

Summary
本文介绍了制备荧光标记的版本,λ噬菌体感染的程序
Abstract
该系统包括lambda和细菌大肠杆菌噬菌体(噬菌体)曾长期担任大肠杆菌细胞命运决定的典范1,2。继由数量噬菌体感染的细胞的同时,选择两种途径之一:裂解(毒力)或溶原性(休眠)3,4。最近,我们开发了一种方法用于荧光标记单个噬菌体,并能够检查实时决定感染后在显微镜下,在单个噬菌体和细胞5的水平。在这里,我们描述的全部程序进行感染实验,在我们前面的工作5。这包括创建的荧光噬菌体感染的细胞,在显微镜下的成像和数据分析。的荧光是一种“混合”,共表达的野生型和YFP-融合版本的衣壳GPD蛋白的噬菌体。根据粗略的噬菌体裂解第一次获得通过诱导溶原GPD-EYFP(ENH均衡的黄色荧光蛋白)的噬菌体,窝藏表达质粒的野生型GPD。然后进行一系列的纯化步骤,然后在显微镜下用DAPI标记和成像。这样做是为了验证的均匀性,DNA的包装效率,荧光信号和结构稳定性的噬菌体库存。进行的初始吸附到细菌的噬菌体在冰上,然后依次由一个短的孵育在35℃至触发病毒DNA注射6。噬菌体/细菌混合物然后移动到的表面上的薄的营养琼脂平板,用盖玻片覆盖,并落射荧光显微镜下成像。感染后处理后4小时,以10分钟的间隔。跟踪多个阶段的位置使得在一个实验中,可以追溯到〜100细胞感染。在每个位置和时间点,获取图像的相位相反,红色和绿色荧光通道。相位对比图像用于以后的自动化CELL的认可,同时荧光通道是用来描述感染的结果:新的荧光噬菌体(绿色),然后通过细胞裂解,或表达的溶源性因素(红色)其次是恢复细胞生长和分裂的生产。收购时间推移电影使用手动和自动相结合的方法进行处理。数据分析的结果,在每个感染事件(例如,感染噬菌体的数量和位置),以及感染的结果(裂解/溶源性)的感染参数的识别。附加参数,如果需要的话,可以提取。
Protocol
1。创作的粗噬菌体溶胞产物(图1)
- 在50ml烧瓶中,接种新鲜菌落LE392(λLZ1)[pPLate * D](有关详细信息,请参阅表1)到6毫升辅以10微克/毫升卡那霉素和100微克/毫升氨苄青霉素的LB培养基中7。用温和摇动(180转),在30℃生长过夜。
- 稀释培养1:100到LBM(LB补充用10mM的用MgSO 4),用温和摇动(180转),并在30℃生长。为了优化噬菌体产率,确保培养体积是不超过将烧瓶的体积容量的十分之一。我们通常准备两个2升或2.5升容量的烧瓶中,并添加到250毫升的LBM介质在每个烧瓶中加入2.5毫升过夜培养。
- 当细胞密度达到OD 600≈0.6(〜2.5 - 3小时),诱导通过移动用温和摇动(180转)的18分钟至42℃水浴摇床培养的溶素原,然后incubatE在37°C(180转),直到溶解有轻微的晃动是可见的(文化变得清晰,在60 - 90分钟)。
- 加入2%氯仿培养,摇用手工混合,然后在室温下孵育15分钟。注意:戴上手套来处理氯仿,避免吸入。
- 培养转移到两个250毫升的离心瓶中,在Sorvall GSA转子中以10,000 rpm离心培养15分钟,在4℃下含有噬菌体颗粒,回收上清液,弃沉淀碎片。进行第二次离心分离,以确保摆脱可见的碎片。
- 使用标准的噬菌体滴定协议的8衡量噬菌体浓度。的噬菌体滴度应为〜5-10×10 9 pfu /毫升。使用supF菌株如LE392作为指示菌株,因为的SAM7突变的荧光噬菌体的基因型中,并使用顶层琼脂和具有丰富NZYM制成琼脂平板上,以获得较大的斑块( 网络连接gure 2)。
2。噬菌体纯化(图1)
- 倒成一个大的(例如,2升)的烧瓶中的溶胞产物,以消化从溶解的细菌的核酸解放添加DNA酶Ⅰ和RNase(1微克/毫升)的溶胞产物,并在室温下孵育1小时。
