

Summary
このプロトコルは、レーザーマイクロ照射によるDNA修復タンパク質の採用測定など、下流顕微鏡研究のためのS相細胞を効率的に同定するための非侵襲的な方法を記述する。
Abstract
DNA損傷修復は、反応性の高い環境で細胞の遺伝的完全性を維持します。細胞は、代謝活動や紫外線などの内因性および外因性の両方の原因による様々なタイプのDNA損傷を蓄積し得る。DNA修復がなければ、細胞の遺伝コードが損なわれ、タンパク質の構造や機能が損なわれ、病気を引き起こす可能性があります。
様々な細胞周期過程における異なるDNA修復経路の時空間的ダイナミクスを理解することは、DNA損傷修復の分野で重要である。現在の蛍光顕微鏡技術は、DNA損傷誘導後の異なる修復タンパク質の採用運動を測定するための優れたツールを提供します。細胞周期のS相中のDNA合成は、DNA修復に関する細胞運命の特異な点である。これは、間違いのためにゲノム全体をスクリーニングするためのユニークなウィンドウを提供します。同時に、DNA合成エラーは、非分裂細胞では検出されないDNA完全性に対する脅威にもなります。したがって、DNA修復プロセスは、細胞周期の他の段階と比較してS相において有意に異なり、それらの違いは十分に理解されていない。
以下のプロトコルは、405nmレーザーラインを搭載したレーザー走査共焦点顕微鏡を用いて、細胞株の調製およびS相におけるDNA修復タンパク質のダイナミクスの測定を記述する。タグ付きPCNA(mPlum付き)は、関心のあるAcGFP標識修復タンパク質(すなわちEXO1b)と組み合わせた細胞周期マーカーとして使用され、S相におけるDNA損傷の採用を測定する。
Introduction
いくつかのDNA修復経路は、細胞内で発生する可能性のあるさまざまなタイプのDNA病変に対処するために進化しており、そのすべてが空間と時間の両方で高度に調節されています。細胞周期の最も脆弱な期間の1つは、DNA合成が起こるS相である。増殖は生命の基本である一方で、大きな課題でもあります。細胞は、突然変異が将来の世代に受け継がれないように、ゲノムの忠実な複製を確実にする必要があります。その結果、増殖は、腫瘍学の分野における治療アプローチの開発に用いられてきた治療的介入点を提供する。
DNA病変におけるタンパク質のリクルートを研究するために使用される主要な技術には、その強みと限界があります。マイクロ照射は、イオン化放射線誘導病巣(IRIF)、クロマチン免疫沈降(ChIP)、または生化学的分別の免疫蛍光イメージングのような代替方法のほとんどよりも、空間的および時間的分解能1 を有する。しかし、マイクロ照射ラックは、同時に多数の細胞をサンプリングすることができる前述の技術の堅牢性をlac.
S相でDNA修復を調べるには、非同期細胞培養集団中のS相細胞を区別できなければならない。これに対処する多くのよく知られた方法は、細胞の同期、または異なる細胞周期の段階の視覚化を含む。しかし、どちらのアプローチも、重大な課題と可能なアーティファクトを導入します。S初期の段階で細胞を濃縮するために広く使用される化学同期方法(例えば、二重チミジンブロック、アブフィジコリン、ヒドロキシ尿素処理)は、複製応力の誘導および最終的にはDNA損傷自体を通じて同期を達成する。これにより、Sフェーズ2でDNA修復プロセスを研究するためのこれらの方法の使用が制限されます。血清飢餓および放出による同期は、非形質細胞株と比較して細胞周期進行の成長因子に依存しない癌細胞株を主に除く、限られた数の細胞株にのみ適用可能である。蛍光ユビキチン細胞周期指標(FUCCI)システムは、細胞周期を研究するのに特に有用なツールであるが、SとG2細胞周期相を分化する場合には基本的な制限がある。
ここでは、蛍光タグ付きPCNAを非侵襲的なS相マーカーとして使用すると、化学細胞サイクル同期方法の欠点を制限し、FUCCIシステムよりも特異性と柔軟性を可能にすることが示されています。単一マーカーとして、PCNAは非同期集団におけるS相細胞を強調できるだけでなく、S相内の細胞の正確な進行(すなわち、早期、中期、後期S相)4を示すことができる。外因性の低い発現レベル、タグ付きPCNAは、細胞周期進行およびDNA修復プロセスの両方に対する最小限の干渉を保証します。重要なことに、PCNAは、複数のDNA病変の修復に関与し、局所的に誘発されたDNA損傷部位1、4に採用される、適切なDNA損傷誘導のための内部制御としても機能する。
ここで示した実験は、S相でEXO1bの採用ダイナミクスを測定する方法と、これが確立されたPARP阻害剤であるオラパリブによってどのように影響を受けるかを示しています。EXO1bヌクレアーゼ活性は、ミスマッチ修復(MMR)、ヌクレオチド切除修復(NER)、および二本鎖破断(DSB)修復を含む幅広いDNA修復経路に関連しています。S相では、EXO1bはDNA切除5中に3'ssDNAオーバーハングの形成を通じて相同組換え(HR)に大きな役割を果たす。EXO1bは、停止したDNAフォークを再開するチェックポイント活性化における役割を持つDNA複製にさらに関与しており、また、複製5におけるストランド変位時の遅れ鎖でのプライマー除去および岡崎フラグメント成熟を再開する。損傷したDNAサイトへのEXO1bのリクルートメントは、ポリ(ADP-リボース)(PAR)6,7との直接相互作用によって調節される。EXO1bの細胞周期特異的な意味が数多くあるため、PCNAを用いたS相特異的採用研究に最適です。
Protocol
1. ヒト骨肉腫由来細胞の培養(U-2 OS)
注:U-2 OS細胞は、平坦な形態、大きな核を有し、ガラスを含むいくつかの表面に強く付着しているため、これらの研究に最適です。同様の特性を持つ他の細胞株も使用できる。
- U-2 OS細胞株の培養には、10%のウシ胎児血清(FBS)と抗生物質(100 U/mLペニシリンおよび100μg/mLストレプトマイシン)を添加したマッコイの5A培地を使用してください。5%CO2を含む加湿雰囲気で37°Cの細胞をインキュベートする。顕微鏡検査の研究では、十分な細胞数を提供するために10cm皿で細胞培養を維持する。
- 細胞が90%の合流度(7 x 106 細胞/10 cmの皿)に近づくと、細胞を分割する。
- 細胞をPBSでリンスし、血清中に含まれるトリプシン阻害剤を洗い流す。
- トリプシン-EDTAを1 mL加え、細胞層が均等に覆われていることを確認します。
- 細胞層がプレートから持ち上げられるまで37°Cでインキュベートする(約6分)。
- 培地を含む血清中のトリプシン化細胞を再懸濁し、1/10分 の体積(約0.7 x106 細胞)を10mLの補充増殖培地を含む新しい10cmプレートに添加します。
- 実験に先立ち、メーカーの推奨に従ってユニバーサルマイコプラズマ検出キットを使用して、マイコプラズマ汚染の細胞を定期的にテストします。
2. レトロウイルス感染
注:BSL-2安全対策および組換えウイルスを扱う際には、次の項をご覧ください: NIHガイドライン、セクション III-D-3: 組織培養における組換えウイルス.
