pour les études de recrutement spécifiques à la phase S utilisant le PCNA.
Protocol
1. Culture de cellules dérivées de l’ostéosarcome humain (U-2 OS)
REMARQUE: Les cellules U-2 OS sont idéales pour ces études car elles ont une morphologie plate, un gros noyau et se fixent fortement à plusieurs surfaces, y compris le verre. D’autres lignées cellulaires présentant des caractéristiques similaires pourraient également être utilisées.
- Pour la culture de lignées cellulaires U-2 OS, utilisez le milieu 5A de McCoy complété par 10% de sérum bovin fœtal (FBS) et des antibiotiques (100 U / mL de pénicilline et 100 μg / mL de streptomycine). Incuber des cellules à 37 °C dans une atmosphère humidifiée contenant 5 % de CO2. Pour les études de microscopie, maintenir la culture cellulaire dans une boîte de 10 cm pour fournir un nombre suffisant de cellules.
- Lorsque les cellules approchent 90% de confluence (~ 7 x 106 cellules / plat de 10 cm), divisez les cellules.
- Rincez les cellules avec du PBS pour éliminer les inhibiteurs de la trypsine contenus dans le sérum.
- Ajouter 1 mL de trypsine-EDTA et s’assurer que la couche cellulaire est également couverte.
- Incuber à 37 °C jusqu’à ce que la couche cellulaire soit soulevée de la plaque (environ 6 min).
- Remettre en service les cellules trypsinisées dans un milieu contenant du sérum pour inactiver la trypsine et ajouter 1/10ème du volume (~0,7 x 106 cellules) dans une nouvelle plaque de 10 cm contenant 10 mL de milieu de croissance supplémenté.
- Avant l’expérimentation, tester régulièrement les cellules pour la contamination par les mycoplasmes à l’aide du kit de détection universelle des mycoplasmes en suivant les recommandations du fabricant.
2. Infection rétrovirale
REMARQUE: Pour les mesures de sécurité BSL-2 et lorsque vous travaillez avec des virus recombinants, veuillez vous référer à: NiH Guidelines, Section III-D-3: Virus recombinants en culture tissulaire.
- Ensemencez 4 x10 6 cellules HEK293T pour obtenir une confluence d’environ 60% en 24 h après le placage dans un plat de culture de 10 cm.
- Pour cultiver HEK293T, veuillez suivre les étapes de culture de U-2 OS décrites dans 1.1-1.3 de ce protocole. Pour HEK293T, remplacez le support 5A de McCoy par le DMEM. Assurez-vous de toujours laver doucement les cellules HEK293T car elles se fixent faiblement aux plaques de culture tissulaire.
- Transfectez les cellules HEK293T à l’aide d’un réactif de transfection à base de lipides pour l’emballage viral des plasmides.
- Pour les vecteurs rétroviraux, combiner 1,5 μg de VSV-G (Addgene #8454) et 1,5 μg de vecteurs d’emballage pUMVC (Addgene #8449) avec 3 μg du vecteur contenant le gène d’intérêt (dans un squelette de vecteur rétroviral résistant à la puromycine) dans 250 μL de milieux sériques réduits Opti-MEM dans un tube de microcentrifuge. Ajouter 1 μL de réactif P3000 pour chaque μg d’ADN ajouté dans le mélange Opti-MEM/ADN (dans ce cas 6 μL) et mélanger doucement en tapotant. Ne pas vortexer ou pipeter de haut en bas.
- Dans un autre tube de microcentrifugation, combiner 2 μL par μg d’ADN (dans ce cas 12 μL) de réactif de transfection avec 250 μL de milieux sériques réduits Opti-MEM.
- Mélanger les deux mélanges (500 μL combinés, ne pas vortex, seulement mélanger par tapotement doux) et laisser incuber pendant 15 min à température ambiante.
- Soigneusement, ajoutez le mélange goutte à goutte aux cellules HEK293T ensemencées sans détacher les cellules. Faites tourbillonner doucement les assiettes.
- Infection virale pour générer des lignées cellulaires stables.
- Retirer le virus contenant un surnageant des cellules HEK293T 72 h après la transfection. Filtrer soigneusement la solution avec un filtre de 0,45 μm pour éliminer les débris cellulaires et les cellules détachées. Éventuellement, ajouter 8 μg/mL de polybrene au surnageant viral pour faciliter l’infection virale.
- Ajouter le virus contenant un surnageant aux cellules U-2 OS à ~ 50% de confluence dans un plat de 10 cm (~ 3 x 106 cellules). Ensemencez les cellules U-2 OS la veille.
- Infecter pendant 6 à 16 h avant d’enlever et d’éliminer le surnageant contenant le virus.
REMARQUE: Pour obtenir la quantité souhaitée de surexpression pour le gène d’intérêt, incuber une série de dilutions virales pendant une durée déterminée. Vérifiez les niveaux d’expression du transgène dans chaque lignée cellulaire nouvellement établie avec le transfert western en le comparant aux niveaux endogènes. - Permettre aux cellules de choisir en présence d’antibiotiques appropriés (pendant 3-4 jours en cas de puromycine à une concentration finale de 2 μg/mL) et vérifier l’expression du gène d’intérêt marqué par la protéine fluorescente au microscope.
- Répétez ces étapes pour générer des lignées cellulaires à double étiquette. Dans les expériences présentées ici, mPlum-PCNA a été exprimé à partir d’un vecteur rétroviral (pBABE) combiné à EXO1B-AcGFP, également exprimé à partir d’un vecteur rétroviral (pRetroQ-AcGFP1-N1).
3. Préparation des cellules pour la micro-irradiation
- Cellules de placage : 24 h avant l’expérience, plaquer un total de 8,0 x10 4 cellules dans un volume compris entre 500 μL et 1 mL de milieu (pour une confluence d’environ 70 %) sur un verre de couverture à quatre puits bien chambré avec un fond en verre borosilicate n° 1,5 qui offre des résultats idéaux pour la microscopie confocale à fort grossissement et la micro-irradiation laser. Une plus grande fluidité cellulaire permet de mesurer plus de cellules dans un seul champ de vision (FOV); cependant, des lames entièrement confluentes introduiront des irrégularités du cycle cellulaire.
- Milieu d’imagerie : Une heure avant la micro-irradiation, échanger un milieu de croissance régulier contre du FluoroBrite DMEM complété par 10 % de FBS, 100 U/mL de pénicilline et 100 μg/mL de streptomycine, 15 mM d’HEPES (pH = 7,4) et 1 mM de pyruvate de sodium. Ce support d’imagerie permet de maximiser le rapport signal/bruit permettant la détection d’une fluorescence très faible. Comme il contient du HEPES, il stabilise également le pH en l’absence d’une atmosphère de CO2 à 5%.
- Appliquez tout traitement supplémentaire avant l’imagerie à cette étape. Dans les expériences présentées ici, les cellules ont été prétraitées une heure avant l’imagerie avec soit de l’olaparib (inhibiteur de PARP, à une concentration finale de 1 μM), soit avec un contrôle du véhicule (DMSO)1,8,9.