- 1M氯化钠的裂解,转移到250毫升离心瓶的裂解,并冰上孵育3小时。离心机在Sorvall GSA的溶胞产物在4℃下以10,000 rpm 15分钟回收上清液。噬菌体滴度应该是相似的,粗的溶胞产物,这是〜5-10×10 9 pfu /毫升。加入NaCl促进解离的噬菌体颗粒从细菌碎片,并用PEG 8所需的有效沉淀噬菌体颗粒。
- 成一个大的烧瓶中倒入溶胞产物,例如2升的烧瓶中,添加10%(w / v的)PEG8000到的溶胞产物中,慢慢地搅拌或摇晃,在室温下溶解PEG8000。的溶胞产物转移到250毫升百分之rifuge瓶,然后在4℃下孵育过夜(〜16小时)离心机在Sorvall GSA转子中的溶胞产物在4℃下以10,000 rpm 15分钟弃上清。
- 浸泡颗粒(沉淀噬菌体颗粒与PEG8000)与噬菌体,SM缓冲液(每250毫升的初始噬菌体裂解液的4毫升SM缓冲液)。孵育非常温和摇动或在4℃下振荡16小时
- 入50毫升的Eppendorf离心管中轻轻取出的细胞裂解液(SM缓冲液与噬菌体颗粒),然后洗其它颗粒0.5 - 1毫升SM缓冲液。
- 的溶胞产物中加入等体积的氯仿。反相上下轻轻晃动混匀,用氯仿裂解液了数十倍。在4,000 rpm下离心15分钟,在4℃在Eppendorf 5804R或类似的台式离心机。
- 重复步骤2.6,以获得更清晰的裂解。噬菌体滴度应为〜1-2×10 11 pfu /毫升。
- 准备SM /氯化铯与三种不同的密度(ρ)为1.3克/毫升,1的解决方案.5克/毫升和1.7克/毫升。测量折射率(η),以得到更精确的密度读数。密度转换图9是ρ=η10.8601 - 13.4974在25℃下有关详细信息,请参阅表3。
- 使用注射器用的长针,一个14毫升的超清晰贝克曼的40Ti超速离心机的管加载的解决方案。以避免混合,以形成更好的密度梯度,垫层的溶液(即分层解决方案增加密度下一个另一)应该被使用,即,在1.3克/毫升,1.5的顺序,轻轻地装入2毫升SM /氯化铯解决方案克/毫升和1.7克/毫升,由插入针与一个3毫升的注射器管的底部。
- 轻轻放入8毫升噬菌体裂解液进行叠加,从在14毫升的超离心管的顶部。编制资产负债管。离心机在24,000 rpm的转速下搅拌4小时,在4℃下在Beckman SW40Ti转子
- 在一个黑暗的房间,轻轻地管和管从顶部照亮在黑色背景上使用自动对焦lashlight。的噬菌体频带应该是清晰可见的在1.3克/毫升和1.5克/毫升SM /氯化铯层( 图3A)之间的界面的位置。通过侧管稍低于21.5用3毫升的注射器的针头的带穿刺术。轻轻的噬菌体悬浮液收集〜500μL。噬菌体滴度应为〜5-10×10 11 pfu /毫升。
- 将噬菌体悬浮液4毫升超清晰Beckman超速SW60Ti转子管。填充管与1.5克/毫升SM / CsCl溶液。编制资产负债管。离心在Beckman SW60Ti转子在4℃下以35,000 rpm离心24小时
- 重复相同的步骤,在步骤2.11,从可见光波段收集噬菌体。应该是可见的,如在图3B中所示的频带。
- 噬菌体悬浮液装入到透析膜盒( 表2)中,对1000倍体积的SM缓冲液中,在4℃下透析三次,持续时间为3小时,3小时和overnigHT(约16小时)。透析的目的是摆脱的CsCl的噬菌体悬浮液中存在的。最终噬菌体滴度应为〜5-10×10 11 pfu /毫升。
3。准备一个琼脂糖凝胶板(图4)
- 清洁6显微镜载物片(75×50毫米,1毫米厚),用70%的乙醇。
- 排列5滑动,并用胶带固定,如在图4中示出。
- 混合0.09克琼脂糖到6毫升培养基覆盖保鲜(产生1.5%的琼脂糖)在小烧杯中。在热板上加热直到溶液变为清晰。
- 琼脂糖溶液倒入到安全的幻灯片。