- 種子4 x 106 HEK293T細胞は、10cm培養皿にめっきした後、24時間以内に〜60%の合流を達成した。
- HEK293Tの栽培については、このプロトコルの1.1-1.3に記載のU-2 OSの栽培手順に従ってください。HEK293Tの場合は、マッコイの5A培地をDMEMに置き換えます。HEK293T細胞は、組織培養プレートに弱く付着する場合は、必ず穏やかに洗浄してください。
- プラスミドのウイルス包装用脂質系トランスフェクション試薬を用いたトランスフェクトHEK293T細胞。
- レトロウイルスベクターの場合、1.5 μgのVSV-G(Addgene #8454)と1.5 μgのpUMVC(Addgene #8449)を、目的の遺伝子を含むベクターの3 μg(ピューロマイシン耐性を有するレトロウイルスベクター骨格)と組み合わせて、250 μLのオプチム培地中の血清培地に組み合わせます。オプティ-MEM/DNA混合物(この場合は6μL)に添加したDNAの各μGに対してP3000試薬1μLを加え、タップして軽く混ぜます。上下に渦やピペットをしないでください。
- 別のマイクロ遠心分離チューブでは、トランスフェクション試薬の2μG DNA(この場合は12 μL)に250 μLのオプティ-MEM還元血清培地を組み合わせます。
- 2つの混合物(500 μLを組み合わせ、渦を混ぜない、穏やかなタッピングのみで混ぜる)を組み合わせ、室温で15分間インキュベートさせます。
- 慎重に、細胞を取り外すことなく、播種HEK293T細胞に滴下して混合物を追加します。プレートをそっと旋回します。
- ウイルス感染は安定した細胞株を生成する。
- トランスフェクション後72時間のHEK293T細胞から上清を含むウイルスを除去する。0.45 μmのフィルターで慎重にフィルターを適用して、細胞の破片や脱離した細胞を除去します。必要に応じて、ウイルス感染を促進するために、ウイルス上清に8μg/mLポリブレンを加える。
- U-2 OS細胞に上清を含むウイルスを10cmの皿(〜3 x 106 細胞)で〜50%の合流度で加える。前日に U-2 OS セルをシードします。
- ウイルス含有上清を除去し、廃棄する前に6-16時間感染する。
注:目的の遺伝子に対して所望の過剰発現量を達成するために、一定の時間、一連のウイルス希釈液をインキュベートします。新たに確立された各細胞株におけるトランスジーンの発現レベルを、内因性レベルと比較してウェスタンブロットで確認する。 - 細胞が適切な抗生物質の存在下で(2μg/mL最終濃度でピューロマイシンの場合は3〜4日間)を選択し、顕微鏡下で目的の蛍光タンパク質タグ付き遺伝子の発現を検証することを許可する。
- これらの手順を繰り返して、二重ラベルのセルラインを生成します。ここで提示した実験ではmPlum-PCNAは、EXO1B-AcGFPと組み合わせたレトロウイルスベクター(pBABE)から発現し、またレトロウイルスベクター(pRetroQ-AcGFP1-N1)からも発現した。
3. 微小照射用細胞の調製
- めっき細胞:実験の24時間前に、合計8.0 x 104 細胞を500 μL-1 mLの間の体積に入れ(約70%の合流率)、高倍化共焦点顕微鏡とレーザーマイクロ照射に理想的な結果をもたらす4つのウェルチャンバーカバーグラスに(約70%の合流度)より高い細胞の合流度は単一視野(FOV)でより多くの細胞を測定することを可能にする;しかし、完全にコンフルなスライドは、細胞周期の不規則性を導入します.
- イメージングメディア:マイクロ照射の1時間前に、フルオロブライトDMEMの定期的な成長培地を交換し、10%FBS、100 U/mLペニシリン、100 μg/mLストレプトマイシン、15 mM HEPES(pH=7.4)、1mMナトリウム-ピルビン酸を補います。このイメージングメディアは、信号対雑音比を最大化し、非常に薄暗い蛍光を検出するのに役立ちます。HEPESを含有するため、5%CO2雰囲気の無い場合にもpHを安定化させる。
- このステップでイメージングの前に任意の追加の治療を適用します。ここで提示した実験では、細胞を、olaparib(PARP阻害剤、1μM最終濃度で)または車両制御(DMSO)1、8、9のいずれかでイメージングする1時間前に前処理した。
4. 顕微鏡を準備し、イメージング用のS相細胞を選択する。
- 最良の結果を得るには、ここで概説するシステムと同様の特性を持つ共焦点システムを使用します。ここで提示した実験は、反転した顕微鏡スタンドに取り付けられた共焦点顕微鏡を用いて行った( 材料表を参照)。
注:ここで使用される顕微鏡は50 mW 405 nm FRAPレーザーモジュールおよび60x 1.4 NAオイルプランアポクロマートの目的が装備されていた。共焦点スキャンヘッドには、ガルバノスキャナ(高解像度用)と共振スキャナ(高速イメージング用)の2つのスキャナオプションがありました。- ソフトウェア制御のXYガルバノデバイスを介して、光漂白(FRAP)レーザー後の蛍光回収をサンプルに導入します。488 nmレーザーラインを使用してAcGFPと561 nmまたは594 nmのレーザーラインを励起してmPlumを励起します。
注:次のフィルタの組み合わせは最適な結果を与える:560 nmのロングパスフィルタを使用して、560 nm未満の波長の発光光はAcGFP用の525/50 nmの発光フィルタを通過し、560nmより高い波長の発光光はmPlum用の595/50 nmの発光フィルタを通過した。蛍光ブリードスルーを最小限に抑える適切なフィルタセット(FITC/TRITC、GFP/mCherry、FITC/TxRedなど)を使用できます。
- ソフトウェア制御のXYガルバノデバイスを介して、光漂白(FRAP)レーザー後の蛍光回収をサンプルに導入します。488 nmレーザーラインを使用してAcGFPと561 nmまたは594 nmのレーザーラインを励起してmPlumを励起します。
- 環境室と顕微鏡部品をオンにします。
- 加熱(可能な場合はステージ、目標、環境室)、CO2供給、湿度調整器を少なくとも4時間前に、安定した画像取得のための熱平衡を確保します。
- 顕微鏡に細胞を移す前に、少なくとも1時間レーザーラインと一緒に光源を初期化します。
- 蛍光タグ付きPCNAをマーカーとして使用して、非同期集団のS相細胞を選択します。以下の手順に従って、これを行います。
- この細胞周期相の同定を可能にするS相でmPlumタグ付きPCNAのユニークなローカリゼーションパターンを探します。PCNAは、核子から除外されながら、細胞周期のG1およびG2相における核内の完全に均質な分布を有する。S相では、PCNAは核内の応答体の位置に病巣を形成する。 図1 は、S相全体におけるPCNA病巣の異なるパターンを示しており、早期、中期、後期S相を区別することさえ可能です。
- 眼を見て、マイクロ照射に十分なS相細胞を持つFOVを選択します。非同期 U-2 OS セルは通常、S フェーズでの人口の 30 ~ 40% を占めています。
- PCNAと関心のあるタンパク質(POI)の両方の発現レベル(明るい細胞と薄暗い細胞)の極端な(同様に明るい細胞)を避けるようにしてください。この場合、EXO1b-AcGFPは実験的なアーティファクトにつながる可能性があります。
- 適切なFOVを見つける場合は、光の漂白や望ましくないDNA損傷を最小限に抑えるために、長時間フィールドをスキャンしないようにしてください。
- マイクロ照射に対して目的の関心領域(ROI)を設定します。関連するソフトウェアを使用して( 表を参照)、最初にバイナリ行を挿入して希望のROIを設定します(必要な行数と間隔を設定します)。[ バイナリ] をクリックし、[行の挿入] をクリック |サークル|楕円 を使用して、目的の数の線を描画します。