4. Préparation du microscope et sélection des cellules de phase S pour l’imagerie.
- Utilisez un système confocal qui possède les mêmes propriétés que le système décrit ici pour de meilleurs résultats. Les expériences présentées ici ont été réalisées à l’aide d’un microscope confocal monté sur un support de microscope inversé (voir Tableau des matériaux).
REMARQUE: Le microscope utilisé ici était équipé d’un module laser FRAP de 50 mW et de 405 nm et d’un objectif apochromate de plan d’huile de 60 x 1,4 NA. La tête de balayage confocale avait deux options de scanner: un scanner galvano (pour une haute résolution) et un scanner résonant (pour une imagerie à grande vitesse).- Introduire le laser de récupération de fluorescence après photobleachage (FRAP) dans l’échantillon via un dispositif galvano XY contrôlé par logiciel. Utilisez une ligne laser de 488 nm pour exciter l’AcGFP et une ligne laser de 561 nm ou 594 nm pour exciter mPlum.
REMARQUE: La combinaison de filtres suivante donne des résultats optimaux: à l’aide d’un filtre passe-long de 560 nm, la lumière d’émission d’une longueur d’onde inférieure à 560 nm a été passée à travers un filtre d’émission de 525/50 nm pour AcGFP, tandis que la lumière d’émission d’une longueur d’onde supérieure à 560 nm a été passée à travers un filtre d’émission de 595/50 nm pour mPlum. Tout ensemble de filtres approprié (p. ex., FITC/TRITC, GFP/mCherry, FITC/TxRed) qui garantit un minimum de purge de fluorescence pourrait être utilisé.
- Introduire le laser de récupération de fluorescence après photobleachage (FRAP) dans l’échantillon via un dispositif galvano XY contrôlé par logiciel. Utilisez une ligne laser de 488 nm pour exciter l’AcGFP et une ligne laser de 561 nm ou 594 nm pour exciter mPlum.
- Allumez la chambre environnementale et les composants du microscope.
- Allumez le chauffage (scène, objectif et chambre environnementale si possible), l’alimentation en CO2 et le régulateur d’humidité au moins 4 h avant le début de l’expérience pour assurer l’équilibrage thermique pour une acquisition d’image stable.
- Initialisez les sources lumineuses avec les lignes laser au moins 1 h avant le transfert des cellules au microscope.
- Sélectionnez des cellules en phase S dans une population asynchrone à l’aide de l’APN marqué par fluorescence comme marqueur. Pour ce faire, suivez les étapes ci-dessous.
- Recherchez le modèle de localisation unique du PCNA marqué mPlum en phase S, ce qui permet d’identifier cette phase du cycle cellulaire. Le PCNA a une distribution complètement homogène dans le noyau dans les phases G1 et G2 du cycle cellulaire, tout en étant exclu des nucléoles. En phase S, le PCNA forme des foyers à l’emplacement des réplisomes dans le noyau. La figure 1 montre les différents modèles de foyers pcNA tout au long de la phase S, ce qui permet même de différencier la phase S précoce, moyenne et tardive.
- Regardez à travers l’oculaire pour sélectionner un champ de vision qui a suffisamment de cellules en phase S pour la micro-irradiation. Les cellules os U-2 asynchrones ont généralement 30 à 40% de leur population en phase S.
- Essayez d’éviter les niveaux d’expression extrêmes (cellules brillantes et sombres) pour le PCNA et la protéine d’intérêt (POI), dans ce cas EXO1b-AcGFP, ce qui pourrait conduire à des artefacts expérimentaux.
- Lorsque vous trouvez un champ de vision approprié, essayez d’éviter de scanner le champ pendant une longue période pour minimiser le photobleaching et les dommages indésirables à l’ADN.
- Définissez la région d’intérêt (ROI) souhaitée pour la micro-irradiation. À l’aide du logiciel associé (voir Tableau des matériaux),définissez le retour sur investissement souhaité en insérant d’abord des lignes binaires (définissez le nombre de lignes et l’espacement souhaités). Cliquez sur Binaire, puis sur Insérer une | de ligne Cercle | Ellipse pour dessiner le nombre de lignes souhaité.
- Convertissez ces lignes binaires en ROI et convertissez finalement ces ROI en ROI de stimulation. Pour ce faire, cliquez d’abord sur ROI, puis cliquez sur Déplacer binary vers ROI, puis faites un clic droit sur l’un des ROI et sélectionnez Utiliser comme ROI de stimulation: S1. Placez ces lignes dans le champ de vision pour traverser le noyau des cellules. Des rois d’une longueur de 1024 pixels couvrant l’ensemble du champ de vision ont été utilisés tout au long du protocole.
5. Micro-irradiation pour la coloration par immunofluorescence ou l’imagerie en accéléré.
- Détermination des paramètres optimaux de micro-irradiation.
- Avant la micro-irradiation des cellules, prenez une image à plus haute résolution du champ de vision pour identifier les foyers de PCNA pour une analyse ultérieure. Au lieu d’un balayage séquentiel, enregistrez simultanément les deux canaux optiques utilisés (vert et rouge), afin d’éviter le mouvement des cellules entre les deux longueurs d’onde. Pour une résolution correcte des foyers, utilisez au moins 1024 x 1024 pixels / résolution de champ avec zoom 1x (taille de pixel 0,29 μm sur le système d’imagerie utilisé ici), avec une vitesse de numérisation de 1/8 image / s (4,85 μs / pixel) avec une moyenne de 2x. Une fois ces paramètres définis dans l’interface graphique A1 LFOV Compact et les fenêtres A1 LFOV Scan Area, appuyez sur le bouton Capture pour enregistrer le champ de vision.
REMARQUE: Il est important de maintenir la même taille de pixel tout au long des expériences pour garantir des résultats comparables. - Pour configurer la micro-irradiation, ouvrez l’onglet Stimulation ND dans le logiciel d’imagerie pour accéder à la fenêtre Calendrier (A1 LFOV / Galvano Device). Cela utilise les scanners galvano pour acquérir une série d’images de pré-stimulation, stimuler (à l’aide du laser FRAP LUN-F 50 mW 405 nm), puis acquérir à nouveau une série d’images post-stimulation à l’aide des scanners galvano. Configurez d’abord trois phases dans la fenêtre Calendrier. Dans la colonne Acq/Stim, sélectionnez Acquisition | | de blanchiment Acquisition pour les trois phases respectivement. Pour la phase de blanchiment, définissez S1 comme roi.
NOTE: Dans l’expérience présentée ici, aucune image n’a été acquise pendant la phase de stimulation. - Dans la fenêtre Galvano XY, configurez les facteurs clés pour la micro-irradiation: puissance de sortie laser de 405 nm, temps de séjour (l’itération est 1 par défaut sur ce système). Dans les expériences présentées ici, les cellules ont été irradiées avec le laser FRAP de 405 nm (50 mW à l’extrémité de la fibre) à 100% de puissance de sortie avec un temps de séjour de 1000-3000 μs.
