

Summary
Denne protokollen beskriver en ikke-invasiv metode for effektivt å identifisere S-faseceller for nedstrøms mikroskopistudier, for eksempel måling av DNA-reparasjonsproteinrekruttering ved lasermikrobestråling.
Abstract
DNA-skadereparasjon opprettholder cellenes genetiske integritet i et svært reaktivt miljø. Celler kan akkumulere ulike typer DNA-skader på grunn av både endogene og eksogene kilder som metabolske aktiviteter eller UV-stråling. Uten DNA-reparasjon blir cellens genetiske kode kompromittert, undergraver strukturer og funksjoner av proteiner og potensielt forårsaker sykdom.
Å forstå den romlige dynamikken i de forskjellige DNA-reparasjonsveiene i ulike cellesyklusfaser er avgjørende innen DNA-skadereparasjon. Nåværende fluorescerende mikroskopiteknikker gir gode verktøy for å måle rekrutteringskinetikken til forskjellige reparasjonsproteiner etter DNA-skadeinduksjon. DNA-syntese i S-fasen av cellesyklusen er et særegent punkt i celle skjebne angående DNA-reparasjon. Det gir et unikt vindu for å screene hele genomet for feil. Samtidig utgjør DNA-syntesefeil også en trussel mot DNA-integritet som ikke oppstår i ikke-splittende celler. Derfor varierer DNA-reparasjonsprosessene betydelig i S-fase sammenlignet med andre faser av cellesyklusen, og disse forskjellene er dårlig forstått.
Følgende protokoll beskriver utarbeidelse av cellelinjer og måling av dynamikk av DNA-reparasjonsproteiner i S-fase på lokalt induserte DNA-skadesteder, ved hjelp av et laserskanningskonfokalt mikroskop utstyrt med en 405 nm laserlinje. Tagged PCNA (med mPlum) brukes som cellesyklusmarkør kombinert med et AcGFP-merket reparasjonsprotein av interesse (dvs. EXO1b) for å måle DNA-skaderekruttering i S-fase.
Introduction
Flere DNA-reparasjonsveier har utviklet seg for å adressere de forskjellige typene DNA-lesjoner som kan oppstå i celler, som alle er sterkt regulert i både rom og tid. En av de mest sårbare periodene i cellesyklusen er S-fase, når DNA-syntese oppstår. Selv om spredning er grunnleggende for livet, gir det også en stor utfordring. Celler må sikre trofast replikasjon av deres genom for å unngå mutasjoner som skal overføres til fremtidige generasjoner. Følgelig gir spredning et terapeutisk intervensjonspunkt som har blitt brukt til utvikling av terapeutiske tilnærminger innen onkologi.
Alle de viktigste teknikkene som brukes til å studere proteinrekruttering ved DNA-lesjoner har sine styrker og begrensninger. Mikrobestråling har bedre romlig og tidsmessig oppløsning1 enn de fleste av de alternative metodene som immunfluoreserende avbildning av ioniserende strålingsindusert foci (IRIF), kromatin-immunoprecipitation (ChIP) eller biokjemisk fraksjonering. Imidlertid snører mikrobestråling robustheten til de nevnte teknikkene som kan prøve et stort antall celler samtidig.
For å undersøke DNA-reparasjon i S-fase må man kunne skille S-faseceller i en asynkron cellekulturpopulasjon. Det er mange kjente metoder for å løse dette, som involverer enten synkronisering av celler eller visualisering av de forskjellige cellesyklusfasene. Begge tilnærmingene introduserer imidlertid betydelige utfordringer og mulige gjenstander. Kjemiske synkroniseringsmetoder som er mye brukt til å berike celler i tidlig S-fase (f.eks. dobbel tymidinblokk, aphidicolin og hydroksyureabehandling) oppnår synkronisering gjennom induksjon av replikeringsstress og til slutt DNA-skade i seg selv. Dette begrenser bruken av disse metodene for å studere DNA-reparasjonsprosesser i S fase2. Synkronisering gjennom serum sult og frigjøring gjelder bare for et begrenset antall cellelinjer, i stor grad unntatt kreftcellelinjer som er mindre avhengige av vekstfaktorer for cellesyklusprogresjon sammenlignet med ikke-transformerte cellelinjer. Fluorescens Ubiquitin Cell Cycle Indicator (FUCCI) -systemet er et spesielt nyttig verktøy for å studere cellesyklusen, men det har en grunnleggende begrensning når man skiller mellom S- og G2 cellesyklusfaser3.
Her er det vist at bruk av fluorescerende merket PCNA som en ikke-invasiv markør for S-fase begrenser ulempene ved kjemiske cellesyklussynkroniseringsmetoder, samtidig som det gir mer spesifisitet og fleksibilitet enn FUCCI-systemet. Som en enkelt markør kan IKKE bare PCNA markere S-faseceller i en asynkron populasjon, men den kan også vise den nøyaktige utviklingen av celler i S-fase (dvs. tidlig, midt eller sen S-fase)4. Lave uttrykksnivåer av eksogene, taggede PCNA sikrer minimal interferens med både cellesyklusprogresjon og DNA-reparasjonsprosesser. Viktigst, PCNA fungerer også som en intern kontroll for riktig DNA-skadeinduksjon som det er involvert i reparasjon av flere DNA-lesjoner og rekrutteres til lokalt induserte DNA-skadesteder1,4.
Eksperimentene som presenteres her viser hvordan man måler rekrutteringsdynamikken til EXO1b i S-fase og hvordan dette påvirkes av den veletablerte PARP-hemmeren olaparib. EXO1b kjerneaktivitet er relevant for et bredt spekter av DNA-reparasjonsveier, inkludert mismatch reparasjon (MMR), nukleotid eksisjonsreparasjon (NER) og dobbeltstrenget pause (DSB) reparasjon. I S-fase spiller EXO1b en viktig rolle i homolog rekombinasjon (HR) gjennom dannelsen av 3' ssDNA-overheng under DNA-reseksjon5. EXO1b har blitt ytterligere implisert i DNA-replikasjon med roller i sjekkpunktaktivering for å starte stallede DNA-gafler på nytt, samt primerfjerning og Okazaki fragmentmodning ved den hengende tråden under strandforskyvning i replikering5. EXO1b rekruttering til skadede DNA-nettsteder er regulert av direkte interaksjon med poly (ADP-ribose) (PAR)6,7. På grunn av de mange cellesyklusspesifikke implikasjonene av EXO1b, er det et utmerket valg for S-fasespesifikke rekrutteringsstudier ved hjelp av PCNA.
Protocol
1. Dyrking av humane osteosarcoma-avledede celler (U-2 OS)
MERK: U-2 OS-celler er ideelle for disse studiene, da de har en flat morfologi, stor kjerne og sterkt festet til flere overflater, inkludert glass. Andre cellelinjer med lignende egenskaper kan også brukes.
- For dyrking av U-2 OS cellelinjer, bruk McCoys 5A medium supplert med 10% foster bovint serum (FBS) og antibiotika (100 U / ml penicillin og 100 μg / ml streptomycin). Inkuber celler ved 37 °C i en fuktig atmosfære som inneholder 5 % CO2. For mikroskopistudier, opprettholde cellekultur i en 10 cm tallerken for å gi tilstrekkelig celleantall.
- Når cellene nærmer seg 90% samløp (~ 7 x 106 celler / 10 cm tallerken), del cellene.
- Skyll celler med PBS for å vaske bort trypsinhemmere som finnes i serumet.