- これらのバイナリラインをROIに変換し、最後にこれらのROIを刺激ROIに変換します。これを行うには、最初に [ROI]をクリックし、[ バイナリを ROI に移動] をクリックし、ROI のいずれかを右クリックして、[ 刺激 ROI: S1 として使用] を選択します。これらの線をFOVに入れ、細胞の核を通過させます。FOV 全体に及ぶ 1024 ピクセルの長さの ROI は、プロトコル全体で使用されました。
5. 免疫蛍光染色または時間経過イメージングのためのマイクロ照射。
- 最適なマイクロ照射設定の決定
- 細胞のマイクロ照射前に、後で分析するために、FOVの高分解能画像を撮ってPCNA病巣を同定してください。逐次スキャンの代わりに、使用される両方の光チャネル(緑と赤)を同時に記録し、2つの波長での走査間の細胞の移動を避ける。fociの適切な解像度のために、1xズーム(ここで使用されるイメージングシステムでは0.29 μmピクセルサイズ)で、1/8フレーム/秒のスキャン速度(4.85 μs/ピクセル)を2倍の平均で使用します。これらのパラメータが A1 LFOV コンパクト GUI および A1 LFOV スキャン エリア ウィンドウで設定されたら、 キャプチャ ボタンを押して FOV を記録します。
注: 比較結果を確実にするためには、実験全体を通して同じピクセル サイズを維持することが重要です。 - マイクロ照射を設定するには、イメージングソフトウェアの ND刺激 タブを開き 、時間スケジュール(A1 LFOV / Galvano Device) ウィンドウにアクセスします。ガルバノスキャナを使用して一連のプレ刺激画像を取得し、刺激(LUN-F 50 mW 405 nm FRAPレーザーを使用)、ガルバノスキャナを使用して一連の刺激後画像を再び取得します。最初に 、[タイム スケジュール ] ウィンドウに 3 つのフェーズを設定します。 Acq/Stim 列で 、取得|を選択します。漂白| 3つの相の取得をそれぞれ行う。漂白フェーズの場合は 、S1 を ROI に設定します。
注: ここで紹介した実験では、刺激段階で画像が取得されませんでした。 - Galvano XY ウィンドウで、マイクロ照射の重要な要素を設定します: 405 nm レーザー出力、ドウェル時間 (このシステムでは、反復はデフォルトで 1)。ここで発表した実験では、細胞に405 nm FRAPレーザー(繊維先端の50mW)を1000-3000μsのドウェルタイムで100%の出力で照射しました。
注: レーザードウェル時間はピクセル単位で行われるため、ピクセルサイズが同じである限り、ドウェル時間と電力密度の関係は異なるFOV間で比較されます。 図2Aは 、特定の損傷誘導のためのレーザーパワー設定を最適化するためのDNA損傷応答(DDR)経路特異的タンパク質(DSB用FBXL10および酸化塩基損傷用NTHL1)の使用を示す。これらの安定した細胞株は、プロトコルのセクション2に続くウイルス感染で生成された。
- 細胞のマイクロ照射前に、後で分析するために、FOVの高分解能画像を撮ってPCNA病巣を同定してください。逐次スキャンの代わりに、使用される両方の光チャネル(緑と赤)を同時に記録し、2つの波長での走査間の細胞の移動を避ける。fociの適切な解像度のために、1xズーム(ここで使用されるイメージングシステムでは0.29 μmピクセルサイズ)で、1/8フレーム/秒のスキャン速度(4.85 μs/ピクセル)を2倍の平均で使用します。これらのパラメータが A1 LFOV コンパクト GUI および A1 LFOV スキャン エリア ウィンドウで設定されたら、 キャプチャ ボタンを押して FOV を記録します。
- タイムラプスイメージング。
- タイムスケジュール、A1 LFOVコンパクトGUI、A1 LFOVスキャンエリアウィンドウを使用して、目的の時間枠と間隔のタイムラプスイメージングを設定します。ここで発表された実験では、EXO1bとPCNAの募集を12分間画像化し、1024 x 1024ピクセル/フィールドで5秒ごとにFOVをスキャンし、1xズーム(ここで使用されるイメージングシステムでは0.29 μmピクセルサイズ)を使用し、0.35フレーム/秒のスキャン速度(1.45μ/pixel)を平均化せずに画像化しました。
- A1 LFOV Compact GUIウィンドウでのイメージング中に、レーザーパワー %、ゲインおよびオフセット設定を最適化して、画像の撮影中に光の漂白を減らします。POIとPCNAの両方を測定することを目指す場合は、シーケンシャルスキャンの代わりに同時スキャンを使用して、2つの別々のフルオロフォアのフィールド間のセルの移動を避けます。
- イメージングシステムは、以下の設定で使用されました。488 nmレーザーライン(20 mW):7%レーザーパワー、ゲイン:45(GaAsP検出器)2のオフセット、561 nmレーザーライン(20 mW):4%レーザーパワー、ゲイン40(GaAsP検出器)の2のオフセット。
- タンパク質の運動に応じて、画像間の間隔または全時間経過の持続時間を延長または短縮する。[ タイム スケジュール ]ウィンドウで、3 番目のフェーズの取得行に必要な 間隔 と 期間 を設定します。
- マイクロ照射とその後の時間経過イメージングを実行するには 、今すぐ実行 を押します。
- タイムラプスイメージングの最後に、刺激ROIを別々の画像として保存し、分析に使用される下流のソフトウェアでマイクロ照射の座標を識別するのに役立ちます。
- 免疫蛍光染色。
注:ステップ5.1.3および図2Aは、マイクロ照射によって導入されたDNA病変の種類を評価するために既知のDNA修復タンパク質を使用することを示しています。特定のDNA病変は、細胞を固定した後に特定の抗体を使用することによっても検出することができる。また、内因性タンパク質の抗体検出によるPOIの採用を検出することもできる。DSB をチェックするための γH2A.X の視覚化を以下に示します (図 2B)。図3は、内因性および外因性タグ付きPCNAの細胞周期全体におけるPCNAの局在および採用の一貫性を示す。- ステップ5.1.3の後、mPlum-PCNAの採用に基づいて適切なFRAPイベントを確実にするために、マイクロ照射後に1つの画像を撮ってください。FOVの正確な座標をメモし、後で免疫蛍光標識の後にフィールドを見つける。
- 細胞培養チャンバーを顕微鏡から取り出し、5〜10分間5%CO2 を含む加湿雰囲気で37°Cの細胞をインキュベートします。
注:パラホルムアルデヒド(PFA)は有毒であり、作業は換気の良いエリアまたはヒュームフードで行われるべきです。すべての後続の洗浄およびインキュベーションは4ウェルチャンバースライドの0.5 mLの容積と行われる。インキュベーション時間の後、0.5 mLのPBS(137 mM NaCl、2.7 mM KCl、8 mM Na2HPO 4、および2 mM KH2PO4)で細胞を洗浄し、室温(RT)で10分間PBSで0.5mLの4%PFAで固定します。 - PBSで細胞を1回洗浄し、50 mM NH4Clで洗浄し、残留PFAを焼き付けます。
- PBSで0.1%トリトンX-100でRTで15分間細胞を透過させます。
- ブロッキングバッファ(PBSで5%FBS、3%BSA、0.05%トリトンX-100)で1時間サンプルをブロックします。
- ブロッキング溶液を取り出し、希釈された一次抗体(抗γH2A.X,1:2000)をRTで1時間ブロックする緩衝液に添加します。
- ブロッキングバッファー3 x 10分でウェルを洗います。
- 希釈された二次抗体(抗マウスAlexa 488 Plusコンジュゲート、1:2000)をRTで1時間ブロックするバッファーに添加します。
- ブロッキングバッファー3 x 10分でウェルを洗います。
- 1 μg/mL DAPI溶液を用いて核に対抗し、PBSで15分間使用します。
- PBSで細胞を一度洗います。撮像は、PBSまたはPBS溶液に対して、抗フェード試薬(例えば、AFR3)で直接行い、光の漂白を低減することができる。
6. 採用分析