- 将最后一张幻灯片在上面,小心地避免气泡。将在上面的重量,并允许冷却〜30分钟。
- 取出4张幻灯片就在身边,包板,连同保鲜的顶部和底部的幻灯片。板坯可以在4℃下储存3天。
4。测试纯化的噬菌体库存
- 准备的PBS-琼脂糖凝胶板如上所述(第3项)。
- 用DAPI染色纯化的噬菌体。混合10微升噬菌体(〜1×10 10 pfu /毫升)用10μl的10微克/毫升的DAPI DAPI(最终浓度为5微克/毫升),温育30分钟,在4℃或在室温下10分钟。
- 将1微升的噬菌体/ DAPI混合物在中心的1号24×50mm的盖玻片上,叠加一小片(〜10×10毫米)的预先准备的PBS-琼脂糖板坯。琼脂糖切片的小片被切断后顶端上滑动除去夹心凝胶用剃刀刀片。通过的YFP和DAPI渠道的落射荧光显微镜下样品的图片。单个噬菌体应该是可见的衍射限制的荧光在两个通道中的“斑点”( 图5)。使用相同的显微镜和相机设置,在下面的步骤6.2。
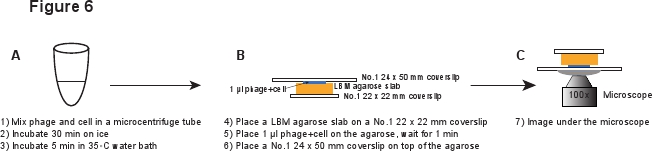
5。感染(图6)
- 在14毫升的Falcon管中,接种一个新的殖民地,LE392 [PP RE-MCHERRY](有关详细信息,请参阅表1)到2毫升的LB培养基中补充有100微克/毫升氨苄青霉素的10mM,用MgSO 4和0.2%麦芽糖。中度摇动(265转)与在37℃下生长过夜。
- 稀释1:1000成LBMM(LB补充用10mM MgSO 4和0.2%麦芽糖)培养,即5毫升LBMM介质,在50毫升烧瓶中加入5μl隔夜培养成。中度摇动(265转),在37℃下生长至OD 600≈0.4。
- 使用LBM介质准备的上述第3节中所描述的LBM-琼脂糖凝胶板。
- 离心1毫升细胞2000克在一个台式微量离心2分钟,在室温下。去除上清,并轻轻悬浮细胞成20微升冰冷LBMM达到OD 600&20。
- 在操纵纯化的噬菌体股票的,使用广泛的移液器吸头或削减常规移液器吸头尖,扩大开放,以避免剪切噬菌体颗粒3。轻轻地中号㈨20μl的细胞与纯化的噬菌体,以达到平均噬菌体细胞比率在0.1 - 5的范围内的20微升。在冰上孵育30分钟,以使噬菌体吸附,然后在35℃水浴中5分钟孵育触发噬菌体DNA注射6。
- 移液器上下分隔任何细胞聚集了几次。再次使用广泛的移液枪枪头,以避免剪切的噬菌体。混合稀释到LBMM 1:10,如5μL混合成45微升LBMM。
- LBM-琼脂糖板坯(〜10×10毫米)上的1号22×22毫米的盖玻片放置一块。预先准备好的LBM-琼脂糖板坯应在室温下放置至少1小时,以确保在使用前,琼脂糖板坯达到室温。将1微升的噬菌体/琼脂糖平板上的细胞混合物,并等待1分钟,从而使混合物吸收到琼脂糖平板。轻轻一级的24×50毫米的盖玻片放置琼脂糖平板上的顶部。这个过程意味着,以避免剪切噬菌体的infected细胞( 图6)。
6。继显微镜下观察细胞的命运
- 小心地安装在舞台上的显微镜的盖玻片。对于成像,使用的高倍率(例如100x)的目标(见显微镜系统在下面的讨论 )。
- 采集的图像设置为初始时间框架。此图片集将被用来表征初始的感染噬菌体的数量和位置。采取了一系列15幅图像,在200nm的z轴方向(垂直方向)的时间间隔。通过YFP通道的图像。此外,通过一个单一的在焦像的相位对比度和mCherry通道。优化光的强度和照射时间,以获得足够的信号,同时最大限度地减少漂白和细胞的损害(见图像采集在下面的讨论 )。
- 收购时间的推移电影后感染的细胞的命运。图片样品相衬,YFP和mCherry在约4小时10分钟的时间间隔的通道。在时间的推移电影,使用一个单一的z位置的图像每个时间点,每通道,以避免不必要的暴露的样品,这可能导致漂白和光毒性。
7。图像分析