- Tilsett 1 ml Trypsin-EDTA og sørg for at cellelaget er likt dekket.
- Inkuber ved 37 °C til cellelaget løftes av platen (ca. 6 min).
- Resuspend de trypsiniserte cellene i serumholdige medier for å inaktivere trypsin og tilsett 1/10th av volumet (~ 0,7 x 106 celler) i en ny 10 cm plate som inneholder 10 ml supplert vekstmedium.
- Før eksperimentering tester du rutinemessig celler for mykoplasmaforurensning ved hjelp av Universal Mycoplasma Detection-settet etter produsentens anbefaling.
2. Retroviral infeksjon
MERK: For BSL-2 sikkerhetstiltak og under arbeid med rekombinante virus, se: NIH Guidelines, Section III-D-3: Rekombinante virus i vevskulturen.
- Frø 4 x 106 HEK293T celler for å oppnå ~ 60% samløp innen 24 timer etter plating i en 10 cm kulturrett.
- For dyrking av HEK293T, følg dyrkingstrinnene til U-2 OS beskrevet i 1.1-1.3 av denne protokollen. For HEK293T erstatter McCoys 5A-medium for DMEM. Pass på at du alltid vasker HEK293T-celler forsiktig når de festes til vevskulturplater svakt.
- Transfekt HEK293T celler ved hjelp av en lipid-basert transfection reagens for viral emballasje av plasmider.
- For retrovirale vektorer kombinerer du 1,5 μg VSV-G (Addgene #8454) og 1,5 μg pUMVC (Addgene #8449) emballasjevektorer sammen med 3 μg av vektoren som inneholder genet av interesse (i en retroviral vektor ryggrad med puromycin motstand) i 250 μL Opti-MEM redusert serum media i et mikrocentrifuge rør. Tilsett 1 μL P3000-reagens for hver μg DNA som tilsetts i Opti-MEM/DNA-blandingen (i dette tilfellet 6 μL) og bland forsiktig ved å tappe. Ikke virvel eller pipette opp og ned.
- I et annet mikrosenterrifugerør kombinerer du 2 μL per μg DNA (i dette tilfellet 12 μL) transfeksjonsreagens med 250 μL Opti-MEM redusert serummedier.
- Kombiner de to blandingene (500 μL kombinert, ikke virvel, bland bare ved forsiktig tapping) og la det inkubere i 15 minutter ved romtemperatur.
- Legg blandingen forsiktig til frøet HEK293T-celler uten å løsne cellene. Virvle platene forsiktig.
- Virusinfeksjon for å generere stabile cellelinjer.
- Fjern viruset som inneholder supernatant fra HEK293T-cellene 72 timer etter transfeksjon. Filtrer forsiktig løsningen med et 0,45 μm filter for å fjerne cellerester og frittliggende celler. Eventuelt, legg 8 μg / ml polybrene til viral supernatant for å lette virusinfeksjon.
- Tilsett virus som inneholder overnatant, i U-2 OS-celler ved ~50 % konfluens i en 10 cm tallerken (~3 x 106 celler). Frø U-2 OS-cellene dagen før.
- Infiser i 6-16 timer før du fjerner og kaster det virusholdige supernatantet.
MERK: For å oppnå ønsket mengde overekspression for genet av interesse, inkuber en rekke virale fortynninger i en fast tid. Kontroller uttrykksnivåene til transgene i hver nyopprettede cellelinje med vestlig blot sammenligner den med endogene nivåer. - Tillat celler å velge i nærvær av passende antibiotika (i 3-4 dager i tilfelle puromycin ved 2 μg / ml endelig konsentrasjon) og verifisere uttrykket av det fluorescerende proteinmerkede genet av interesse under et mikroskop.
- Gjenta disse trinnene for å generere dobbeltmerkede cellelinjer. I forsøkene som presenteres her ble mPlum-PCNA uttrykt fra en retroviral vektor (pBABE) kombinert med EXO1B-AcGFP, også uttrykt fra en retroviral vektor (pRetroQ-AcGFP1-N1).
3. Fremstilling av celler for mikrobestråling
- Plating celler: 24 h før eksperimentet, plate totalt 8,0 x10 4 celler i et volum mellom 500 μL-1 ml medier (for omtrent 70% konfluens) på en fire godt kamret coverglass med en No. 1.5 borosilikat glassbunn som gir ideelle resultater for høy forstørrelse konfokal mikrokopi og laser-istrosilicate glassbunn som gir ideelle resultater for høy forstørrelse konfokal mikrokopi og laser-iradilering. En høyere cellesamlestyrke gir mulighet for flere celler målt i ett enkelt synsfelt (FOV). Sammenfallende lysbilder vil imidlertid introdusere uregelmessigheter i cellesyklusen.
- Bildemedier: En time før mikrobestråling, bytt regelmessig vekstmedium for FluoroBrite DMEM supplert med 10% FBS, 100 U / ml penicillin og 100 μg / ml streptomycin, 15 mM HEPES (pH = 7,4) og 1 mM natrium-pyruvat. Dette bildemediet bidrar til å maksimere signal-til-støy-forholdet, slik at det oppdages svært svak fluorescens. Siden den inneholder HEPES, stabiliserer den også pH i fravær av en 5% CO2-atmosfære.
- Påfør ytterligere behandling før avbildningen på dette trinnet. I forsøkene som presenteres her, ble cellene forhåndsbehandlet en time før avbildning med enten olaparib (PARP-hemmer, ved 1 μM endelig konsentrasjon) eller en kjøretøykontroll (DMSO)1,8,9.
4. Klargjøring av mikroskopet og valg av S-faseceller for avbildning.
- Bruk et konfokalt system som har lignende egenskaper som systemet som er beskrevet her for best resultat. Eksperimentene som presenteres her ble utført ved hjelp av et konfokalt mikroskop montert på et invertert mikroskopstativ (se Materialliste).
MERK: Mikroskopet som ble brukt her var utstyrt med en 50 mW 405 nm FRAP lasermodul, og et 60x 1,4 NA oljeplan-apochromat mål. Det konfokale skannehodet hadde to skanneralternativer: en galvano-skanner (for høy oppløsning) og resonansskanner (for høyhastighetsbilder).- Introduser fluorescensgjenvinning etter fotobleking (FRAP) laser til prøven via en programvarestyrt XY galvano-enhet. Bruk en 488 nm laserlinje for å begeistre AcGFP og en 561 nm eller 594 nm laserlinje for å begeistre mPlum.
MERK: Følgende filterkombinasjon gir optimale resultater: Ved hjelp av et 560 nm langt passfilter ble utslippslys med en bølgelengde lavere enn 560 nm passert gjennom et 525/50 nm utslippsfilter for AcGFP, mens utslippslys med en bølgelengde høyere enn 560 nm ble passert gjennom et 595/50 nm utslippsfilter for mlumP. Ethvert passende filtersett (f.eks. FITC/TRITC, GFP/mCherry, FITC/TxRed) som sikrer minimal fluorescensblødning kan brukes.
- Introduser fluorescensgjenvinning etter fotobleking (FRAP) laser til prøven via en programvarestyrt XY galvano-enhet. Bruk en 488 nm laserlinje for å begeistre AcGFP og en 561 nm eller 594 nm laserlinje for å begeistre mPlum.
- Slå på miljøkammeret og mikroskopkomponentene.
- Slå på oppvarmingen (stadium, objektivt og miljøkammer når det er mulig), CO2-tilførsel og fuktighetsregulatoren minst 4 timer før starten av eksperimentet for å sikre termisk likevekt for stabilt bildeanskaffelse.