注:図 4Aは、DMSO または OLAParib が存在する場合の Exo1b および PCNA の採用の代表的なイメージを示しています。図4Bは、データ分析のための代表的な画像を示す。平均蛍光値は、フィジーを使用して異なるタイムポイントにわたってmPlum-PCNA(A、黄色の長方形)によって強調されたレーザートラックに沿って長方形を使用して平均AcGFP強度を測定することによって計算されました。PCNA は、ROI 座標に沿った照射を正常に行うことを強調する内部制御として機能します。同様に、平均AcGFP蛍光値も、核の損傷のない領域(B、青い長方形)について計算した。バックグラウンド信号強度は、未設定の領域(C、赤い長方形)で測定され、平均蛍光値から差し引かれた(図AおよびB)。従って、各データ収集点に対する相対平均蛍光単位(RFU)を、RFU=(A−C)/(B−C)8,9の式により計算した。マイクロ照射領域の得られたRFU値は、マイクロ照射前のRFU値に正規化される。
- マイクロ照射部位の領域Aを定義するためには、測定から細胞のヌクレオラー領域、複製病巣、および不規則核領域を除外する。フィジーで 2 つの ROI を描画する間に シフト キーを押し、2 つのリージョンを 1 つにグループ化します。
注:タンパク質の採用は、異なる遺伝子や照射条件によって異なります。したがって、領域Aのサイズは個別に決定されなければならない。領域 A のピクセル幅が決定されると、比較採用の場合は一定の値を維持する必要があります。ここで示した実験では、7ピクセル幅の長方形が使用された。 - 記録されたビデオの実行中に移動したセルを分析から除外します。高度に移動性の細胞を含めるには、説明した分析をフレームごとに行う必要があります。
- 採用プロファイルを視覚化するには、統計ソフトウェアを使用して正規化された RFU 値を時間に対してプロットします。
- マン・ホイットニー検定を使用して、DMSOとオラパリブ(n=31)処理の間の指示された時点での差を計算します。
Representative Results
細胞は、それぞれのタイプのDNA病変に特定の方法で対処し、それらがどの細胞周期段階にあるかによっても異なる。例えば、マイクロ照射後、二本鎖破断(DSB)は、非相同末端接合(NHEJ)または細胞周期相に応じてHRのいずれかによって処理される。細胞周期のSおよびG2段階の間に最も広範囲に作用するヌクレアーゼは、適切なHRにとって極めて重要なDNA突出しを作り出す。S期における細胞の評価を促進するために、PCNAを単色細胞周期マーカーとして採用した。 図1A は、細胞周期進行中のmPlum-PCNAの局在プロファイルを示す。PCNAは、G1およびG2相の核内で完全に均質な分布を有する(核子からもほとんど除外されている)。S相では、PCNAはDNA複製の部位に局部化し、核内の明るいスポットとして視覚化することができる。初期S相細胞では、スポットは比較的小さく、細胞の核全体に均等に分布している。S期中に進み、斑点がぼやけ、核と核長体の周囲に向かってより多くの局部化する。後期Sフェーズでは、スポットは数が減少しますが、PCNAが後期複製サイトに集中するにつれてますます大きくなっていきます(図1B)。重要なことに、pBABEベクター骨格からの外因性PCNA発現は内因性レベルよりも低かったが、細胞周期進行およびDDRにおける潜在的なアーティファクトを最小限に抑える顕微鏡による検出には十分であった。 図1C は内因性レベルと比較したPCNA過剰発現の程度を示す。mPlum-PCNAに対応するバンドは、サイズが大きいため、移行速度が遅くなりますのでご注意ください。
我々は、S相におけるこれらの病変へのEXO1bのPARP1/2依存的な採用を調査するために、マイクロ照射中にDSBを導入することを目指した。図2Aは、低用量のエネルギー(1000μs滞住時間)が、DSB応答者(FRUCC複合体8の成分)であるEGFP-FBXL10の採用を誘導しないことを示すが、NTHL1-mCherryの募集を誘導するのに十分であったが、塩基切除修復(BER)経路である、タンパク質リクルートの部位への酸化DNA損傷の部位への採用3000 μsの滞納時間で、EGFP-FBXL10とNTHL1-mCherryの両方がリクルートし、酸化病変とDSBの両方を生成するレーザー出力を実証します。 PCNAは、両方のレーザードウェル時間設定で十分に募集するので、細胞周期マーカーとマイクロ照射を成功させるためのマーカーの両方として機能します。重要なのは、外因性および/または内因性蛍光タンパク質タグ付きPCNAの両方が、同様に動作するようにこのレポーター機能に使用できることです(図3)。内因的にタグ付けされたPCNAは、PCNA遺伝子座13の1つのアレスルに最初のエキソンを持つフレームにmRubyを挿入することによって設計されました(細胞株はヨルグ・マンスフェルトの一種の贈り物でした)。
図4Aおよび図4Cは、S相細胞におけるAcGFPタグ付きEXO1bの採用を示す。EXO1bは約1分程度の微小照射部位で蓄積の最大レベルに達し、その後ゆっくりとDNA病変から離脱し始める。マイクロ照射部位における濃縮物は、グラフ上の>1の相対蛍光単位で示される。オラパリブの存在下では、1分でのレーザストライプでのEXO1bの蓄積は、車両制御と比較して有意に少ない。これらの結果は、文献6、7と一致しています。図4Bは、プロトコルのポイント6で説明されているように、定量のための代表的領域(A、B、およびC)を示しています。図4Dは、マイクロ照射に用いられる細胞における内因性EXO1bおよび外因性EXO1b-AcGFPの同等の発現レベルを示す。
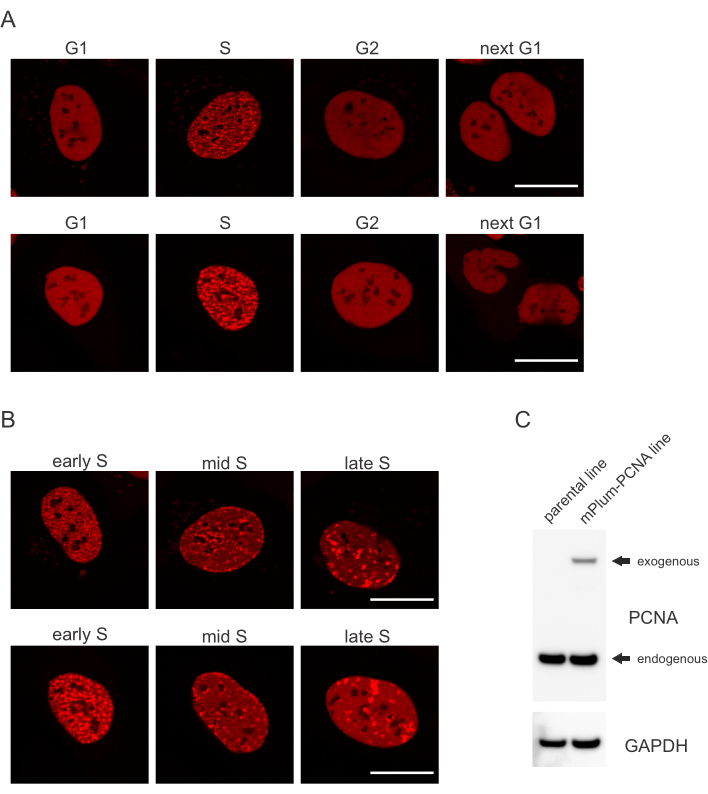
図1:PCNAのローカリゼーションパターン(A)画像は、U-2 OSセルの細胞周期全体で安定的に統合された外因性PCNAの局在パターンを示す。(B) 画像は、U-2 OS セルの S 段階 (初期、中、および後期) の異なる段階で PCNA 病巣パターンを示します。(C)イメージングに使用されるU-2 OS細胞における内因性および外因性レベルのPCNAを示すウェスタンブロット。スケールバーは20 μmを表します。

図2: 最適化されたレーザー出力によるDSBの誘導. (A) レーザー設定を最適化して、異なる形態のDNA損傷を誘発することができる。EGFP-FBXL10とNTHL1-mCherryの両方を安定的に発現するU-2 OS細胞を、それぞれDSBおよび酸化病変部位を同定するために使用した。405 nmレーザーラインによるマイクロ照射は、1000 μsまたは3000 μsのデュエルタイムを有する非同期U-2 OSセルで行った。スケールバーは、20μm(B)γH2A.Xに対する免疫蛍光染色を、mRubyタグ付き内因性PCNAを有するヒト網膜色素上皮細胞(hTERT RPE-1)に対して行った。細胞を固定し、マイクロ照射後5分で1000 μsまたは3000 μsのドウェル時間で処理した。スケールバーは20 μmを表します。

図3:1000μsまたは3000μsレーザードウェルタイムでのマイクロ照射部位への内因性mRuby-PCNAおよび外生mPlum-PCNAの同等の募集 S相中の内因性および外因性タグ付きPCNAフォーム複製病巣。 この図の大きなバージョンを表示するには、ここをクリックしてください。

図4:PARP1/2依存性EXO1bのS相採用EXO1b-AcGFPおよびmPlum-PCNAを安定的に発現するU-2 OS細胞は、3000 μsの住み込み時間を用いて405 nm FRAPレーザーラインをマイクロ照射した。(A)車両制御(DMSO)またはオラパリブ(1μM)のいずれかで前処理した後の示された時点でのマイクロ照射細胞の代表的な画像。スケールバーは、20 μm(B) 採用分析用のA、B、およびC領域の定義された領域の代表的な画像を表します。スケールバーは、生細胞イメージングによって捕捉された20μm(C)DNA損傷リクルートメントダイナミクスを表す。相対平均蛍光値および画像は、12分間5s毎に取得した。条件ごとに、≥30個の細胞を評価した。平均蛍光値(黒い実線)と標準誤差(シェーディング領域で視覚化された範囲)を時間に対してプロットした。破線は、マイクロ照射後1分の採用値を示す。DMSO(n=32)とオラパリブ(n=31)の間の差は、マン・ホイットニー検定を用いて計算された。アステリックスはp<0.0001を示す。(D)ウェスタンブロットは、微量照射に用いられる細胞における内因性EXO1bおよび外因性EXO1b-AcGFPの発現レベルを比較する。この図の大きなバージョンを表示するには、ここをクリックしてください。