- 手动计算噬菌体的数量和记录的噬菌体的位置和细胞长度在最初的时间框架。这是可以做到使用软件例如作为的MetaMorph或ImageJ的。记录的细胞的命运(裂解,溶原性或未受感染的),溶解时间,以及任何其他所需的信息通过播放时间推移电影。为了识别不同的细胞命运,看到时间的推移电影的代表性成果下文。
- 除了上面的人工分析,可以提取更多的量化信息(例如,随着时间的推移在个体细胞中)的荧光水平使用自动细胞识别和谱系追踪算法。我们用一个家庭的Matlab程序为TRacing细胞谱系和荧光水平,与Schnitzcell Matlab代码,用于细胞分割Elowitz组在加州理工学院(书面)。
8。代表性的成果:
噬菌体电镀:
荧光标记的噬菌体(在步骤1.6和第2节)的噬斑显着小于野生型( 图2)。因此,我们孵化板至少12小时,在37°C孵化器的斑块是可见的。
噬菌体超速离心法:
噬菌体样本梯度(步骤2.10)与在CsCl步骤超速离心后,两个频带应该是可见的( 图3A)。顶带,噬菌体悬浮液和SM /氯化铯1.3克/毫升层之间的界面处,含有细胞碎片和空噬菌体衣壳。底部带,在SM /氯化铯1.3克/毫升和1.5克/毫升层之间的界面处,是噬菌体频带。氏S波段出现绿色的荧光噬菌体λLZ2。带为野生型噬菌体λIG2903出现偏蓝5。在步骤2.12 CsCl平衡梯度超速离心后,一个噬菌体频带应该是在管的中间部分( 图3B)中可见。由于荧光λ噬菌体LZ2包含GPD-EYFP和GPD衣壳,蛋白质-DNA的比例的混合物,是高于野生型。因此,带的荧光λ噬菌体LZ2稍轻(似乎是在一个较高的位置,在管内)比野生型λIG290310。
DAPI染色:
图5显示了典型的图像进行标记的噬菌体后,用DAPI(第4节)。一个成功纯化的噬菌体,YFP和DAPI信号应当具有接近100%对应。我们通常观察不到1%的YFP点的s不包含的DAPI(表示无病毒基因组的衣壳)。小于1%的DAPI斑点不包含YFP(相应非荧光噬菌体)5。
时间推移电影:
确认所积累的YFP荧光(绿色通道)进入细胞内,细胞裂解裂解细胞。溶源细胞是公认的积累在细胞内,并恢复正常细胞的生长和分裂的统一mCherry荧光(红色)。未感染的细胞(或细胞感染已失败的地方)将不显示任何上述的表型,将正常的生长和分裂。 图7示出了几个相对比度的图像集,YFP和mCherry通道,以及相应的重叠图像,这些三个渠道,从一个典型的时间间隔短片(第6章)。单个噬菌体(绿色斑点)是清晰可见的帧( 图7A)在初始时间。通常情况下,一些在细胞表面上(大概是感染这些细胞),而其他的噬菌体未吸附的噬菌体,在图7B(左面板)中所示。的感染结果随着时间的推移变得区分的。裂解周期表示由细胞内生产的新的噬菌体(绿色, 图7C),然后通过细胞裂解(分解细胞与释放绿色噬菌体, 图7D)。的溶原性表示的mCherry从生产的P的RE启动子(红, 图7C)和恢复细胞生长和分裂(红色, 图7D)。




图1。 AB)。通过一系列的步骤(面板CL)的纯化的噬菌体。

图2。噬菌体斑块。的荧光的噬菌体的噬斑(左)是小于野生型(右)的12小时孵育后板在37℃下

图3。噬菌体带超速离心后A)可见超速离心在氯化铯步骤梯度后两个带。顶一个对应的细胞碎片和空噬菌体衣壳的底部区域包含所需的噬菌体。左:荧光噬菌体,右:野生型。B )单噬菌体带CsCl平衡梯度超速离心后可见。该荧光噬菌体乐队的(左)是绿色,一个蓝色的带为野生型噬菌体(右)。

图4的过程制备琼脂糖凝胶板坯。

图5。DAPI染色后的噬菌体的荧光图像。个人噬菌体很容易分辨的,和YFP和DAPI信号共同定位非常好。

图6描述。原理噬菌体感染和成像设置。 点击这里查看全尺寸的版本,对本图片。