REMARQUE: Étant donné que le temps de séjour laser est par pixel, tant que la taille des pixels reste la même, la relation entre le temps de séjour et la densité de puissance sera comparable entre différents FOV. La figure 2A montre l’utilisation de protéines spécifiques à la voie de réponse aux dommages à l’ADN (DDR) (FBXL10 pour les DSB et NTHL1 pour les dommages à la base oxydative) pour optimiser les paramètres de puissance laser pour l’induction de dommages spécifiques. Ces lignées cellulaires stables ont été générées avec une infection virale à la suite de la section 2 du protocole.
- Avant la micro-irradiation des cellules, prenez une image à plus haute résolution du champ de vision pour identifier les foyers de PCNA pour une analyse ultérieure. Au lieu d’un balayage séquentiel, enregistrez simultanément les deux canaux optiques utilisés (vert et rouge), afin d’éviter le mouvement des cellules entre les deux longueurs d’onde. Pour une résolution correcte des foyers, utilisez au moins 1024 x 1024 pixels / résolution de champ avec zoom 1x (taille de pixel 0,29 μm sur le système d’imagerie utilisé ici), avec une vitesse de numérisation de 1/8 image / s (4,85 μs / pixel) avec une moyenne de 2x. Une fois ces paramètres définis dans l’interface graphique A1 LFOV Compact et les fenêtres A1 LFOV Scan Area, appuyez sur le bouton Capture pour enregistrer le champ de vision.
- Imagerie en accéléré.
- Configurez l’imagerie en accéléré pour la fenêtre de temps et les intervalles souhaités à l’aide des fenêtres Calendrier horaire, A1 LFOV Compact GUI et Zone de numérisation A1 LFOV. Dans les expériences présentées ici, le recrutement d’EXO1b et de PCNA a été imagé pendant 12 min, scannant le champ de vision toutes les 5 secondes, à 1024 x 1024 pixels / champ, en utilisant un zoom 1x (résultant en une taille de pixel de 0,29 μm sur le système d’imagerie utilisé ici) avec une vitesse de numérisation de 0,35 image / s (1,45 μs / pixel) sans moyenne pour réduire le photo-blanchiment.
- Optimisez les paramètres de pourcentage de puissance laser, de gain et de décalage pour réduire le photo-blanchiment pendant l’imagerie dans la fenêtre A1 LFOV Compact GUI. Si l’on vise à mesurer à la fois le POI et le PCNA, utilisez le balayage simultané au lieu du balayage séquentiel pour éviter le mouvement cellulaire entre le balayage du champ pour les deux fluorophores distincts.
- Le système d’imagerie a été utilisé avec les paramètres suivants. Pour la ligne laser 488 nm (20 mW) : 7 % de puissance laser, gain : 45 (détecteur GaAsP) avec et décalage de 2, pour la ligne laser 561 nm (20 mW) : 4 % de puissance laser, gain 40 (détecteur GaAsP) avec et décalage de 2.
- Selon la cinétique de la protéine, prolongez ou raccourcissez l’intervalle entre les images ou la durée du laps de temps total. Dans la fenêtre Calendrier, définissez l’intervalle et la durée souhaités pour la ligne d’acquisition de la troisième phase.
- Appuyez sur Exécuter maintenant pour exécuter la micro-irradiation et l’imagerie accélérée ultérieure.
- À la fin de l’imagerie en accéléré, enregistrez les rois de stimulation sous forme d’images séparées, ce qui sera une aide utile pour identifier les coordonnées de la micro-irradiation dans tout logiciel en aval utilisé pour l’analyse.
- Coloration par immunofluorescence.
REMARQUE : L’étape 5.1.3 et la figure 2A illustrent l’utilisation de protéines de réparation de l’ADN connues pour évaluer les types de lésions d’ADN introduites par la micro-irradiation. Certaines lésions d’ADN peuvent également être détectées en utilisant des anticorps spécifiques après avoir fixé les cellules. Il est également possible de détecter le recrutement du POI par la détection d’anticorps de la protéine endogène. La visualisation de γH2A.X pour vérifier la recherche de DSB est illustrée ci-dessous(Figure 2B). La figure 3 montre la cohérence de la localisation et du recrutement de l’APN tout au long du cycle cellulaire pour l’APN marqué endogène et exogène.- Après l’étape 5.1.3, prenez une seule image après micro-irradiation pour assurer un événement FRAP approprié en fonction du recrutement de mPlum-PCNA. Prenez note des coordonnées exactes du champ de vision pour retrouver le champ plus tard après l’étiquetage immunofluorescent.
- Sortez la chambre de culture cellulaire du microscope et incubez les cellules à 37 °C dans une atmosphère humidifiée contenant 5 % de CO2 pendant 5 à 10 min.
REMARQUE: Le paraformaldéhyde (PFA) est toxique et le travail doit être effectué dans un endroit bien ventilé ou dans une hotte aspirante. Tout le lavage et l’incubation ultérieurs seront effectués avec des volumes de 0,5 mL dans la glissière de la chambre de 4 puits. Après le temps d’incubation, laver les cellules avec 0,5 mL de PBS (137 mM NaCl, 2,7 mM KCl, 8 mM Na2HPO4et 2 mM KH2PO4) et fixer avec 0,5 mL de PFA à 4% dans PBS pendant 10 min à température ambiante (RT). - Lavez les cellules une fois avec du PBS, puis lavez-les avec 50 mM NH4Cl pour éteindre le PFA résiduel.
- Perméabiliser les cellules pendant 15 min à TA avec 0,1% de Triton X-100 dans PBS.
- Bloquez les échantillons pendant 1 h avec un tampon bloquant (5% FBS, 3% BSA, 0,05% Triton X-100 en PBS).
- Retirer la solution bloquante et ajouter l’anticorps primaire dilué (anti-γH2A.X, 1:2000) dans le tampon bloquant pendant 1 h à TA.
- Lavez les puits avec un tampon bloquant 3 x 10 min.
- Ajouter un anticorps secondaire dilué (conjugué anti-souris Alexa 488 Plus, 1:2000) dans un tampon bloquant pendant 1 h à TA.
- Lavez les puits avec un tampon bloquant 3 x 10 min.
- Contre-maintenir le noyau avec une solution de DAPI de 1 μg/mL dans du PBS pendant 15 min.
- Lavez les cellules une fois avec PBS. L’imagerie peut être réalisée directement dans du PBS ou une solution de PBS avec des réactifs antifade (par exemple, AFR3) pour réduire le photobleaching.