Discussion
重要なステップと、プロトコルのトラブルシューティング/変更の可能性
マイクロ照射のための適切な組織培養容器は、成功のために重要です。ほとんどの高解像度イメージングシステムは、0.17 mm のカバーガラスの厚さに最適化されています。より高いまたはより低い厚さのイメージ投射室またはプラスチックポリマーから作られたもの(405 nmのイメージ投射のために最大限に活用されない)を使用して、著しくイメージの質を減らすことができる。ガラス表面を使用する場合は、細胞接着を強化するために処理された組織培養であることを確認してください。それらが組織培養処理されていない場合、これらのチャンバーは、例えば、細胞を播種する前にポリD-リジンでコーティングする必要があります。チャンバーカバーグラスに細胞をめっきする場合、細胞周期の不規則性や細胞への追加応力を避けるために理想的な細胞密度が最優先されます。安定した温度を維持するために実験する前に顕微鏡部品の適切な熱平衡は、時間経過イメージングを通じて焦点を維持するために重要であり、また、時間とサンプルにわたって均質なDDRを確保するために必要です。
細胞は、アーテフの実測データを減らすために、マイクロ照射の前に健康な状態にあることが重要です。細胞が感染後/選択した不規則な形態を有する場合、形態が正常に戻るまで細胞が複数の通路を進むことを可能にする。使用する細胞ラインにマイコプラズマ汚染が含まれるのを常に確認してください。マイコプラズマ感染の多くの有害作用の中で、それはまた、宿主細胞にDNA損傷を引き起こし、それらのDDR経路14、15に影響を与える可能性がある。細胞培養でマイコプラズマを検出する最も敏感な方法は、PCR(DAPIまたはHoechstによる検出に対する)を介して行います。
目的の修復タンパク質の最適な過剰発現は、しかし、検出するのに十分な高い内因性レベルに匹敵する必要があります。ウイルスベクターに使用されるプロモーター、感染時のウイルスの引き量体、および感染時間の長さはすべて、理想的な発現レベルに合わせて調整することができる。一貫した結果を得るために、個々の細胞クローンを分離して、均質な発現レベルと正常な細胞形態を確保する。適切な細胞周期およびDNA修復マーカー機能のために、内因性レベルよりも高いタグ付きPCNAを過剰発現しないベクター構築物を使用することが推奨される。PCNA過剰発現の低レベルであっても、S相細胞を判別するのに十分である。レトロウイルス pBABE ベクターは、この目的のために正常に使用されています (Addgene #1764, #1765, #1766, #1767).PCNAは、任意の単量体赤色(例えば、mPlum、mCherry、mRuby など)または単量体緑色蛍光タンパク質(例えば、mEGFP、AcGFP、mWasabi、mNeonGreen、mEmeraldなど)でタグ付けすることができ、交互にタグ付けされたPOIと組み合わせることができます。蛍光タグ付き POI を過剰表現する場合、いくつかの制限と考慮事項があります。蛍光タグは、正常なタンパク質機能および局在化を妨げる可能性があります。したがって、タグの位置(NまたはC末端)を考慮する必要があります。非単量体変異体のオリゴマー化がPOIの機能に影響を与える可能性があるとして、常に単量体蛍光タンパク質を使用する。
光路の多くのコンポーネントが細胞に送達される実際の電力に影響を与えるため、レーザー設定は各イメージングシステムごとに決定する必要があります。レーザー微小照射は、励起波長、FRAPレーザーの出力、および任意のプレ感作剤(ブロモデキシウリジンまたはホーチストのような)が使用された場合に、いくつかのタイプのDNA病変を引き起こす可能性があります。405 nmレーザーは、酸化的DNA損傷、単鎖および二本鎖破断16、17を引き起こす可能性があります。レーザー出力設定値を高くすることで、DSBの量が増加します。このプロトコルでは、予感作法は利用されなかったが、これらの技術は文献で大きくカバーされ、以下の議論で再びキャップされている。我々の意見では、所望の病変が生成されるかどうかをテストする最良の方法は、既知のDNA損傷経路特異的遺伝子の同定を試験することによってである。NTHL1またはOGG1の募集は、BER経路の成分、酸化DNA塩基10、11、17、18、19の誘導を示唆し、FBXL10またはXRCC5はDSBs8、20、21の存在を示す。XRCC1の採用は、酸化DNA塩基と単一鎖破断(SSB)22,23の両方の存在を示すことができる。XPC(すなわち、RAD4)は、紫外線(UV)17、24によって生成されるかさばるDNA付加物を除去するNERの良い指標である。外因性タンパク質をリクルートすると、ある種の不規則性を導入する可能性があるため、内因性DNA修復タンパク質またはマーカー(二本鎖破断のγH2A.Xなど)の免疫蛍光染色は、特定のDNA病変の存在を確認することができる。あるいは、特定のタイプのDNA病変に対して提起された抗体も使用できる。送達されたレーザーパワーを調整するために、ドウェル時間とレーザーパワーの両方を変更することができます。
数学的モデリングの助けを借りて、POIの採用特性(例えば、複数のDNA結合ドメインの寄与、異なるシグナル伝達事象に対する感受性)に関する貴重な洞察を提供できる詳細な運動分析を行うことができる。自動採用評価と細胞追跡を組み合わせて、堅牢なワークフロー 1,25を作成できます。
DNAプリリシタイズの利点と限界
マイクロ照射前のDNAの予感作は、DNA修復タンパク質募集16,17に一般的に用いられるツールである。微量照射前の感作DNAは、DSBの影響を受けやすくなります。DNAの予感作のための2つの最も一般的な方法は、ブロモデキシウリジン(BrdU)またはHoechst色素のいずれかによる細胞の前処理です。高いレーザーパワーでマイクロ照射ができないシステムでは、これらの方法はDSBのようなDNA病変を誘発するために必要である可能性があります。しかし、DNAの予感作は重大な合併症を導入する可能性がある。BrdU(10 μMの最終濃度で使用される)は、DNAに適切に組み込むために細胞に24時間(または使用される細胞株の全細胞周期に相当する時間)に添加する必要があり、細胞周期干渉を引き起こす可能性がある26。Hoechst 33342(1 μg/mLの最終濃度で使用される)は、長いインキュベーション期間の後に細胞毒性を有するが、色素で核を飽和するのに十分な時間を必要とする。したがって、マイクロ照射の15〜20分前にのみ適用する必要があります。それ以外の場合、採用データは一貫していません。このように染色された細胞は、27,28時間以上培養中に保てることができない。Hoechst 33342染料ほど透過性の細胞ではないHoechst 33358を使用しないでください。事前感作は実験間で不要な分散を導入し、細胞密度の違いにより敏感な実験を行う(これは組み込まれた色素/細胞の量に影響を与えるため)。
共焦点顕微鏡の利点と限界