gure 7“SRC =”/ files/ftp_upload/3363/3363fig7.jpg“/>
图7。随时间推移电影的噬菌体感染的典型形象。显示的内容是相位对比,YFP和mCherry渠道,以及作为叠加的三个通道。 (A)的从初始时间帧中,YFP-通道的图像。左,YFP的图像在不同的z轴位置的总和。三个右侧图像样本YFP的图像在不同的z轴位置,对应于细胞表面的不同区域。 (B),(C)和(D)的相位对比(中间左),YFP(中间偏右)和mCherry(右)信道在不同的时间框架中的,重叠的图像(左)。 (B)在t = 0时,两个细胞被视为一个单一的噬菌体(绿色斑点),和一个单元,每个感染感染由3的噬菌体。同时我们也发现有一些未被吸附的噬菌体。 (C)在t = 80分钟,由单个噬菌体感染的细胞,每一个进入裂解途径,指示d的细胞内生产的新的噬菌体(绿色)。 3噬菌体感染的细胞溶源性途径,如由生产mCherry从PRE启动子(红色)表示已经进入。 (D)在t = 2小时,溶解途径导致细胞溶解(细胞分解),,而溶原性的细胞分为§。
§图7所示的左侧面板(C)及(D)转载细胞,141,蓝鹰,塞缪尔·O.斯金纳曾,Chenghang宗,让吸管,迈克尔·Feiss和IDO戈尔丁,在亚细胞水平的决策将决定结果噬菌体感染管理研究所版权所有(2010),爱思唯尔的许可。
| 应变名称 | 相关基因 | 资料来源/参考 |
| 菌株 | ||
| LE392 | 支持F | ,伊利诺伊大学的约翰·Cronan |
| 噬菌体菌株 | ||
| λLZ1 | GPD-EYFP,cI857 SAM7 D-EYFP b :: KanR表 | Zeng 等。 |
| λLZ2 | GPD马赛克,同样的基因型为λLZ1 | Zeng 等。 |
| 质粒 | ||
| PP RE - mCherry | mCherry的P RE的控制下, 放大器 ŕ | Zeng 等。 |
| pPLate * D | GPDλ晚期启动子的控制下, 放大器ŕ | Zeng 等。 |
表1细菌菌株,在这项工作中所使用的噬菌体和质粒。
| 密度ρ(克/毫升) | 中海集运(G) | SM(毫升) | 折射率η |
| 1.30 | 39 | 86 | 1.3625 |
| 1.50 | 67 | 82 | 1.3815 |
| 1.70 | 95 | 75 | 1.3990 |
表3中。在SM缓冲液(100毫升)中的氯化铯溶液的步骤梯度。
Discussion
菌株,噬菌体和质粒:
菌株LE392就是supF。这是选择抑制的噬菌体基因组的的SAM7突变在(有关详细信息,请参阅表1)。因此,诱导细胞溶素原最终会裂解,并释放噬菌体颗粒,将受感染的细胞,所选择的裂解途径。溶源细胞生长在30℃,由于温度敏感的cI 857的噬菌体基因组中的等位基因的存在。热诱导后,GPD-EYFP和野生型GPD共同从基因组为λLZ1和质粒pPlate的* D分别表示。其结果是,新创建的λ噬菌体LZ2衣壳包含GPD-EYFP和GPD蛋白的混合物。这马赛克噬菌体是结构稳定和足够的荧光,以允许检测单个噬菌体5。 PP RE - mCherry是记者的质粒用于检测到选择的溶原性pathwa的Y。启动子P RE被激活由CII期间1,11溶源性成立。来自PE-GFP 11 12 mCherry更换绿色荧光蛋白基因与PP RE - mCherry 5。有关详细信息,请参阅我们早期的工作5。
生长条件的参数:
(第1节),在溶原菌的诱导轻微的晃动在180rpm提供了一个很好的病毒产量13。在生长培养基中使用的葡萄糖应该避免,因为葡萄糖代谢产生酸性代谢产物,并在酸性pH值13的成熟的lambda颗粒是不稳定的。用MgSO 4的另外的目的是稳定的噬菌体衣壳3。携带野生型CI(而不是857 词 )的噬菌体,溶原可诱导的DNA损伤剂丝裂霉素C 3。步骤1.3中,在37°C的潜伏期一般应不超过90分钟。这是大家有用微升通过OD 600每30分钟检查细胞密度。有一个良好的溶胞产物中,OD 600下降到约0.2或更低,和剩余的OD 600是一个结果的细胞碎片。孵化过长,可能导致在一个较低的噬菌体的产率,因为新创建的噬菌体可以开始他们的DNA注入到细胞碎片。为了获得一个可见的噬菌体带(至少1×10 11噬菌体颗粒),在步骤2.11和2.13,增长至少500毫升的文化,在第1.2步。此外,0.2%的麦芽糖培养基中生长,在5.1和5.2的目的是诱导表达的羔羊,λ噬菌体的受体吸附3,14。记者质粒PP RE - mCherry旨在减少mCherry背景水平,而不是步骤5.2的100倍,1000倍稀释液。在步骤5.5中的噬菌体DNA注入触发,35°C的选择,以避免诱导的温度敏感cI857等位基因。
噬菌体净化:
jove_content“>噬菌体纯化步骤(步骤2.1到2.11)可以与其它净化协议5取代,但最终超速离心通过CsCl平衡梯度(步骤2.12及2.13)是不可避免的。摇摆铲斗转子需要在步骤2.10和2.12至确保锐利可见噬菌体带。获得纯噬菌体可以很容易地采取了一个星期,所以它是必要的检查沿途的噬菌体滴度,以确保没有出错过程中的中间步骤。噬菌体处理:
在第2纯化过程中,关键是处理噬菌体裂解液轻轻地从噬菌体头,以避免剪切噬菌体的尾巴。在第5节(例如,步骤5.5到5.7)中的细胞的感染,它也是很关键的,以避免从被感染的细胞中的噬菌体颗粒的剪切。请注意,如果噬菌体被剪切其DNA注入后从被感染的细胞,其结果是一个“暗”感染,即在fection结果将在实验中观察到,但不会感染噬菌体。为了尽量减少这样的问题,我们使用了一个广泛的移液器吸头时处理的噬菌体或噬菌体/细胞混合物。
DAPI测试:
染色噬菌体与DAPI(第4),是一种快速,有效的方法来检查产品的纯度的噬菌体。它也可以被用于测试可能降解的一个现有的噬菌体库存中随着时间的推移。对于一个纯粹的股票,在荧光显微镜下YFP和DAPI信号的定位应该是接近100%。通常,我们观察,YFP的斑点,小于1%的不包含DAPI(表示无病毒基因组的衣壳),这表明这些颗粒没有成功打包病毒DNA,或已经注入了他们的DNA别处。小于1%的DAPI斑点不包含YFP(相应非荧光噬菌体)。如果不是这种情况下,步骤2.12至2.14需要被重复在邻刻申,净化一次。成像参数方面,在显微镜安装在步骤4.3是至关重要的,因为在第5,在这里,因为没有长期活细胞成像。但是如在第5部分,保持相同的显微镜设置是有用的,如果一个人希望校准一个单一的噬菌体颗粒的荧光强度。如果也不是很干净,或太多的DAPI染料用于PBS-琼脂糖切片,一些DAPI染色斑点相应的噬菌体DNA可以用“卤素”包围。如果使用太少的DAPI染料的DAPI信道的信号,可以是非常弱的。
显微镜系统:
对于成像在第6条中,我们利用了倒落射荧光显微镜(ECLIPSE TE2000-E,尼康)100倍的目标(计划FLUO,数值孔径1.40,浸油)和标准过滤器集(尼康)。的荧光的光源是一个电弧灯与光强度的控制。下面的功能是电脑控制:X,Y和Z宝sition明场和荧光百叶窗和荧光过滤器的选择。一个自动对焦的功能是必需的。否则,焦点可以很容易地渐行渐远在时间的推移电影(通常4小时之久)。在每个时间点的能力获得多个位置(X,Y)是有用的,因为它允许遵循在平行的多重感染事件。我们通常在每部电影,获得8级职位跟进到100感染事件。我们使用的相机是CCD与冷却的512×512 16×16微米像素摄像头,支持16的位(Cascade512,光度)的动态范围。使用MetaMorph软件(Molecular Devices公司)进行收购事项。显微镜时,应被放置在一个温度受控室,或者,在显微镜载物台应通过一个温度控制的室包围。