- Initialiser lyskilder sammen med laserlinjene minst 1 time før overføring av cellene til mikroskopet.
- Velg S-faseceller i en asynkron populasjon ved hjelp av fluorescerende kodet PCNA som markør. Gjør dette ved å følge trinnene nedenfor.
- Se etter det unike lokaliseringsmønsteret til den mPlum-taggede PCNA i S-fasen, noe som gjør identifisering av denne cellesyklusfasen mulig. PCNA har en helt homogen fordeling i kjernen i G1- og G2-faser av cellesyklusen, samtidig som den utelukkes fra nukleolene. I S-fase danner PCNA foci på plasseringen av replisomer i kjernen. Figur 1 viser de ulike mønstrene til PCNA-foci gjennom hele S-fasen, noe som gjør det mulig å skille mellom tidlig, midt og sen S-fase.
- Se gjennom okulæren for å velge en FOV som har nok S-faseceller til mikrobestråling. Asynkrone U-2 OS-celler har vanligvis 30-40% av befolkningen i S-fase.
- Prøv å unngå ekstremer i uttrykksnivåer (både lyse og svake celler) for både PCNA og proteinet av interesse (POI), i dette tilfellet EXO1b-AcGFP, noe som kan føre til eksperimentelle gjenstander.
- Når du finner en passende FOV, prøv å unngå å skanne feltet i lang tid for å minimere fotobleaching og uønsket DNA-skade.
- Angi ønsket interesseområde (ROI) for mikrobestråling. Bruk den tilknyttede programvaren (se Materialliste), angi ønsket avkastning ved først å sette inn binære linjer (angi ønsket antall linjer og avstand). Klikk Binær, og klikk deretter Sett inn linje | Sirkel | Ellipse for å tegne ønsket antall linjer.
- Konverter disse binære linjene til ROIer, og konverter til slutt disse ROIene til stimulerings-ROIer. Dette gjør du ved først å klikke avkastning, deretter klikke Flytt binær til avkastning, høyreklikk deretter en av ROIene og velg Bruk som stimulerings-AVKASTNING: S1. Plasser disse linjene i FOV for å passere gjennom kjernen av cellene. ROIer med en lengde på 1024 piksler som strakte seg over hele FOV ble brukt i hele protokollen.
5. Mikrobestråling for immunfluorescensfarging eller tidsforløpavbildning.
- Bestemme optimale mikrobestrålingsinnstillinger.
- Før mikrobestråling av cellene, ta et bilde med høyere oppløsning av FOV for å identifisere PCNA-foci for senere analyse. I stedet for sekvensiell skanning registrerer du samtidig begge de optiske kanalene som brukes (grønn og rød), for å unngå cellebevegelser mellom skanning ved de to bølgelengdene. For riktig oppløsning av foci, bruk minst 1024 x 1024 piksler / feltoppløsning med 1x zoom (0,29 μm pikselstørrelse på bildesystemet som brukes her), med 1/8 ramme / skannehastighet (4,85 μs / piksel) med 2x gjennomsnitt. Når disse parametrene er angitt i A1 LFOV Compact GUI og A1 LFOV Scan Area-vinduene, trykker du på Capture-knappen for å spille inn FOV.
MERK: Det er viktig å opprettholde samme pikselstørrelse gjennom eksperimenter for å sikre sammenlignbare resultater. - Hvis du vil konfigurere mikrobestrålingen, åpner du kategorien ND-stimulering i bildeprogramvaren for å få tilgang til vinduet Tidsplan (A1 LFOV / Galvano-enhet). Dette bruker galvano-skannerne til å skaffe seg en rekke pre-stimuleringsbilder, stimulere (ved hjelp av LUN-F 50 mW 405 nm FRAP-laser), og deretter skaffe seg en rekke bilder etter stimulering igjen ved hjelp av galvano-skannerne. Først definerer du tre faser i Tidsplan-vinduet. I Acq/Stim-kolonnen velger du Anskaffelse | Bleking | Anskaffelse for de tre fasene. For blekefasen setter du S1 som avkastning.
MERK: I eksperimentet som presenteres her, ble det ikke samlet inn bilder under stimuleringsfasen. - I Galvano XY-vinduetkonfigurerer du nøkkelfaktorene for mikrobestråling: 405 nm lasereffekt, oppholdstid (gjentakelse er 1 som standard på dette systemet). I forsøkene som ble presentert her, ble cellene bestrålet med 405 nm FRAP-laser (50 mW ved fiberspissen) ved 100% effekt med en 1000-3000 μs oppholdstid.
MERK: Fordi laserboetiden er per pikselbasis, så lenge pikselstørrelsen forblir den samme, vil forholdet mellom oppholdstid og strømtetthet være sammenlignbart mellom forskjellige FOVer. Figur 2A viser bruk av DNA-skaderespons (DDR) banespesifikke proteiner (FBXL10 for DSB-er og NTHL1 for oksidativ baseskade) for å optimalisere lasereffektinnstillinger for spesifikk skadeinduksjon. Disse stabile cellelinjene ble generert med virusinfeksjon etter paragraf 2 i protokollen.
- Før mikrobestråling av cellene, ta et bilde med høyere oppløsning av FOV for å identifisere PCNA-foci for senere analyse. I stedet for sekvensiell skanning registrerer du samtidig begge de optiske kanalene som brukes (grønn og rød), for å unngå cellebevegelser mellom skanning ved de to bølgelengdene. For riktig oppløsning av foci, bruk minst 1024 x 1024 piksler / feltoppløsning med 1x zoom (0,29 μm pikselstørrelse på bildesystemet som brukes her), med 1/8 ramme / skannehastighet (4,85 μs / piksel) med 2x gjennomsnitt. Når disse parametrene er angitt i A1 LFOV Compact GUI og A1 LFOV Scan Area-vinduene, trykker du på Capture-knappen for å spille inn FOV.
- Tidsforløp bildebehandling.
- Definer tidsforløpavbildning for ønsket tidsvindu og intervaller ved hjelp av vinduene Tidsplan, A1 LFOV Compact GUI og A1 LFOV Scan Area. I forsøkene som presenteres her, ble rekrutteringen av EXO1b og PCNA avbildet i 12 min, skanning av FOV hvert 5. sekund, ved 1024 x 1024 piksler/felt, ved hjelp av 1x zoom (noe som resulterer i 0,29 μm pikselstørrelse på bildesystemet som brukes her) med 0,35 bilde-/skannehastighet (1,45 μs/piksel) uten gjennomsnitt for å redusere fotobleking.
- Optimaliser innstillingene for lasereffekt %, forsterkning og forskyvning for å redusere fotobleking under bildet i A1 LFOV Compact GUI-vinduet. Hvis man tar sikte på å måle både POI og PCNA, bruk samtidig skanning i stedet for sekvensiell skanning for å unngå cellebevegelse mellom skanning av feltet for de to separate fluoroforene.
- Bildesystemet ble brukt med følgende innstillinger. For 488 nm laserlinje (20 mW): 7% lasereffekt, forsterkning: 45 (GaAsP-detektor) med og offset på 2, for 561 nm laserlinje (20 mW): 4% lasereffekt, få 40 (GaAsP-detektor) med og offset på 2.
- Avhengig av proteinets kinetikk, utvid eller forkort intervallet mellom bilder eller varigheten av den totale tidsforløpet. I Vinduet Tidsplan angir du ønsket intervall og varighet for den tredje faseanskaffelsesraden.