共焦点顕微鏡のイメージング速度は、広視野顕微鏡と比較して制限される可能性があります。しかし、共振スキャナを搭載した共焦点顕微鏡は、回転ディスク顕微鏡の速度に近づく(分解能を犠牲にして)イメージング速度を大幅に向上させることができます。3つの特徴はA1R HD25の共焦点システムをここに提示される議定書のための優秀な選択にする。まず、システムの25mm FOVは、単一のスキャンフィールド(通常の設定では5〜10セル対5〜10セル)の15〜20個の細胞間で画像を作成し、統計分析に十分な細胞を得るために必要な取得数を制限することを可能にします。第二に、FRAPモジュールと2つのスキャンヘッドは、連続的にだけでなく、同時に細胞を画像化し、マイクロ照射することを可能にする。最後に、共振とガルバノスキャナの両方を持つ柔軟性は、蛍光葉の消光を最小限に抑える例外的な速度で高テンポラル解像度イメージングを簡単に切り替える機能と、より低速なスキャン速度を利用して高い信号対ノイズ比で画像を生成する高空間分解能イメージングを容易に行うことができます。上記の柔軟性を可能にする使用システムでは、より広く利用可能な共焦点顕微鏡構成に似たが、ガルバノスキャナのみが提示された実験(マイクロ照射および後続のイメージングの両方に対して)に使用された。
マイクロ照射の利点と限界
マイクロ照射は比類のない空間的および時間的分解能を提供するが、それは制限がないわけではない。レーザーマイクロ照射によるDNA損傷は、天然の損傷剤と比較して、核の特定の部分に非常にクラスター化されています。したがって、マイクロ照射によるクロマチン応答は、均質に分散した損傷と比較して異なる場合がある。さらに、微量照射は時間がかかり、数十個の細胞にしか行われないかもしれないが、大規模な集団ベースの生化学的方法(クロマチン分別、免疫沈降、ChIP)は、一度に何千もの細胞を研究することによって、より高い堅牢性を提供することができる。従来の生化学的手法によるマイクロ照射による観測の検証は、信頼できる結論を得るための効果的な戦略です。あるFOV内の多くの細胞に対して同時にマイクロ照射が可能であるが、撮像システムは、この作業を行うためにより多くの時間を必要とするだろう。したがって、DNA病変に非常に迅速にリクルートするタンパク質のダイナミクスを測定すると、同時に使用されるマイクロ照射に対する可能なROIの数が制限されます。このプロトコルに使用されるイメージングシステムでは、1024ピクセルの長いROIのマイクロ照射は、1000 μsのドウェル時間を使用して1032 ms、3000 μsのドウェル時間を使用して3088 msを使用して完了します。複数のROIラインを使用すると、マイクロ照射を完了するのに必要な時間が大幅に増加します(例えば、7 x 1024ピクセルの長さのROIは1000 μsのドウェル時間を使用して14402 ms、3000 μsのドウェル時間を使用して21598 msかかります)。この時間は画像取得から失われ、考慮する必要があります。迅速な採用イベントを撮影する場合は、可能な限り最短のROIを使用し、一度に1つのセルのみをマイクロ照射します。
同期方式に対する利点と制限
細胞周期特異的研究では、既存の方法は、細胞を特定の細胞周期相に同期させるか、蛍光レポーターを使用して細胞の特定の細胞周期段階を同定することを含む。ただし、これらの各方法には、独自の課題と制限があります。
FUCCIシステム3(CDT1およびジェミニンの切り捨てられた形の蛍光タンパク質に依存する)は細胞周期の研究のために特に有用な用具であるが、細胞周期のSおよびG2相の間の分化に関しては限界がある。ジェミニンレベルは、すでに中期S相から高く、M相まで高く保たれ、これらの相を分離することは困難です。FUCCIシステムを使用すると、2つの光学チャネルを使用して、POIをイメージングすることはできません。
非癌細胞株は、血清中に見られる成長因子(血清飢餓)を除去することによってG0に同期させることができ、細胞に対するDNA損傷はほとんどまたは全く起こさなかった。しかし、ほとんどの癌細胞株は、培地に十分な量の血清がなくても、細胞周期を経て部分的に進行し続けるだろう。さらに、セルは部分的に G1 後期、初期 S フェーズまでに同期を失い始めます。血清飢餓に加えて、細胞周期の同期を達成するために多数の化学的方法がある。ヒドロキシ尿素、アブフィジコリン、チミジンブロックは、DNA複製を停止して細胞を初期S相に同期させる方法です。これらの方法は安価でシンプルですが、複製ストレスを引き起こし、DNA損傷をもたらします。これらのDNA複製阻害剤は、H2Aのリン酸化を誘導することが示されている。X は、DSBs2,29のよく知られたマーカーです。S相細胞のマーカーとしてタグ付きPCNAを用いる方法は、化学的な同期によって引き起こされるアーティファクトの可能性を低減し、血清飢餓と比較して広範囲の細胞株に適用することができる。
結論
DNA損傷は、変異原性病変が細胞の悪性形質転換につながる可能性のある遺伝性疾患の原動力です。DNA合成機を標的とすることは、がんのような超増殖性疾患の治療における基本的な治療戦略である。これらの疾患をより標的にした方法で治療するためには、DNA病変を修復するタンパク質をよりよく理解する必要があります。ここで説明するプロトコルは、従来の同期手法によって提示される課題を最小限に抑え、可能なアーティファクトを低減し、実験の再現性を高めることによって、S段階でのマイクロ照射ベースの研究に役立ちます。
Disclosures
著者らは、提示された作品の出版はニコン株式会社が後援したと述べている。著者らは、競合する利益は存在しない、と宣言している。
Acknowledgments
著者らは、M.パガーノの継続的な支援に感謝し、D.シモネスキ、A.マルツィオ、G.タンが原稿の批判的なレビューを行ってくれたことに感謝しています。B.三和谷ミンターは、R.三和谷とB.ミンターの継続的なサポートに感謝します。G・ロナは、K・ロナネ・ジュラシュとG・ロナの継続的な支援に感謝します。
Materials
| Name | Company | Catalog Number | Comments |
| Ammonium chloride | Sigma-Aldrich | A9434-500G | For quenching formaldehyde |
| Anti-EXO1 Rabbit Polyclonal Antibody | Proteintech | 16253-1-AP | primary antibody |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | 05-636 | primary antibody |
| Bovine Serum Albumin | Sigma-Aldrich | 3117332001 | BSA for blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Merck | 19-160 | pre-sensitizing agent |
| Citifluor™ Mountant Solution AFR3 | Electron Microscopy Sciences | 17973-10 | antifade containing PBS solution for imaging |
| DAPI | Sigma-Aldrich | D9542-1MG | nucleic acid stain |
| DMEM Medium | Thermo Fisher Scientific | 10569010 | Cell culture medium for HEK293T cells |
| DMSO | Sigma-Aldrich | D2650-100ML | Vehichle control and dissolution solvent |
| EGFP-FBXL10 | Addgene | #126542 | viral expression vector for EGFP-FBXL10 |
| EXO1b-AcGFP (in pRetroQ) | custom cloning | na | EXO1b cDNA was cloned in the NheI, BamHI sites of pRetroQ-AcGFP1-N1 vector. |