6. Analyse du recrutement
REMARQUE : La figure 4A montre des images représentatives du recrutement d’Exo1b et de PCNA en présence de DMSO ou d’olaparib. La figure 4B montre une image représentative pour l’analyse des données. Les valeurs moyennes de fluorescence ont été calculées en mesurant les intensités moyennes d’AcGFP à l’aide d’un rectangle le long de la piste laser mis en évidence par le mPlum-PCNA (A, rectangles jaunes) à différents points temporels en utilisant Fidji. PcNA peut servir de contrôle interne pour mettre en évidence l’irradiation réussie le long des coordonnées du retour sur investissement. De même, les valeurs moyennes de fluorescence AcGFP ont également été calculées pour les régions non endommagées du noyau (B, rectangles bleus). L’intensité du signal de fond a été mesurée dans des zones non peuplées (C, rectangles rouges) et a été soustraite des valeurs fluorescentes moyennes(figures A et B). Ainsi, l’unité fluorescente moyenne relative (RFU) pour chaque point de collecte de données a été calculée par l’équation RFU = (A − C)/ (B − C)8,9. Les valeurs RFU résultantes de la région micro-irradiée sont normalisées aux valeurs RFU avant la micro-irradiation.
- Pour définir la région A du site micro-irradié, exclure de la mesure les régions nucléolaires, les foyers de réplication et les régions nucléaires irrégulières de la cellule. Maintenez la touche Maj entre le dessin de deux rois aux Fidji pour regrouper deux régions distinctes en une seule.
REMARQUE: Le recrutement des protéines varie selon les gènes et les conditions d’irradiation; ainsi, la taille de la région A doit être déterminée individuellement. Une fois la largeur de pixel de la région A déterminée, elle doit rester constante pour tout recrutement comparatif. Dans les expériences présentées ici, des rectangles de 7 pixels de largeur ont été utilisés. - Exclure de l’analyse les cellules qui se sont déplacées pendant la durée des vidéos enregistrées. Pour inclure des cellules très mobiles, l’analyse décrite doit être effectuée image par image.
- Pour visualiser le profil de recrutement, tracez les valeurs RFU normalisées par rapport au temps à l’aide d’un logiciel statistique.
- Calculer la différence à un point temporel indiqué entre le traitement par DMSO et olaparib (n = 31) à l’aide d’un test de Mann-Whitney.
Representative Results
Les cellules traitent chaque type de lésion d’ADN d’une manière spécifique qui dépend également de la phase du cycle cellulaire dans laquelle elles se trouvent. Par exemple, après micro-irradiation, les cassures double brin (DSB) seront traitées soit par jointure d’extrémité non homologue (NHEJ), soit hr en fonction de la phase du cycle cellulaire. Les nucléases agissant le plus largement pendant les phases S et G2 du cycle cellulaire créent des surplombs d’ADN qui sont cruciaux pour une BONNE HR. Pour favoriser l’évaluation des cellules en phase S, le PCNA a été utilisé comme marqueur de cycle cellulaire monocolore. La figure 1A montre le profil de localisation de mPlum-PCNA au cours de la progression du cycle cellulaire. Le PCNA a une distribution complètement homogène dans le noyau en phase G1 et G2 (tout en étant principalement exclu des nucléoles). En phase S, le PCNA se localise sur les sites de réplication de l’ADN, qui peuvent être visualisés comme des points lumineux dans le noyau. Dans les cellules de la phase S précoce, les taches sont relativement petites et également réparties dans tout le noyau de la cellule. Progressant dans la phase S moyenne, les taches deviennent floues et se localisent davantage vers le périmètre du noyau et des nucléoles. À la fin de la phase S, les taches diminuent en nombre mais deviennent de plus en plus grandes à mesure que le PCNA se concentre sur les sites de réplication tardive(Figure 1B). Il est important de se faire une indication importante que l’expression exogène du PCNA à partir de l’épine dorsale du vecteur pBABE était inférieure aux niveaux endogènes, mais était suffisante pour la détection par microscopie, ce qui minimise les artefacts potentiels dans la progression du cycle cellulaire et la DDR. La figure 1C montre l’étendue de la surexpression du PCNA par rapport aux niveaux endogènes. Veuillez noter que la bande correspondant à mPlum-PCNA migre plus lentement en raison de sa plus grande taille.
Nous avions pour objectif d’introduire des DSB lors de la micro-irradiation afin d’étudier le recrutement parp1/2 d’EXO1b pour ces lésions en phase S. La figure 2A montre que de faibles doses d’énergie (1000 μs de temps de séjour) n’induisent pas le recrutement d’EGFP-FBXL10, un répondeur DSB (composant du complexe FRUCC 8),alors qu’il était suffisant pour induire le recrutement de NTHL1-mCherry, une protéine de la voie de réparation de l’excision de base (BER), recrutant vers des sites de dommages oxydatifs à l’ADN10,11,12. À 3000 μs de temps de séjour, EGFP-FBXL10 et NTHL1-mCherry recrutent, démontrant une sortie laser qui génère à la fois des lésions oxydatives et des DSB. Renforçant ces résultats, la figure 2B montre une coloration par immunofluorescence contre γH2A.X (marqueur DSB), ce qui est clairement plus apparent lors de l’utilisation de doses d’énergie plus élevées. Le PCNA sert à la fois de marqueur du cycle cellulaire et de marqueur pour une micro-irradiation réussie, car il recrute de manière adéquate avec les deux réglages de temps de séjour au laser. Il est important de savoir que le PCNA exogène et/ou endogène marqué par une protéine fluorescente peut être utilisé pour cette fonction rapporteure, car ils se comportent de manière similaire(figure 3). Le PCNA marqué de manière endogène a été conçu en insérant mRuby dans le cadre avec le premier exon dans un allèle du locus13 du PCNA (la lignée cellulaire était un cadeau aimable de Jörg Mansfeld).
La figure 4A et la figure 4C montrent le recrutement d’EXO1b marqué AcGFP dans les cellules de phase S. EXO1b atteint le niveau maximal d’accumulation sur les sites de micro-irradiation vers 1 minute, puis commence lentement à se désengager des lésions d’ADN par la suite. Les enrichissements sur les sites de micro-irradiation sont désignés par une unité de fluorescence relative > 1 sur le graphique. En présence d’olaparib, l’accumulation d’EXO1b au niveau de la bande laser à 1 minute est nettement inférieure à celle du contrôle du véhicule. Ces résultats sont en accord avec la littérature6,7. La figure 4B montre des régions représentatives pour la quantification (zones A, B et C) telles que décrites au point 6 du protocole. La figure 4D montre les niveaux d’expression comparables d’EXO1b endogène et d’EXO1b-AcGFP exogène dans les cellules utilisées pour la micro-irradiation.