- Trykk kjør nå for å utføre mikrobestrålingen og den påfølgende tidsforløpavbildningen.
- På slutten av tidsforløpet lagrer du stimulerings-ROIene som separate bilder, noe som vil være et nyttig hjelpemiddel for å identifisere koordinatene for mikrobestråling i all nedstrøms programvare som brukes til analyse.
- Immunfluorescensfarging.
MERK: Trinn 5.1.3 og figur 2A viser bruk av kjente DNA-reparasjonsproteiner for å vurdere hvilke typer DNA-lesjoner som innføres ved mikrobestråling. Visse DNA-lesjoner kan også oppdages ved å bruke spesifikke antistoffer etter å ha fikset cellene. Det er også mulig å oppdage rekruttering av POI ved antistoffdeteksjon av det endogene proteinet. Visualiseringen av γH2A.X for å se etter DSBer er vist nedenfor (Figur 2B). Figur 3 viser konsistensen av PCNA-lokalisering og rekruttering gjennom hele cellesyklusen for både endogene og eksogene tagget PCNA.- Etter trinn 5.1.3, ta bare ett bilde etter mikrobestråling for å sikre riktig FRAP-hendelse basert på rekruttering av mPlum-PCNA. Legg merke til de nøyaktige koordinatene til FOV for å finne feltet senere etter immunfluorescerende merking.
- Ta cellekulturkammeret ut av mikroskopet og inkuber celler ved 37 °C i en fuktig atmosfære som inneholder 5 % CO2 i 5–10 minutter.
MERK: Paraformaldehyd (PFA) er giftig, og arbeidet bør gjøres i et godt ventilert område eller en avtrekkshette. All etterfølgende vask og inkubasjon vil bli gjort med 0,5 ml volumer i 4 brønnkammersklie. Etter inkubasjonstiden, vask cellene med 0,5 ml PBS (137 mM NaCl, 2,7 mM KCl, 8 mM Na2HPO4og 2 mM KH2PO4) og fest med 0,5 ml 4% PFA i PBS i 10 ved min romtemperatur (RT). - Vask cellene en gang med PBS, vask dem deretter med 50 mM NH4Cl for å slukke gjenværende PFA.
- Permeabiliser cellene i 15 min ved RT med 0,1% Triton X-100 i PBS.
- Blokker prøvene i 1 time med blokkeringsbuffer (5 % FBS, 3 % BSA, 0,05 % Triton X-100 i PBS).
- Fjern blokkeringsløsningen og tilsett det fortynnede primære antistoffet (anti-γH2A.X, 1:2000) i blokkeringsbuffer i 1 time ved RT.
- Vask brønnene med blokkeringsbuffer 3 x 10 min.
- Tilsett fortynnet sekundært antistoff (antimus Alexa 488 Plus conjugate, 1:2000) i blokkeringsbuffer i 1 t ved RT.
- Vask brønnene med blokkeringsbuffer 3 x 10 min.
- Motvirke kjernen med 1 μg/ml DAPI-oppløsning i PBS i 15 min.
- Vask cellene en gang med PBS. Avbildningen kan utføres direkte i PBS eller en PBS-løsning med antifadereagenser (f.eks. AFR3) for å redusere fotobleking.
6. Rekrutteringsanalyse
MERK: Figur 4A viser representative bilder av Exo1b- og PCNA-rekruttering i nærvær av DMSO eller olaparib. Figur 4B viser et representativt bilde for dataanalyse. Gjennomsnittlige fluorescensverdier ble beregnet ved å måle gjennomsnittlige AcGFP-intensiteter ved hjelp av et rektangel langs lasersporet fremhevet av mPlum-PCNA (A, gule rektangler) på tvers av forskjellige tidspunkter ved hjelp av Fiji. PCNA kan fungere som en intern kontroll for å markere vellykket bestråling langs ROI-koordinatene. Tilsvarende ble gjennomsnittlige AcGFP fluorescensverdier også beregnet for uskadede områder av kjernen (B, blå rektangler). Bakgrunnssignalintensiteten ble målt i ubefolkede områder (C, røde rektangler) og ble trukket fra de gjennomsnittlige fluorescerende verdiene (figur A og B). Dermed ble den relative gjennomsnittlige fluorescerende enheten (RFU) for hvert datainnsamlingspunkt beregnet av ligningen RFU = (A − C)/ (B − C)8,9. De resulterende RFU-verdiene i det mikrobestrålede området normaliseres til RFU-verdiene før mikrobestråling.
- For å definere regionen A på det mikrobestrålede stedet, ekskludere nukleolarregioner, replikeringsfokus og uregelmessige kjernefysiske regioner i cellen fra måling. Hold nede Skift-tasten mellom å tegne to ROIer i Fiji for å gruppere to separate regioner som ett.
MERK: Proteinrekruttering vil variere mellom ulike gener og bestrålingsforhold; Dermed må størrelsen på region A bestemmes individuelt. Når pikselbredden i område A er bestemt, bør den forbli konstant for eventuelle komparative rekrutteringer. I eksperimentene som ble presentert her, ble det brukt 7 pikselbredderektangler. - Utelat celler som beveget seg i løpet av varigheten av de innspilte videoene fra analysen. For å inkludere svært mobile celler må den beskrevne analysen utføres ramme for ramme.
- For å visualisere rekrutteringsprofilen, plott de normaliserte RFU-verdiene mot tiden ved hjelp av en statistisk programvare.
- Beregn forskjellen ved et angitt tidspunkt mellom DMSO- og olaparib-behandling (n=31) ved hjelp av en Mann-Whitney-test.
Representative Results
Celler adresserer hver type DNA-lesjon på en bestemt måte som også avhenger av hvilken cellesyklusfase de er i. For eksempel, etter mikrobestråling, vil dobbeltstrengede pauser (DSB) bli behandlet enten ved ikke-homolog sluttkobling (NHEJ) eller HR avhengig av cellesyklusfasen. Nukleaser som virker mest omfattende i S- og G2-fasene av cellesyklusen, skaper DNA-overheng som er avgjørende for riktig HR. For å fremme evalueringen av celler i S-fase, ble PCNA ansatt som en enfarget cellesyklusmarkør. Figur 1A viser lokaliseringsprofilen til mPlum-PCNA under cellesyklusprogresjon. PCNA har en helt homogen fordeling i kjernen i G1- og G2-fasen (samtidig som den for det meste utelukkes fra kjernen). I S-fase lokaliserer PCNA til steder for DNA-replikasjon, som kan visualiseres som lyse flekker i kjernen. I tidlige S-faseceller er flekkene relativt små og like fordelt gjennom cellens kjerne. Fremover i midten av S-fasen blir flekkene uskarpe og lokaliserer mer mot omkretsen av kjernen og kjernen. I sen S-fase reduseres flekkene i antall, men blir stadig større etter hvert som PCNA konsentrerer seg på områder med sen replikering (Figur 1B). Viktigst, eksogen PCNA uttrykk fra pBABE vektor ryggraden var mindre enn endogene nivåer, men var nok for deteksjon av mikroskopi som minimerer potensielle gjenstander i celle syklus progresjon og DDR. Figur 1C viser omfanget av PCNA-overekspression sammenlignet med endogene nivåer. Vær oppmerksom på at båndet som tilsvarer mPlum-PCNA migrerer langsommere på grunn av sin større størrelse.