| Fetal Bovine Serum | Gibco | 16140071 | Media supplement |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 | Phenol red free medium for microscopy |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A32723 | secondary antibody |
| HEK293T cells | ATCC | ATCC CRL-3216 | Cell line for viral packaging |
| HEPES | Sigma-Aldrich | H0887-100ML | Buffering agent to supplement live cell imaging medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | pre-sensitizing agent |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 | Transfection reagent |
| McCoy’s 5A (Modified) Medium | Life Technologies | 16600-108 | Cell culture medium for U-2 OS cells |
| mCherry-PCNA | Addgene | #55117 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA | Addgene | #55994 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA (in pBABE) | custom cloning | na | mPlum-PCNA cDNA was cloned from Addgene #55994 in the BamHI, SalI sites of pBABE (puro) |
| Nikon A1R-HD25 Confocal Scanhead and Controller | Nikon | na | confocal imaging system |
| Nikon LUN4 laser unit | Nikon | na | excitation system |
| Nikon LUN-F 50 mW 405 nm FRAP laser unit | Nikon | na | FRAP laser unit |
| Nikon NIS Elements Confocal Controller Software | Nikon | na | Confocal controlling software |
| Nikon Ti2-E Inverted Microscope | Nikon | na | inverted epifluorescent microscope base |
| Nikon Ti2-LAPP Modular Illumination System | Nikon | na | illumination system |
| NTHL1-mCherry (in pRetroQ) | custom cloning | na | NTHL1 cDNA was cloned in the NheI, SalI sites of pRetroQ-mCherry-N1 vector. |
| Nunc Lab-Tek II Chambered Coverglass (4 well) | Thermo Fisher Scientific | 155382PK | Live cell microscopy cell culture chamber |
| Olaparib | Selleck Chemicals | S1060 | PARP inhibitor |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985062 | Dilution medium for transient transfection |
| Paraformaldehyde aqueous solution (32%) | Thermo Fisher Scientific | 50-980-494 | Fixative |
| pBABE (hygro) | Addgene | #1765 | retroviral expression vector (for low expression levels) |
| pBABE (neo) | Addgene | #1767 | retroviral expression vector (for low expression levels) |
| pBABE (puro) | Addgene | #1764 | retroviral expression vector (for low expression levels) |
| pBABE (zeo) | Addgene | #1766 | retroviral expression vector (for low expression levels) |
| PCNA Antibody (PC10) | Santa Cruz | sc-56 | primary antibody |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco | 10378016 | Media supplement |
| polybrene | Sigma-Aldrich | TR-1003 | Increase viral infection efficiency |
| pRetroQ-AcGFP-C1 | Takara | 632506 | retroviral expression vector |
| pRetroQ-AcGFP-N1 | Takara | 632505 | retroviral expression vector |
| pRetroQ-mCherry-C1 | Takara | 632567 | retroviral expression vector |
| pRetroQ-mCherry-N1 | Takara | 632568 | retroviral expression vector |
| pUMVC | Addgene | #8449 | Viral packaging vector |
| Sodium-pyruvate | Thermo Fisher Scientific | 11360070 | Supplement for live cell imaging medium |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | Dilute in PBS for cell permeabilization buffer |
| Trypsin-EDTA Solution 10X | Sigma-Aldrich | 59418C-100ML | Dilute in PBS to split cells |
| U-2 OS Cells | ATCC | HTB-96 | Optimal cell line for microscopy experiments |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K | PCR based Mycoplasma detection kit |
| VSV-G | Addgene | #8454 | Viral protein envelope vector |
References
- Aleksandrov, R., et al.