图像采集:
活细胞成像,这是至关重要的样品,以避免不必要的暴露,这可能导致漂白,河粉totoxicity。因此,最好是先描述您的系统找到一个最佳的光线照射,它允许用于荧光检测,而不会导致过多的漂白剂或抑制细胞生长。为了获得一个良好的荧光图像,发挥激发光强度,暴露时间和摄像机的增益。在步骤6.2-6.3,在10分钟的帧间隔被选择曝光最小化的目的。在每个帧中,只有一个单一的在焦像需要在相位对比(细胞识别)和荧光通道(用于确定细胞的命运)。在第一时间点,但是,多个通过YFP通道的z位置的图像必需能够捕获所有感染噬菌体在细胞表面上。 YFP的曝光时间的初始帧中可能还需要要高于用于在稍后的时间帧中的时间间隔短片。
图像分析:
非常仔细地计算噬菌体颗粒周围的细胞表面的步骤7.1。如如上所述,我们采取了一系列步骤6.2 YFP通道的Z-栈通过。然而,这仍可能留下一些荧光噬菌体颗粒的焦点,挑战计数。细胞在初始时间帧的长度测量使用Metamorph软件。也可以测定细胞长度ImageJ的或其他软件工具。此外,自动化家居的Matlab程序是非常有用的,如沿细胞系,荧光随时间变化的信息。
Disclosures
没有利益冲突的声明。
Acknowledgments
我们感谢迈克尔·Feiss和Jean鸭嘴的指导噬菌体创建和净化。我们感谢迈克尔Elowitz提供细胞识别软件,Schnitzcell。在戈尔丁实验室工作的支持,补助金从国家卫生研究院(R01GM082837),美国国家科学基金会(082265,PFC:活细胞的物理中心),韦尔奇基金会(批准Q-1759)和人类前沿科学计划(RGY二千〇八分之七十)。
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform | Fisher Scientific | C298-500 | |
| NaCl | Fisher Scientific | S271-3 | |
| DNase I | Sigma-Aldrich | D4527-10KU | |
| RNase | Sigma-Aldrich | R4642-10MG | |
| PEG8000 | Fisher Scientific | BP233-1 | |
| SM buffer | TEKnova, Inc. | S0249 | |
| NZYM | TEKnova, Inc. | N2062 | |
| CsCl | Sigma-Aldrich | C3011-250G | |
| Syringe | BD Biosciences | 309585 | |
| Needle | BD Biosciences | 305176 | |
| Dialysis cassette | Thermo Fisher Scientific, Inc. | 66333 | |
| Microscope slide | Corning | 2947-75x50 | |
| Agarose | Fisher Scientific | BP160-100 | |
| SW40Ti ultra-clear tube | Beckman Coulter Inc. | 344060 | |
| SW60Ti ultra-clear tube | Beckman Coulter Inc. | 344062 | |
| SW40Ti rotor | Beckman Coulter Inc. | 331302 | |