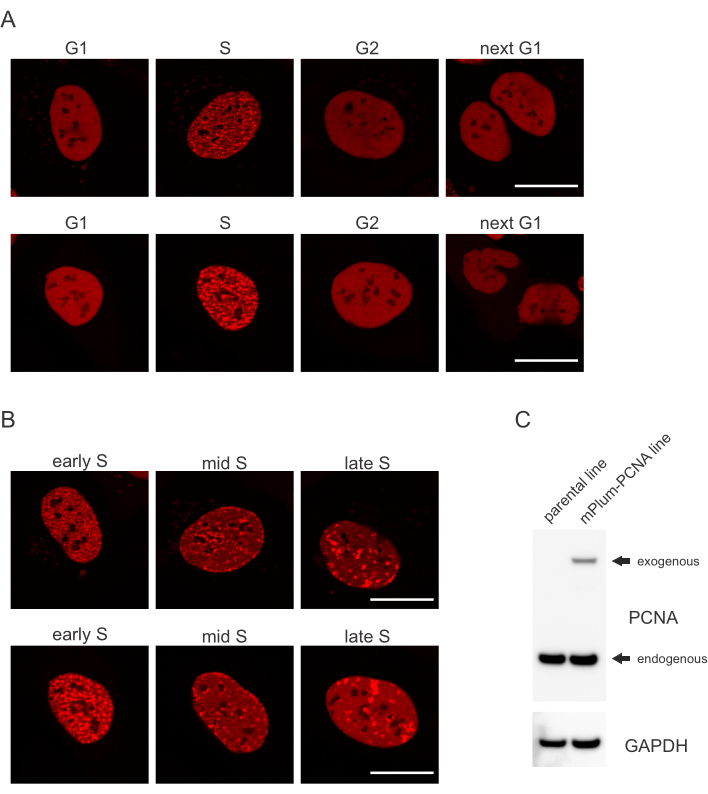
Figure 1: Schéma de localisation du PCNA. (A) Les images montrent le modèle de localisation du PCNA exogène intégré de manière stable tout au long du cycle cellulaire dans les cellules U-2 OS. (B) Les images montrent des modèles de foyers PCNA à différents stades de la phase S (précoce, moyen et tardif) dans les cellules U-2 OS. (C) Transfert western montrant des niveaux endogènes et exogènes de PCNA dans les cellules U-2 OS utilisées pour l’imagerie. La barre d’échelle représente 20 μm. Veuillez cliquer ici pour afficher une version agrandie de cette figure.

Figure 2: Induction des DSB grâce à une puissance de sortie laser optimisée. (A) Les réglages laser peuvent être optimisés pour induire différentes formes de dommages à l’ADN. Des cellules U-2 OS exprimant de manière stable à la fois EGFP-FBXL10 et NTHL1-mCherry ont été utilisées pour identifier les DSB et les sites de lésions oxydatives, respectivement. La micro-irradiation avec une ligne laser de 405 nm a été réalisée sur des cellules OS U-2 asynchrones avec un temps de séjour de 1000 μs ou 3000 μs. La barre d’échelle représente 20 μm. (B) Une coloration immunofluorescente contre γH2A.X a été effectuée sur des cellules épithéliales pigmentaires rétiniennes humaines (hTERT RPE-1) ayant un PCNA endogène marqué mRuby. Les cellules ont été fixées et traitées 5 minutes après la micro-irradiation avec un temps de séjour de 1000 μs ou 3000 μs. La barre d’échelle représente 20 μm. Veuillez cliquer ici pour afficher une version agrandie de cette figure.

Figure 3: Recrutement comparable de mRuby-PCNA endogène et de mPlum-PCNA exogène sur des sites de micro-irradiation à un temps de séjour laser de 1000 μs ou 3000 μs. L’APN marqué endogène et exogène forme des foyers de réplication pendant la phase S. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 4: Recrutement dépendant du PARP1/2 d’EXO1b en phase S. Les cellules U-2 OS exprimant de manière stable EXO1b-AcGFP et mPlum-PCNA ont été micro-irradiées avec une ligne laser FRAP de 405 nm en utilisant un temps de séjour de 3000 μs. (A) Images représentatives de cellules micro-irradiées aux points temporels indiqués après prétraitement avec contrôle du véhicule (DMSO) ou olaparib (1 μM). La barre d’échelle représente 20 μm. (B) Images représentatives de régions définies de zones A, B et C pour l’analyse de recrutement. La barre d’échelle représente 20 μm. (C) La dynamique de recrutement des dommages à l’ADN a été capturée par imagerie de cellules vivantes. Des valeurs de fluorescence moyennes relatives et des images ont été acquises toutes les 5 s pendant 12 minutes. Pour chaque condition, ≥30 cellules ont été évaluées. Les valeurs de fluorescence relatives moyennes (lignes noires pleines) et l’erreur-type (plage visualisée par une zone ombrée) ont été tracées par rapport au temps. La ligne pointillée indique les valeurs de recrutement à 1 min après la micro-irradiation. La différence entre le traitement par DMSO (n = 32) et l’olaparib (n = 31) a été calculée à l’aide d’un test de Mann-Whitney. Astérix désigne p<0,0001. (D) Western blot compare les niveaux d’expression d’EXO1b endogène et d’EXO1b-AcGFP exogène dans les cellules utilisées pour la micro-irradiation. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Discussion
Étapes critiques et dépannage/modifications potentielles du protocole
Un récipient de culture tissulaire approprié pour la micro-irradiation est essentiel au succès. La plupart des systèmes d’imagerie haute résolution sont optimisés pour une épaisseur de verre de couverture de 0,17 mm. L’utilisation de chambres d’imagerie d’épaisseur supérieure ou inférieure ou de chambres en polymères plastiques (non optimisées pour l’imagerie 405 nm) peut réduire considérablement la qualité de l’image. Lorsque vous utilisez des surfaces en verre, assurez-vous qu’elles sont traitées par culture tissulaire pour améliorer l’adhérence cellulaire. Si elles ne sont pas traitées par culture tissulaire, ces chambres devront être recouvertes, par exemple, de poly-D-lysine avant d’ensemencer les cellules. Lors du placage des cellules dans le verre de couverture chambré, la densité cellulaire idéale est primordiale pour éviter les irrégularités du cycle cellulaire et le stress supplémentaire pour les cellules. Un bon équilibre thermique des composants du microscope avant l’expérimentation pour maintenir une température stable est crucial pour maintenir la mise au point tout au long de l’imagerie en accéléré et est également nécessaire pour assurer un DDR homogène à travers le temps et les échantillons.
Il est essentiel que les cellules soient en bonne santé avant la micro-irradiation afin de réduire les données artéfactuelles. Si les cellules ont une morphologie irrégulière après l’infection / sélection, laissez les cellules progresser à travers plusieurs passages jusqu’à ce que la morphologie revienne à la normale. Assurez-vous toujours que les lignées de cellules utilisées sont exemptes de contamination par les mycoplasmes. Parmi les nombreux effets indésirables de l’infection à mycoplasmes, il provoque également des dommages à l’ADN des cellules hôtes et pourrait affecter leurs voies DDR14,15. La façon la plus sensible de détecter les mycoplasmes dans la culture cellulaire est par PCR (par opposition à la détection avec DAPI ou Hoechst).