- Darzynkiewicz, Z., Halicka, H. D., Zhao, H., Podhorecka, M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: Analysis by flow cytometry. Methods in Molecular Biology. 761, 85-96 (2011).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Herce, H. D., Rajan, M., Lattig-Tunnemann, G., Fillies, M., Cardoso, M. C. A novel cell permeable DNA replication and repair marker. Nucleus. 5 (6), 590-600 (2014).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in dna replication with putative roles in cancer. International Journal of Molecular Sciences. 20 (1), (2018).
- Cheruiyot, A., et al. Poly(ADP-ribose)-binding promotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amsterdam). 35, 106-115 (2015).
- Zhang, F., Shi, J., Chen, S. H., Bian, C., Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Research. 43 (22), 10782-10794 (2015).
- Rona, G., et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. elife. 7, (2018).
- Young, L. M., et al. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the dna damage response. Cell Reports. 13 (3), 451-459 (2015).
- Kong, X., et al. Laser microirradiation to study in vivo cellular responses to simple and complex dna damage. Journal of Visualized Experiments. (131), e56213 (2018).
- Kong, X., et al. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS One. 6 (8), 23548 (2011).
- Lan, L., et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Research. 42 (4), 2330-2345 (2014).
- Zerjatke, T., et al. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Reports. 19 (9), 1953-1966 (2017).
- Ji, Y., Karbaschi, M., Cooke, M. S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutation Research. 845, 403054 (2019).
- Sun, G., et al. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infection and Immunity. 76 (10), 4405-4413 (2008).
- Gassman, N. R., Wilson, S. H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amsterdam). 31, 52-63 (2015).
- Muster, B., Rapp, A., Cardoso, M. C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics. 4 (1), 47-68 (2017).
- Ikeda, S., et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 273 (34), 21585-21593 (1998).
- Rosenquist, T. A., Zharkov, D. O., Grollman, A. P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proceedings of the National Academy of Science U. S. A. 94 (14), 7429-7434 (1997).
- Reid, D. A., et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Science U. S. A. 112 (20), 2575-2584 (2015).
- Taccioli, G. E., et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 265 (5177), 1442-1445 (1994).
- Marsin, S., et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 278 (45), 44068-44074 (2003).
- Thompson, L. H., Brookman, K. W., Jones, N. J., Allen, S. A., Carrano, A. V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cell Biology. 10 (12), 6160-6171 (1990).
- Scharer, O. D.
- Oeck, S., et al. High-throughput evaluation of protein migration and localization after laser micro-irradiation. Science Reports. 9 (1), 3148 (2019).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Science Reports. 6, 19567 (2016).
- Durand, R. E., Olive, P. L. Cytotoxicity, mutagenicity and dna damage by hoechst 33342. Journal of Histochemistry and Cytochemistry. 30 (2), 111-116 (1982).
- Tobey, R. A., Oishi, N., Crissman, H. A. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proceedings of the National Academy of Science U. S. A. 87 (13), 5104-5108 (1990).
- Podhorecka, M., Skladanowski, A., Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. Journal of Nucleic Acids. 2010, (2010).