Vi hadde som mål å introdusere DSB-er under mikrobestråling for å undersøke PARP1/2-avhengig rekruttering av EXO1b til disse lesjonene i S-fase. Figur 2A viser at lave doser energi (1000 μs oppholdstid) ikke induserer rekrutteringen av EGFP-FBXL10, en DSB-responder (komponent i FRUCC-komplekset 8), mens det var tilstrekkelig å indusere rekrutteringen av NTHL1-mCherry, en base excision repair (BER) baneprotein, rekruttering til steder med oksidativ DNA-skade10,11,12. Ved 3000 μs oppholdstid, både EGFP-FBXL10 og NTHL1-mCherry rekrutt, demonstrerer en laser utgang som genererer både oksidative lesjoner og DSBs. Styrking av disse resultatene, Figur 2B viser immunfluorescence farging mot γH2A.X (DSB markør), som er tydeligere når du bruker høyere energi doser. PCNA fungerer som både en cellesyklusmarkør og en markør for vellykket mikrobestråling, da den tilstrekkelig rekrutterer med begge laserboende tidsinnstillinger. Det er viktig at både eksogene og/eller endogene fluorescerende proteinmerkede PCNA kan brukes til denne reporterfunksjonen, da de oppfører seg på samme måte (figur 3). Endogenet merket PCNA ble konstruert ved å sette inn mRuby i ramme med den første exon i ett smug av PCNA locus13 (cellelinjen var en slags gave av Jörg Mansfeld).
Figur 4A og figur 4C viser rekrutteringen av AcGFP-merket EXO1b i S-faseceller. EXO1b når maksimalt akkumuleringsnivå på mikrobestrålingssteder rundt 1 minutt og begynner deretter sakte å løsne fra DNA-lesjonene etterpå. Berikelser på mikrobestrålingssteder er betegnet med en > 1 relativ fluorescensenhet på grafen. I nærvær av olaparib er akkumulering av EXO1b ved laserstripen på 1 minutt betydelig mindre sammenlignet med kjøretøykontrollen. Disse resultatene er i samsvar med litteraturen6,7. Figur 4B viser representative regioner for kvantifisering (område A, B og C) som beskrevet i punkt 6 i protokollen. Figur 4D viser sammenlignbare uttrykksnivåer av endogen EXO1b og eksogen EXO1b-AcGFP i celler som brukes til mikrobestråling.
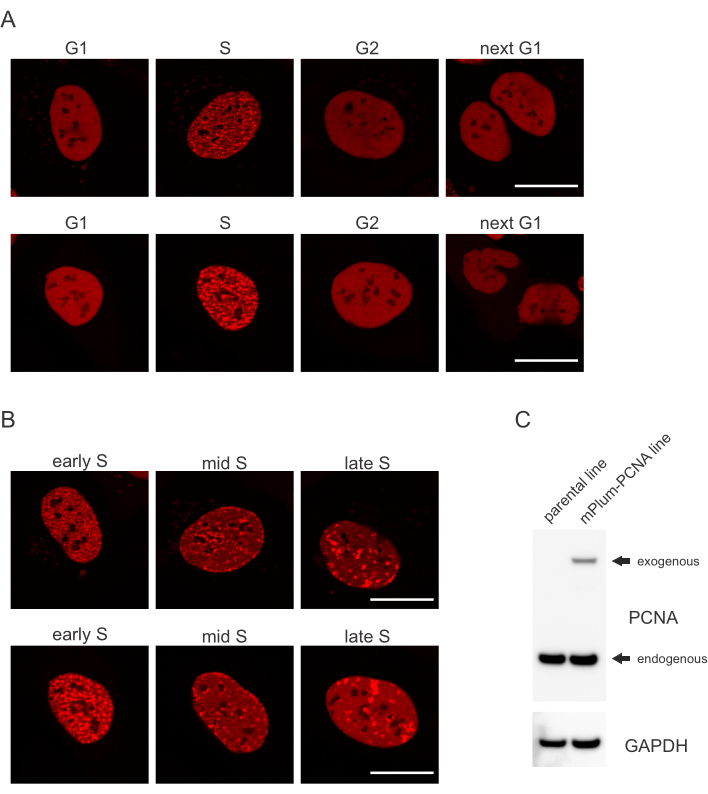
Figur 1: Lokaliseringsmønsteret til PCNA. (A) Bilder viser lokaliseringsmønster av stabilt integrert, eksogen PCNA gjennom hele cellesyklusen i U-2 OS-celler. (B) Bilder viser PCNA-fokusmønstre i ulike faser av S-fasen (tidlig, midt og sent) i U-2 OS-celler. (C) Vestlig blott som viser endogene og eksogene nivåer av PCNA i U-2 OS-cellene som brukes til avbildning. Skalalinjen representerer 20 μm. Klikk her for å se en større versjon av denne figuren.

Figur 2: Induksjon av DSBer gjennom optimalisert lasereffekt. (A) Laserinnstillinger kan optimaliseres for å indusere ulike former for DNA-skade. U-2 OS-celler som stabilt uttrykker henholdsvis EGFP-FBXL10 og NTHL1-mCherry, ble brukt til å identifisere DSBer og steder med oksidative lesjoner. Mikrobestråling med en 405 nm laserlinje ble utført på asynkrone U-2 OS-celler med enten 1000 μs eller 3000 μs botid. Skala bar representerer 20 μm. (B) Immunofluorescent farging mot γH2A.X ble gjort på humane retinal pigment epitelceller (hTERT RPE-1) har mRuby-merket endogen PCNA. Celler ble fikset og behandlet 5 minutter etter mikrobestråling med enten 1000 μs eller 3000 μs oppholdstid. Skalalinjen representerer 20 μm. Klikk her for å se en større versjon av denne figuren.

Figur 3: Sammenlignbar rekruttering av endogen mRuby-PCNA og eksogen mPlum-PCNA til mikrobestrålingssteder ved 1000 μs eller 3000 μs laserbotid. Både endogene og eksogene taggede PCNA danner replikasjonsfokus under S-fase. Klikk her for å se en større versjon av denne figuren.

Figur 4: PARP1/2-avhengig rekruttering av EXO1b i S-fase. U-2 OS-celler som stabilt uttrykte EXO1b-AcGFP og mPlum-PCNA ble mikrobestrålet med 405 nm FRAP laserlinje ved hjelp av 3000 μs oppholdstid. (A) Representative bilder av mikrobestrålede celler på de angitte tidspunktene etter forbehandling med enten kjøretøykontroll (DMSO) eller olaparib (1 μM). Skalalinjen representerer 20 μm. (B) Representative bilder av definerte områder i A-, B- og C-områder for rekrutteringsanalysen. Skala bar representerer 20 μm. (C) DNA skade rekrutteringsdynamikk ble fanget opp av live celle avbildning. Relative gjennomsnittlige fluorescensverdier og bilder ble anskaffet hver 5 s i 12 min. For hver betingelse ble ≥30 celler evaluert. Gjennomsnittlige relative fluorescensverdier (heltrukne svarte linjer) og standardfeil (område visualisert av et skyggelagt område) ble plottet mot tiden. Stiplet linje viser rekrutteringsverdier etter mikrobestråling. Forskjellen mellom DMSO (n=32) og olaparib (n=31) behandling ble beregnet ved hjelp av en Mann-Whitney-test. Asterix betegner p<0.0001. (D) Vestlig flekk sammenligner uttrykksnivåene til endogene EXO1b og eksogene EXO1b-AcGFP i celler som brukes til mikrobestråling. Klikk her for å se en større versjon av denne figuren.