La surexpression optimale de la protéine de réparation d’intérêt doit être comparable à des niveaux endogènes, mais suffisamment élevée pour être détectée. Le promoteur utilisé sur les vecteurs viraux, le titre viral pendant l’infection et la durée de l’infection peuvent tous être ajustés pour des niveaux d’expression idéaux. Pour des résultats cohérents, isolez des clones cellulaires individuels pour assurer des niveaux d’expression homogènes et une morphologie cellulaire normale. Il est recommandé d’utiliser des constructions vectorielles qui ne surexpriment pas le PCNA marqué à des niveaux plus élevés que les niveaux endogènes pour une fonction appropriée de marqueur de réparation du cycle cellulaire et de l’ADN. Même de faibles niveaux de surexpression de PCNA sont suffisants pour discriminer les cellules en phase S. Les vecteurs rétroviraux pBABE ont été utilisés avec succès à cette fin (Addgene #1764, #1765, #1766, #1767). Le PCNA peut être marqué avec n’importe quelle protéine rouge monomère(par exemple, mPlum, mCherry, mRuby, etc.) ou vert monomère (par exemple, mEGFP, AcGFP, mWasabi, mNeonGreen, mEmerald, etc.) qui pourraient ensuite être combinées avec un POI alternativement marqué. La surexpression d’un poI marqué par fluorescence présente certaines limites et considérations. Les étiquettes fluorescentes peuvent perturber la fonction et la localisation normales des protéines. Ainsi, l’emplacement de l’étiquette (terminal N ou C) doit être pris en compte. Utilisez toujours des protéines fluorescentes monomères, car l’oligomérisation de variantes non monomères peut affecter la fonction du POI.
Les réglages laser doivent être déterminés pour chaque système d’imagerie, car de nombreux composants du chemin optique affecteront la puissance réelle délivrée dans les cellules. La micro-irradiation laser peut provoquer plusieurs types de lésions de l’ADN en fonction de la longueur d’onde d’excitation, de la puissance de sortie du laser FRAP et si des agents pré-sensibilisants (comme la bromodésoxyuridine ou Hoechst) ont été utilisés. Les lasers 405 nm peuvent causer des dommages oxydatifs à l’ADN, des cassures simple et double brin16,17. En utilisant des paramètres de sortie laser plus élevés, la quantité de DSB augmente. Dans ce protocole, les méthodes de pré-sensibilisation n’ont pas été utilisées, mais ces techniques sont largement couvertes dans la littérature et récapitulent dans la discussion ci-dessous. À notre avis, la meilleure façon de tester si la lésion souhaitée est générée est de tester le recrutement de gènes spécifiques connus de la voie de dommages à l’ADN. Le recrutement de NTHL1 ou OGG1, composants de la voie BER, suggère l’induction de bases d’ADN oxydées10,11,17,18,19, tandis que FBXL10 ou XRCC5 indiquent la présence de DSB8,20,21. Le recrutement de XRCC1 peut indiquer à la fois la présence de bases d’ADN oxydées et de cassures simple brin (SSB)22,23. XPC (c’est-à-dire RAD4) est un bon indicateur de NER qui élimine les adducts d’ADN encombrants générés par la lumière ultraviolette (UV)17,24. Parce que le recrutement de protéines exogènes peut introduire certaines irrégularités, la coloration immunofluorescente des protéines ou des marqueurs endogènes de réparation de l’ADN (comme γH2A.X pour les cassures double brin) peut confirmer la présence de lésions d’ADN spécifiques. Alternativement, des anticorps élevés contre des types spécifiques de lésions d’ADN pourraient également être utilisés. Pour ajuster la puissance laser fournie, le temps de séjour et la puissance du laser peuvent être modifiés.
À l’aide de la modélisation mathématique, une analyse cinétique détaillée pourrait être effectuée qui pourrait fournir des informations précieuses sur les propriétés de recrutement du POI (par exemple, contribution de plusieurs domaines de liaison à l’ADN, sensibilité à différents événements de signalisation, etc.). L’évaluation automatisée du recrutement et le suivi des cellules pourraient être combinés pour créer des flux de travail robustes 1,25.
Avantages et limites de la pré-sensibilisation à l’ADN
La pré-sensibilisation de l’ADN avant la micro-irradiation est un outil couramment utilisé pour le recrutement des protéines de réparation de l’ADN16,17. La sensibilisation de l’ADN avant la micro-irradiation le rend plus sensible aux DSB. Les deux méthodes les plus courantes de pré-sensibilisation de l’ADN sont le prétraitement des cellules avec du colorant Bromodeoxyuridine (BrdU) ou Hoechst. Pour les systèmes qui ne sont pas capables de micro-irradiation à des puissances laser élevées, ces méthodes peuvent être nécessaires pour induire des lésions d’ADN comme les DSB. De plus, en l’absence d’un détecteur de lumière transmise ou d’un signal fluorescent mettant en évidence le noyau cellulaire (par exemple, lors de l’étude du recrutement de protéines endogènes de réparation de l’ADN non marquées), Hoechst agit à la fois comme un outil de pré-sensibilisation et une coloration nucléaire fluorescente. Cependant, la pré-sensibilisation de l’ADN peut introduire des complications importantes. BrdU (utilisé à une concentration finale de 10 μM) doit être ajouté aux cellules 24 heures (ou équivalent à un cycle cellulaire complet dans la lignée cellulaire utilisée) pour s’incorporer correctement dans l’ADN et peut provoquer une interférence du cycle cellulaire26. Hoechst 33342 (utilisé à une concentration finale de 1 μg/mL) est cytotoxique après de longues périodes d’incubation mais nécessite suffisamment de temps pour saturer le noyau avec le colorant. Par conséquent, il ne doit être appliqué que 15 à 20 minutes avant la micro-irradiation; sinon, les données de recrutement ne seront pas cohérentes. Les cellules colorées de cette façon ne peuvent pas être conservées en culture pendant plus de quelques heures27,28. Assurez-vous de ne pas utiliser Hoechst 33358, qui n’est pas aussi perméable aux cellules que le colorant Hoechst 33342. La pré-sensibilisation peut également introduire une variance inutile entre les expériences et rendre l’expérience encore plus sensible aux différences de densité cellulaire (car cela affectera la quantité de colorant / cellule incorporé).
Avantages et limites de la microscopie confocale
La vitesse d’imagerie de la microscopie confocale peut être limitative par rapport à la microcopie à grand champ. Cependant, un microscope confocal équipé d’un scanner résonant peut considérablement améliorer la vitesse d’imagerie (au détriment de la résolution) en se rapprochant des vitesses de la microscopie à disque rotatif. Trois caractéristiques font du système confocal A1R HD25 un excellent choix pour le protocole présenté ici. Tout d’abord, le champ de vision de 25 mm du système permet d’imager entre 15 et 20 cellules dans un seul champ scanné (contre 5 à 10 cellules dans des configurations régulières), limitant ainsi le nombre d’acquisitions nécessaires pour obtenir suffisamment de cellules pour l’analyse statistique. Deuxièmement, le module FRAP et deux têtes de balayage permettent d’imager et de micro-irradier les cellules simultanément, et pas seulement séquentiellement. Enfin, la flexibilité d’avoir à la fois les scanners résonants et galvano permet de basculer facilement entre l’imagerie à haute résolution temporelle avec une vitesse exceptionnelle qui minimise la trempe des fluorophores, et l’imagerie à haute résolution spatiale qui utilise des vitesses de numérisation plus lentes pour produire des images avec un rapport signal / bruit plus élevé. Alors que le système utilisé permettait la flexibilité susmentionnée, pour ressembler à des configurations de microscope confocal plus largement disponibles, seul le galvano scanner a été utilisé dans les expériences présentées (à la fois pour la micro-irradiation et l’imagerie ultérieure).