| SW60Ti rotor | Beckman Coulter Inc. | 335649 | |
| Refractometer | Fisher Scientific | 13-947 | |
| Epifluorescence microscope | Nikon Instruments | Eclipse TE2000-E | |
| Table 2. Reagents and equipment. | |||
References
- Oppenheim, A. B., Kobiler, O., Stavans, J., Court, D. L., Adhya, S.
- Ptashne, M. A genetic switch : phage lambda revisited. , 3rd edn, Cold Spring Harbor Laboratory Press. (2004).
- Hendrix, R. W. Lambda II. , Cold Spring Harbor Laboratory. (1983).
- Hershey, A. D. The Bacteriophage lambda. , Cold Spring Harbor Laboratory. (1971).
- Zeng, L. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell. 141, 682-691 (2010).
- Edgar, R. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. , (2008).
- Ausubel, F. M. Current protocols in molecular biology. , John Wiley & Sons. (1994).
- Sambrook, J., Russell, D. W. Molecular cloning : a laboratory manual. , 3rd edn, Cold Spring Harbor Laboratory Press. (2001).
- Fasman, G. D. Practical handbook of biochemistry and molecular biology. , CRC press. (1989).
- Kaiser, A. D. On the internal structure of bacteriophage lambda. J. Gen. Physiol. 49, 171-178 (1966).
- Kobiler, O. Quantitative kinetic analysis of the bacteriophage lambda genetic network. Proc Natl Acad Sci. 102, 4470-4475 (2005).
- Shaner, N. C. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572 (2004).
- Personal communication with M. Feiss. , Forthcoming.
- Schwartz, M. The adsorption of coliphage lambda to its host: effect of variations in the surface density of receptor and in phage-receptor affinity. J. Mol. Biol. 103, 521-536 (1976).

{kind=link}