Discussion
Kritiske trinn og potensiell feilsøking/modifikasjon av protokoller
Riktig vevskulturfartøy for mikrobestråling er avgjørende for å lykkes. De fleste bildesystemer med høy oppløsning er optimalisert for tykkelse på 0,17 mm dekselglass. Bruk av bildekamre med høyere eller lavere tykkelse eller de som er laget av plastpolymerer (ikke optimalisert for 405 nm avbildning), kan redusere bildekvaliteten betydelig. Når du bruker glassflater, må du sørge for at de er vevskultur behandlet for å forbedre celleadhesjonen. Hvis de ikke er vevskulturbehandlet, må disse kamrene belegges, for eksempel med poly-D-lysin før du sår cellene. Når du plating celler inn i kammeret coverglass, ideell celle tetthet er avgjørende for å unngå celle syklus uregelmessigheter og ekstra stress til cellene. Riktig termisk likevekt av mikroskopkomponentene før eksperimentering for å opprettholde en stabil temperatur er avgjørende for både å opprettholde fokuset gjennom hele tidsforløpavbildningen og er også nødvendig for å sikre en homogen DDR over tid og prøver.
Det er kritisk at cellene er i en sunn tilstand før mikrobestråling for å redusere artefaktuelle data. Hvis cellene har uregelmessig morfologi etter infeksjon/seleksjon, la cellene gå gjennom flere passasjer til morfologien blir normal igjen. Sørg alltid for at cellelinjene som brukes er fri for mykoplasmaforurensning. Blant de mange bivirkningene av mykoplasmainfeksjon forårsaker det også DNA-skade på vertscellene og kan påvirke DERES DDR-veier14,15. Den mest følsomme måten å oppdage mykoplasma i cellekulturen er gjennom PCR (versus. deteksjon med DAPI eller Hoechst).
Optimal overekspression av reparasjonsproteinet av interesse bør være sammenlignbart med endogene nivåer, men høyt nok til deteksjon. Promotoren som brukes på virale vektorer, viral titer under infeksjon, og lengden på infeksjonstiden kan alle justeres for ideelle uttrykksnivåer. For konsistente resultater isolerer du individuelle cellekloner for å sikre homogene uttrykksnivåer og normal cellemorfologi. Det anbefales å bruke vektorkonstruksjoner som ikke overekspresserer tagget PCNA på høyere enn endogene nivåer for riktig cellesyklus og DNA-reparasjonsmarkørfunksjon. Selv lave nivåer av PCNA-overekspressering er tilstrekkelig til å diskriminere S-faseceller. Retrovirale pBABE-vektorer har blitt brukt til dette formålet (Addgene #1764, #1765, #1766, #1767). PCNA kan merkes med monomeriske røde(f.eks. mPlum, mCherry, mRuby, etc.) eller monomeriske grønne fluorescerende proteiner (f.eks. mEGFP, AcGFP, mWasabi, mNeonGreen, mEmerald, etc.) som deretter kan kombineres med en vekselvis merket POI. Overekspressering av et fluorescerende merket POI har noen begrensninger og hensyn. Fluorescerende koder kan forstyrre normal proteinfunksjon og lokalisering. Dermed må plasseringen av brikken (N eller C-terminalen) vurderes. Bruk alltid monomeriske fluorescerende proteiner, da oligomerisering av ikke-monomeriske varianter kan påvirke POI-funksjonen.
Laserinnstillingene må bestemmes for hvert bildebehandlingssystem, da mange komponenter i den optiske banen vil påvirke den faktiske effekten som leveres inn i cellene. Lasermikrobestråling kan forårsake flere typer DNA-lesjoner avhengig av eksitasjonsbølgelengden, utgangseffekten til FRAP-laseren og om noen pre-sensibiliserende midler (som Bromodeoxyuridine eller Hoechst) ble brukt. 405 nm lasere kan forårsake oksidativ DNA-skade, enkle og doble strandede pauser16,17. Ved å bruke høyere laserutgangsinnstillinger øker mengden DSBer. I denne protokollen ble pre-sensibiliseringsmetoder ikke brukt, men disse teknikkene er sterkt dekket i litteraturen og gjeninnført i diskusjonen nedenfor. Etter vår mening er den beste måten å teste om ønsket lesjon genereres ved å teste for rekruttering av kjente DNA-skadeveispesifikke gener. Rekruttering av NTHL1 eller OGG1, komponenter i BER-banen, antyder induksjon av oksidert DNA-base10,11,17,18,19, mens FBXL10 eller XRCC5 indikerer tilstedeværelsen av DSBer8,20,21. Rekruttering av XRCC1 kan indikere både tilstedeværelsen av oksiderte DNA-baser og enkeltstrengede pauser (SSB)22,23. XPC (dvs. RAD4) er en god indikator på NER som fjerner de store DNA-adducts generert av ultrafiolett lys (UV)17,24. Fordi rekruttering av eksogene proteiner kan introdusere visse uregelmessigheter, kan immunfluorescerende farging av endogene DNA-reparasjonsproteiner eller markører (som γH2A.X for dobbeltstrengede pauser) bekrefte tilstedeværelsen av spesifikke DNA-lesjoner. Alternativt kan antistoffer som er reist mot bestemte typer DNA-lesjoner også brukes. For å justere den leverte laserkraften kan både oppholdstiden og laserkraften endres.
Ved hjelp av matematisk modellering kan det utføres en detaljert kinetisk analyse som kan gi verdifull innsikt i rekrutteringsegenskapene til INTERESSE (f.eks. bidrag av flere DNA-bindende domener, følsomhet mot ulike signalhendelser, etc.). Automatisert rekrutteringsevaluering og cellesporing kan kombineres for å skape robuste arbeidsflyter 1,25.
Fordeler og begrensninger ved DNA-pre-sensibilisering
Pre-sensibilisering av DNA før mikrobestråling er et vanlig brukt verktøy for DNA-reparasjon proteinrekruttering16,17. Sensibiliserende DNA før mikrobestråling gjør det mer utsatt for DSB-er. De to vanligste metodene for DNA pre-sensibilisering er forbehandling av celler med enten Bromodeoxyuridine (BrdU) eller Hoechst fargestoff. For systemer som ikke er i stand til mikrobestråling ved høye laserkrefter, kan disse metodene være nødvendige for å indusere DNA-lesjoner som DSB-er. I tillegg, i fravær av en overført lysdetektor eller et fluorescerende signal som fremhever cellekjernen (for eksempel når du studerer rekruttering av ukodede endogene DNA-reparasjonsproteiner), fungerer Hoechst som både et pre-sensibiliserende verktøy og en fluorescerende kjerneflekk. DNA pre-sensibilisering kan imidlertid introdusere betydelige komplikasjoner. BrdU (brukt ved en endelig konsentrasjon på 10 μM) må legges til celler 24 timer (eller tid tilsvarende en full cellesyklus i cellelinjen som brukes) for å kunne innlemmes riktig i DNA og kan forårsake cellesyklusinterferens26. Hoechst 33342 (brukes ved en endelig konsentrasjon på 1 μg/ml) er cytotoksisk etter lange inkubasjonsperioder, men krever tilstrekkelig tid til å mette kjernen med fargestoffet. Derfor bør den bare påføres 15-20 minutter før mikrobestråling; Ellers vil ikke rekrutteringsdataene være konsistente. Cellene farget på denne måten kan ikke holdes i kultur i mer enn noen få timer27,28. Pass på at du ikke bruker Hoechst 33358, som ikke er så cellegjennomtrengelig som Hoechst 33342 fargestoffet. Pre-sensibilisering kan også introdusere unødvendig varians blant eksperimenter og gjør eksperimentet enda mer følsomt for forskjeller i celletetthet (da dette vil påvirke mengden innlemmet fargestoff / celle).