Avantages et limites de la micro-irradiation
Bien que la micro-irradiation offre une résolution spatiale et temporelle inégalée, elle n’est pas sans limites. Les dommages à l’ADN causés par la micro-irradiation laser sont fortement regroupés dans des parties spécifiques du noyau par rapport aux agents nocifs naturels. Ainsi, la réponse de la chromatine due à la micro-irradiation peut différer par rapport aux dommages répartis de manière homogène. De plus, la micro-irradiation prend du temps et ne peut être menée que sur quelques dizaines de cellules, tandis que les méthodes biochimiques basées sur une grande population (fractionnement de la chromatine, immunoprécipitation, ChIP) peuvent fournir une robustesse accrue en étudiant des milliers de cellules à la fois. La vérification des observations faites par micro-irradiation avec des techniques biochimiques traditionnelles est une stratégie efficace pour des conclusions fiables. Bien que la micro-irradiation simultanée de nombreuses cellules dans un certain champ de vision soit possible, le système d’imagerie aura besoin de plus de temps pour effectuer la tâche. Par conséquent, la mesure de la dynamique des protéines qui se recrutent très rapidement dans les lésions de l’ADN limite le nombre de RETOURS possibles pour la micro-irradiation utilisée simultanément. Sur le système d’imagerie utilisé pour ce protocole, la micro-irradiation d’un seul ROI de 1024 pixels de long prend 1032 ms en utilisant un temps de séjour de 1000 μs et 3088 ms en utilisant un temps de séjour de 3000 μs pour compléter. L’utilisation de plusieurs lignes de retour sur investissement augmentera considérablement le temps nécessaire pour terminer la micro-irradiation (par exemple, un retour sur investissement de 7 x 1024 pixels prend 14402 ms en utilisant un temps de séjour de 1000 μs et 21598 ms en utilisant un temps de séjour de 3000 μs). Ce temps est perdu de l’acquisition d’images et doit être pris en considération. Lorsque vous imaginez des événements de recrutement rapides, utilisez le retour sur investissement le plus court possible et ne micro-irradiez qu’une cellule à la fois.
Avantages et limites par rapport aux méthodes de synchronisation
Pour les études spécifiques au cycle cellulaire, les méthodes existantes impliquent soit la synchronisation des cellules en phases spécifiques du cycle cellulaire, soit l’utilisation de rapporteurs fluorescents pour identifier la phase spécifique du cycle cellulaire de la cellule. Cependant, chacune de ces méthodes présente ses propres défis et limites.
Le système FUCCI3 (s’appuyant sur des formes tronquées marquées par des protéines fluorescentes de CDT1 et Geminin) est un outil particulièrement utile pour les études du cycle cellulaire, mais présente des limites lorsqu’il s’agit de différencier les phases S et G2 du cycle cellulaire. Les niveaux de Géminiens sont déjà élevés à partir de la phase S moyenne et restent élevés jusqu’à la phase M, ce qui rend ces phases difficiles à séparer. L’utilisation du système FUCCI signifie également que deux canaux optiques du microscope ne peuvent pas être utilisés pour l’imagerie du POI.
Les lignées cellulaires non cancéreuses pourraient être synchronisées en G0 par l’élimination des facteurs de croissance présents dans le sérum (famine sérique) causant peu ou pas de dommages à l’ADN aux cellules. Cependant, la plupart des lignées cellulaires cancéreuses continueront partiellement à progresser tout au long du cycle cellulaire, même sans quantités adéquates de sérum dans leur milieu. De plus, les cellules commencent partiellement à perdre leur synchronisation à la fin de la phase G1, au début de la phase S. En plus de la famine sérique, il existe de nombreuses méthodes chimiques pour obtenir la synchronisation du cycle cellulaire. Les blocs d’hydroxyurée, d’aphidicoline et de thymidine sont des méthodes d’arrêt de la réplication de l’ADN pour synchroniser les cellules dans la phase S précoce. Bien que ces méthodes soient peu coûteuses et simples, elles introduisent un stress de réplication qui entraîne des dommages à l’ADN. Il a été démontré que ces inhibiteurs de la réplication de l’ADN induisent la phosphorylation de H2A. X, un marqueur bien connu des DSB2,29. La méthode d’utilisation du PCNA marqué comme marqueur pour les cellules en phase S réduit le potentiel d’artefacts causés par la synchronisation chimique et peut être appliquée à un large éventail de lignées cellulaires par rapport à la famine sérique.
Conclusion
Les dommages à l’ADN sont une force motrice pour les maladies génétiques où les lésions mutagènes peuvent conduire à la transformation maligne des cellules. Cibler la machinerie de synthèse de l’ADN est une stratégie thérapeutique fondamentale dans le traitement des maladies hyperprolifératives comme le cancer. Afin de traiter ces maladies de manière plus ciblée, nous avons besoin d’une meilleure compréhension des protéines qui réparent les lésions de l’ADN. Le protocole décrit ici aide les études basées sur la micro-irradiation en phase S en minimisant les défis présentés par les méthodes de synchronisation traditionnelles pour réduire les artefacts possibles et augmenter la reproductibilité des expériences.
Disclosures
Les auteurs affirment que la publication de l’œuvre présentée a été parrainée par Nikon Corporation. Les auteurs déclarent qu’il n’existe pas d’intérêts concurrents.
Acknowledgments
Les auteurs remercient M. Pagano pour son soutien continu ainsi que D. Simoneschi, A. Marzio et G. Tang pour leur critique du manuscrit. B. Miwatani-Minter remercie R. Miwatani et B. Minter pour leur soutien continu. G. Rona remercie K. Ronane Jurasz et G. Rona pour leur soutien continu.