Fordeler og begrensninger ved konfektmikroskopi
Bildehastigheten til konfektmikroskopi kan begrenses sammenlignet med widefieldmikrokopi. Imidlertid kan et konfokalt mikroskop utstyrt med en resonansskanner forbedre bildehastigheten (på bekostning av oppløsning) som kommer nær hastighetene til spinning-diskmikroskopi. Tre funksjoner gjør A1R HD25 konfokalt system til et utmerket valg for protokollen som presenteres her. For det første gjør systemets 25 mm FOV det mulig å bilde mellom 15-20 celler i et enkelt skannet felt (vs. 5-10 celler i vanlige oppsett), noe som begrenser antall oppkjøp som er nødvendige for å få nok celler til statistisk analyse. For det andre gjør FRAP-modulen og to scanheads det mulig å avbilde og mikrobestråle cellene samtidig, ikke bare sekvensielt. Til slutt gir fleksibiliteten ved å ha både resonans- og galvanoskannere muligheten til enkelt å bytte mellom høy temporal oppløsningsavbildning med eksepsjonell hastighet som minimerer slukking av fluoroforer, og bildebehandling med høy romlig oppløsning som bruker langsommere skannehastigheter for å produsere bilder med høyere signal-til-støy-forhold. Mens det brukte systemet tillot den nevnte fleksibiliteten, å ligne mer allment tilgjengelige konfokale mikroskopkonfigurasjoner, ble bare galvano-skanneren brukt i de presenterte eksperimentene (for både mikrobestråling og påfølgende avbildning).
Fordeler og begrensninger ved mikrobestråling
Mens mikrobestråling gir uovertruffen romlig og tidsmessig oppløsning, er den ikke uten begrensninger. DNA-skade ved lasermikrobestråling er svært gruppert til bestemte deler av kjernen sammenlignet med naturlig forekommende skadelige midler. Dermed kan kromatinrespons på grunn av mikrobestråling variere sammenlignet med homogent distribuert skade. I tillegg er mikrobestråling tidkrevende og kan bare utføres på noen få dusin celler, mens store befolkningsbaserte biokjemiske metoder (kromatinfraksjonering, immunoprecipitation, ChIP) kan gi økt robusthet ved å studere tusenvis av celler om gangen. Verifisering av observasjoner gjort ved mikrobestråling med tradisjonelle biokjemiske teknikker er en effektiv strategi for pålitelige konklusjoner. Selv om samtidig mikrobestråling av mange celler i en bestemt FOV er mulig, vil bildesystemet trenge mer tid til å utføre oppgaven. Derfor begrenser måling av dynamikken i proteiner som rekrutterer veldig raskt til DNA-lesjoner antall mulige ROIer for mikrobestråling som brukes samtidig. På bildesystemet som brukes til denne protokollen, tar mikrobestrålingen av en enkelt 1024 piksler lang avkastning 1032 ms ved hjelp av 1000 μs oppholdstid og 3088 ms ved hjelp av 3000 μs dveler tid å fullføre. Bruk av flere linjer med ROIer vil øke tiden det tar å fullføre mikrobestråling betydelig (f.eks. 7 x 1024 piksler lang avkastning tar 14402 ms ved hjelp av 1000 μs oppholdstid og 21598 ms ved bruk av 3000 μs oppholdstid). Denne gangen går tapt fra bildeinnhenting og må tas i betraktning. Ved avbildning av raske rekrutteringshendelser, bruk kortest mulig avkastning og bare mikrobestråle en celle om gangen.
Fordeler og begrensninger i forhold til synkroniseringsmetoder
For cellesyklusspesifikke studier innebærer de eksisterende metodene enten synkronisering av celler i bestemte cellesyklusfaser eller bruk av fluorescerende reportere for å identifisere cellens spesifikke cellesyklusfase. Hver av disse metodene gir imidlertid sine egne utfordringer og begrensninger.
FUCCI-systemet3 (avhengig av fluorescerende proteinmerkede avkortede former for CDT1 og Geminin) er et spesielt nyttig verktøy for cellesyklusstudier, men har begrensninger når det gjelder å skille mellom S- og G2-faser av cellesyklusen. Geminin-nivåene er allerede høye fra midten av S-fasen og holder seg høye til M-fasen, noe som gjør disse fasene vanskelige å skille. Bruk av FUCCI-systemet betyr også at to optiske kanaler i mikroskopet ikke kan brukes til avbildning av POI.
Ikke-kreftcellelinjer kan synkroniseres til G0 ved fjerning av vekstfaktorer som finnes i serumet (serum sult) forårsaker liten eller ingen DNA-skade på cellene. Imidlertid vil de fleste kreftcellelinjer delvis fortsette å utvikle seg gjennom cellesyklusen selv uten tilstrekkelige mengder serum i media. I tillegg begynner celler delvis å miste synkroniseringen innen slutten av G1, tidlig S-fase. I tillegg til serum sult, er det mange kjemiske metoder for å oppnå cellesyklussynkronisering. Hydroksyurea, afidolin og tymidinblokker er metoder for å stoppe DNA-replikasjon for å synkronisere celler inn i tidlig S-fase. Selv om disse metodene er billige og enkle, introduserer de replikeringsstress som resulterer i DNA-skade. Disse DNA-replikasjonshemmerne har vist seg å indusere fosforylering av H2A. X, en velkjent markør for DSBer2,29. Metoden for å bruke tagged-PCNA som markør for S-faseceller reduserer potensialet for gjenstander forårsaket av kjemisk synkronisering og kan brukes på et bredt spekter av cellelinjer sammenlignet med serum sult.
Konklusjon
DNA-skade er en drivkraft for genetiske sykdommer der mutagene lesjoner kan føre til ondartet transformasjon av celler. Målretting mot DNA-syntesemaskineriet er en grunnleggende terapeutisk strategi i behandling av hyperproliferative sykdommer som kreft. For å behandle disse sykdommene på en mer målrettet måte, trenger vi en bedre forståelse av proteinene som reparerer DNA-lesjoner. Protokollen som beskrives her hjelper mikrobestrålingsbaserte studier i S-fase ved å minimere utfordringene ved tradisjonelle synkroniseringsmetoder for å redusere mulige gjenstander og øke reproduserbarheten til forsøkene.
Disclosures
Forfatterne oppgir at utgivelsen av det presenterte arbeidet ble sponset av Nikon Corporation. Forfatterne erklærer at det ikke finnes konkurrerende interesser.
Acknowledgments
Forfatterne takker M. Pagano for hans kontinuerlige støtte samt D. Simoneschi, A. Marzio og G. Tang for deres kritiske gjennomgang av manuskriptet. B. Miwatani-Minter takker R. Miwatani og B. Minter for deres fortsatte støtte. G. Rona takker K. Ronane Jurasz og G. Rona for deres fortsatte støtte.