Materials
| Name | Company | Catalog Number | Comments |
| Ammonium chloride | Sigma-Aldrich | A9434-500G | For quenching formaldehyde |
| Anti-EXO1 Rabbit Polyclonal Antibody | Proteintech | 16253-1-AP | primary antibody |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | 05-636 | primary antibody |
| Bovine Serum Albumin | Sigma-Aldrich | 3117332001 | BSA for blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Merck | 19-160 | pre-sensitizing agent |
| Citifluor™ Mountant Solution AFR3 | Electron Microscopy Sciences | 17973-10 | antifade containing PBS solution for imaging |
| DAPI | Sigma-Aldrich | D9542-1MG | nucleic acid stain |
| DMEM Medium | Thermo Fisher Scientific | 10569010 | Cell culture medium for HEK293T cells |
| DMSO | Sigma-Aldrich | D2650-100ML | Vehichle control and dissolution solvent |
| EGFP-FBXL10 | Addgene | #126542 | viral expression vector for EGFP-FBXL10 |
| EXO1b-AcGFP (in pRetroQ) | custom cloning | na | EXO1b cDNA was cloned in the NheI, BamHI sites of pRetroQ-AcGFP1-N1 vector. |
| Fetal Bovine Serum | Gibco | 16140071 | Media supplement |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 | Phenol red free medium for microscopy |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A32723 | secondary antibody |
| HEK293T cells | ATCC | ATCC CRL-3216 | Cell line for viral packaging |
| HEPES | Sigma-Aldrich | H0887-100ML | Buffering agent to supplement live cell imaging medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | pre-sensitizing agent |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 | Transfection reagent |
| McCoy’s 5A (Modified) Medium | Life Technologies | 16600-108 | Cell culture medium for U-2 OS cells |
| mCherry-PCNA | Addgene | #55117 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA | Addgene | #55994 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA (in pBABE) | custom cloning | na | mPlum-PCNA cDNA was cloned from Addgene #55994 in the BamHI, SalI sites of pBABE (puro) |
| Nikon A1R-HD25 Confocal Scanhead and Controller | Nikon | na | confocal imaging system |
| Nikon LUN4 laser unit | Nikon | na | excitation system |
| Nikon LUN-F 50 mW 405 nm FRAP laser unit | Nikon | na | FRAP laser unit |
| Nikon NIS Elements Confocal Controller Software | Nikon | na | Confocal controlling software |
| Nikon Ti2-E Inverted Microscope | Nikon | na | inverted epifluorescent microscope base |
| Nikon Ti2-LAPP Modular Illumination System | Nikon | na | illumination system |
| NTHL1-mCherry (in pRetroQ) | custom cloning | na | NTHL1 cDNA was cloned in the NheI, SalI sites of pRetroQ-mCherry-N1 vector. |
| Nunc Lab-Tek II Chambered Coverglass (4 well) | Thermo Fisher Scientific | 155382PK | Live cell microscopy cell culture chamber |
| Olaparib | Selleck Chemicals | S1060 | PARP inhibitor |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985062 | Dilution medium for transient transfection |
| Paraformaldehyde aqueous solution (32%) | Thermo Fisher Scientific | 50-980-494 | Fixative |
| pBABE (hygro) | Addgene | #1765 | retroviral expression vector (for low expression levels) |
| pBABE (neo) | Addgene | #1767 | retroviral expression vector (for low expression levels) |
| pBABE (puro) | Addgene | #1764 | retroviral expression vector (for low expression levels) |
| pBABE (zeo) | Addgene | #1766 | retroviral expression vector (for low expression levels) |
| PCNA Antibody (PC10) | Santa Cruz | sc-56 | primary antibody |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco | 10378016 | Media supplement |
| polybrene | Sigma-Aldrich | TR-1003 | Increase viral infection efficiency |
| pRetroQ-AcGFP-C1 | Takara | 632506 | retroviral expression vector |
| pRetroQ-AcGFP-N1 | Takara | 632505 | retroviral expression vector |
| pRetroQ-mCherry-C1 | Takara | 632567 | retroviral expression vector |
| pRetroQ-mCherry-N1 | Takara | 632568 | retroviral expression vector |
| pUMVC | Addgene | #8449 | Viral packaging vector |
| Sodium-pyruvate | Thermo Fisher Scientific | 11360070 | Supplement for live cell imaging medium |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | Dilute in PBS for cell permeabilization buffer |
| Trypsin-EDTA Solution 10X | Sigma-Aldrich | 59418C-100ML | Dilute in PBS to split cells |
| U-2 OS Cells | ATCC | HTB-96 | Optimal cell line for microscopy experiments |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K | PCR based Mycoplasma detection kit |
| VSV-G | Addgene | #8454 | Viral protein envelope vector |
References
- Aleksandrov, R., et al. Protein dynamics in complex DNA lesions. Molecular Cell. 69 (6), 1046-1061 (2018).
- Darzynkiewicz, Z., Halicka, H. D., Zhao, H., Podhorecka, M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: Analysis by flow cytometry. Methods in Molecular Biology. 761, 85-96 (2011).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Herce, H. D., Rajan, M., Lattig-Tunnemann, G., Fillies, M., Cardoso, M. C. A novel cell permeable DNA replication and repair marker. Nucleus. 5 (6), 590-600 (2014).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in dna replication with putative roles in cancer. International Journal of Molecular Sciences. 20 (1), (2018).
- Cheruiyot, A., et al. Poly(ADP-ribose)-binding promotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amsterdam). 35, 106-115 (2015).
- Zhang, F., Shi, J., Chen, S. H., Bian, C., Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Research. 43 (22), 10782-10794 (2015).
- Rona, G., et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. elife. 7, (2018).
- Young, L. M., et al. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the dna damage response. Cell Reports. 13 (3), 451-459 (2015).
- Kong, X., et al. Laser microirradiation to study in vivo cellular responses to simple and complex dna damage. Journal of Visualized Experiments. (131), e56213 (2018).
- Kong, X., et al. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS One. 6 (8), 23548 (2011).
- Lan, L., et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Research. 42 (4), 2330-2345 (2014).
- Zerjatke, T., et al. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Reports. 19 (9), 1953-1966 (2017).
- Ji, Y., Karbaschi, M., Cooke, M. S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutation Research. 845, 403054 (2019).
- Sun, G., et al. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infection and Immunity. 76 (10), 4405-4413 (2008).
- Gassman, N. R., Wilson, S. H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amsterdam). 31, 52-63 (2015).
- Muster, B., Rapp, A., Cardoso, M. C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics. 4 (1), 47-68 (2017).
- Ikeda, S., et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 273 (34), 21585-21593 (1998).
- Rosenquist, T. A., Zharkov, D. O., Grollman, A. P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proceedings of the National Academy of Science U. S. A. 94 (14), 7429-7434 (1997).
- Reid, D. A., et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Science U. S. A. 112 (20), 2575-2584 (2015).
- Taccioli, G. E., et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 265 (5177), 1442-1445 (1994).
- Marsin, S., et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 278 (45), 44068-44074 (2003).
- Thompson, L. H., Brookman, K. W., Jones, N. J., Allen, S. A., Carrano, A. V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cell Biology. 10 (12), 6160-6171 (1990).
- Scharer, O. D. Nucleotide excision repair in eukaryotes. Cold Spring Harbor Perspective Biology. 5 (10), 012609 (2013).
- Oeck, S., et al. High-throughput evaluation of protein migration and localization after laser micro-irradiation. Science Reports. 9 (1), 3148 (2019).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Science Reports. 6, 19567 (2016).
- Durand, R. E., Olive, P. L. Cytotoxicity, mutagenicity and dna damage by hoechst 33342. Journal of Histochemistry and Cytochemistry. 30 (2), 111-116 (1982).
- Tobey, R. A., Oishi, N., Crissman, H. A. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proceedings of the National Academy of Science U. S. A. 87 (13), 5104-5108 (1990).
- Podhorecka, M., Skladanowski, A., Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. Journal of Nucleic Acids. 2010, (2010).