Materials
| Name | Company | Catalog Number | Comments |
| Ammonium chloride | Sigma-Aldrich | A9434-500G | For quenching formaldehyde |
| Anti-EXO1 Rabbit Polyclonal Antibody | Proteintech | 16253-1-AP | primary antibody |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | 05-636 | primary antibody |
| Bovine Serum Albumin | Sigma-Aldrich | 3117332001 | BSA for blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Merck | 19-160 | pre-sensitizing agent |
| Citifluor™ Mountant Solution AFR3 | Electron Microscopy Sciences | 17973-10 | antifade containing PBS solution for imaging |
| DAPI | Sigma-Aldrich | D9542-1MG | nucleic acid stain |
| DMEM Medium | Thermo Fisher Scientific | 10569010 | Cell culture medium for HEK293T cells |
| DMSO | Sigma-Aldrich | D2650-100ML | Vehichle control and dissolution solvent |
| EGFP-FBXL10 | Addgene | #126542 | viral expression vector for EGFP-FBXL10 |
| EXO1b-AcGFP (in pRetroQ) | custom cloning | na | EXO1b cDNA was cloned in the NheI, BamHI sites of pRetroQ-AcGFP1-N1 vector. |
| Fetal Bovine Serum | Gibco | 16140071 | Media supplement |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 | Phenol red free medium for microscopy |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A32723 | secondary antibody |
| HEK293T cells | ATCC | ATCC CRL-3216 | Cell line for viral packaging |
| HEPES | Sigma-Aldrich | H0887-100ML | Buffering agent to supplement live cell imaging medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | pre-sensitizing agent |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 | Transfection reagent |
| McCoy’s 5A (Modified) Medium | Life Technologies | 16600-108 | Cell culture medium for U-2 OS cells |
| mCherry-PCNA | Addgene | #55117 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA | Addgene | #55994 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA (in pBABE) | custom cloning | na | mPlum-PCNA cDNA was cloned from Addgene #55994 in the BamHI, SalI sites of pBABE (puro) |
| Nikon A1R-HD25 Confocal Scanhead and Controller | Nikon | na | confocal imaging system |
| Nikon LUN4 laser unit | Nikon | na | excitation system |
| Nikon LUN-F 50 mW 405 nm FRAP laser unit | Nikon | na | FRAP laser unit |
| Nikon NIS Elements Confocal Controller Software | Nikon | na | Confocal controlling software |
| Nikon Ti2-E Inverted Microscope | Nikon | na | inverted epifluorescent microscope base |
| Nikon Ti2-LAPP Modular Illumination System | Nikon | na | illumination system |
| NTHL1-mCherry (in pRetroQ) | custom cloning | na | NTHL1 cDNA was cloned in the NheI, SalI sites of pRetroQ-mCherry-N1 vector. |
| Nunc Lab-Tek II Chambered Coverglass (4 well) | Thermo Fisher Scientific | 155382PK | Live cell microscopy cell culture chamber |
| Olaparib | Selleck Chemicals | S1060 | PARP inhibitor |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985062 | Dilution medium for transient transfection |
| Paraformaldehyde aqueous solution (32%) | Thermo Fisher Scientific | 50-980-494 | Fixative |
| pBABE (hygro) | Addgene | #1765 | retroviral expression vector (for low expression levels) |
| pBABE (neo) | Addgene | #1767 | retroviral expression vector (for low expression levels) |
| pBABE (puro) | Addgene | #1764 | retroviral expression vector (for low expression levels) |
| pBABE (zeo) | Addgene | #1766 | retroviral expression vector (for low expression levels) |
| PCNA Antibody (PC10) | Santa Cruz | sc-56 | primary antibody |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco | 10378016 | Media supplement |
| polybrene | Sigma-Aldrich | TR-1003 | Increase viral infection efficiency |
| pRetroQ-AcGFP-C1 | Takara | 632506 | retroviral expression vector |
| pRetroQ-AcGFP-N1 | Takara | 632505 | retroviral expression vector |
| pRetroQ-mCherry-C1 | Takara | 632567 | retroviral expression vector |
| pRetroQ-mCherry-N1 | Takara | 632568 | retroviral expression vector |
| pUMVC | Addgene | #8449 | Viral packaging vector |
| Sodium-pyruvate | Thermo Fisher Scientific | 11360070 | Supplement for live cell imaging medium |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | Dilute in PBS for cell permeabilization buffer |
| Trypsin-EDTA Solution 10X | Sigma-Aldrich | 59418C-100ML | Dilute in PBS to split cells |
| U-2 OS Cells | ATCC | HTB-96 | Optimal cell line for microscopy experiments |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K | PCR based Mycoplasma detection kit |
| VSV-G | Addgene | #8454 | Viral protein envelope vector |
References
- Aleksandrov, R., et al.
- Darzynkiewicz, Z., Halicka, H. D., Zhao, H., Podhorecka, M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: Analysis by flow cytometry. Methods in Molecular Biology. 761, 85-96 (2011).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Herce, H. D., Rajan, M., Lattig-Tunnemann, G., Fillies, M., Cardoso, M. C. A novel cell permeable DNA replication and repair marker. Nucleus. 5 (6), 590-600 (2014).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in dna replication with putative roles in cancer. International Journal of Molecular Sciences. 20 (1), (2018).
- Cheruiyot, A., et al. Poly(ADP-ribose)-binding promotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amsterdam). 35, 106-115 (2015).
- Zhang, F., Shi, J., Chen, S. H., Bian, C., Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Research. 43 (22), 10782-10794 (2015).
- Rona, G., et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. elife. 7, (2018).
- Young, L. M., et al. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the dna damage response. Cell Reports. 13 (3), 451-459 (2015).
- Kong, X., et al. Laser microirradiation to study in vivo cellular responses to simple and complex dna damage. Journal of Visualized Experiments. (131), e56213 (2018).
- Kong, X., et al. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS One. 6 (8), 23548 (2011).
- Lan, L., et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Research. 42 (4), 2330-2345 (2014).
- Zerjatke, T., et al. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Reports. 19 (9), 1953-1966 (2017).
- Ji, Y., Karbaschi, M., Cooke, M. S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutation Research. 845, 403054 (2019).
- Sun, G., et al. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infection and Immunity. 76 (10), 4405-4413 (2008).
- Gassman, N. R., Wilson, S. H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amsterdam). 31, 52-63 (2015).
- Muster, B., Rapp, A., Cardoso, M. C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics. 4 (1), 47-68 (2017).
- Ikeda, S., et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 273 (34), 21585-21593 (1998).
- Rosenquist, T. A., Zharkov, D. O., Grollman, A. P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proceedings of the National Academy of Science U. S. A. 94 (14), 7429-7434 (1997).
- Reid, D. A., et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Science U. S. A. 112 (20), 2575-2584 (2015).
- Taccioli, G. E., et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 265 (5177), 1442-1445 (1994).
- Marsin, S., et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 278 (45), 44068-44074 (2003).
- Thompson, L. H., Brookman, K. W., Jones, N. J., Allen, S. A., Carrano, A. V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cell Biology. 10 (12), 6160-6171 (1990).
- Scharer, O. D.
- Oeck, S., et al. High-throughput evaluation of protein migration and localization after laser micro-irradiation. Science Reports. 9 (1), 3148 (2019).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Science Reports. 6, 19567 (2016).
- Durand, R. E., Olive, P. L. Cytotoxicity, mutagenicity and dna damage by hoechst 33342. Journal of Histochemistry and Cytochemistry. 30 (2), 111-116 (1982).
- Tobey, R. A., Oishi, N., Crissman, H. A. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proceedings of the National Academy of Science U. S. A. 87 (13), 5104-5108 (1990).
- Podhorecka, M., Skladanowski, A., Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. Journal of Nucleic Acids. 2010, (2010).
