

Summary
この記事では、バクテリオファージラムダの感染の蛍光標識されたバージョンを準備する手順を説明します
Abstract
バクテリオファージ(ファージ)ラムダと細菌大腸菌を含むシステム大腸菌は長い細胞運命決定の1,2のためのパラダイムを務めている。ファージの数によって、細胞の同時感染後、2つの経路のものが選ばれる:溶解(強毒性)または溶原(休止状態)3,4。我々は最近、蛍光個々のファージを標識するための方法を開発し、個々のファージとセル5のレベルでは、顕微鏡下でリアルタイムで感染後の決定を調べることができました。ここで、我々は以前の仕事5に記載された感染実験を行うための完全な手順について説明します。これは蛍光ファージの作成、細胞の感染、顕微鏡下でのイメージングおよびデータ分析が含まれています。蛍光ファージは、カプシドGPDタンパク質の "ハイブリッド"、共発現する野生型およびYFP融合バージョンです。原油ファージ溶菌液は最初GPD-EYFP(ENHの溶原菌を誘導することによって得られるプラスミドを発現する野生型GPDを宿す、黄色蛍光タンパク質)ファージをとれた。精製工程のシリーズは、その後、顕微鏡下でのDAPI標識やイメージング、続いて行われます。これは、均一性、DNAパッケージング効率、蛍光シグナル及びファージストックの構造安定性を確認するために行われます。細菌にファージの初期吸着は、その後、35℃で短時間のインキュベーションに続いて、氷の上で実行される°Cは、ウイルスDNA注入6をトリガする。ファージ/細菌混合物を、次いで、薄い栄養寒天平板の表面に移動したカバーガラスで覆われており、落射蛍光顕微鏡下で撮像される。感染後の処理は10分間隔で、4時間続きます。複数のステージの位置は約100細胞の感染は、単一の実験でトレースすることができるように追跡されます。それぞれの位置と時間の時点で、画像は位相コントラストと赤と緑の蛍光チャネルで取得されています。位相コントラスト画像は、自動化されたセルのために後で使用されている新しい蛍光ファージの生産(緑)細胞溶解、または再開細胞の増殖と分裂に続いて溶原性因子の発現(赤色)が続いています。蛍光チャンネルが感染の転帰を特徴付けるために使用されるのに対し、Lは認識。取得したタイムラプスムービーは、手動と自動の方法を組み合わせて使用して処理されます。各感染事象(感染ファージの例数と位置)だけでなく、感染症の転帰(溶解/溶原性)のための感染パラメータの同定におけるデータ解析結果。必要に応じて追加のパラメータを抽出することができる。
Protocol
1。原油ファージ溶解液(図1)の作成
- 50ミリリットルのフラスコに、LE392の新鮮なコロニー(λLZ1)[pPLate * D](詳細は表1を参照してください)10μg/ mlのカナマイシンおよび100μg/ mlのアンピシリンを補充したLB培地7の6ミリリットルに接種する。 ℃の軽度の振とう(180 rpm)を用いて30℃で一晩培養します。
- LBM(LBは、10mM MgSO 4で補充)に文化1:100に希釈し、軽度の振とう(180 rpm)しながら30℃で成長します。ファージ収率を最適化するために、培養液量がもうフラスコボリューム容量の10分の1以下であることを確認してください。我々は通常2リットルまたは2.5リットルの容量の2つのフラスコを用意し、各フラスコ内の250mlのLBM培地中に2.5ミリリットル一晩培養した培養液を追加します。
- 細胞密度がOD 600に達したとき≈0.6(〜2.5から3時間)、incubat、マイルドな振とう(180 rpm)しながら18分間42℃のウォーターバスシェーカーに文化を移動することによって溶原菌を誘導し、37のe℃で溶解するまで穏やかな振とう(180回転)で表示されている(文化が〜60で、明らかになる - 90分)。
- 培養液に2%のクロロホルムを加え、混合し、次いで、室温で15分間インキュベートし、手で振る。注意:クロロホルムを扱うために手袋を着用し、それを呼吸しないようにしてください。
- 2 250mlの遠心ボトルに文化を移し、4℃で15分間、10,000 rpmでソーバルGSAローターの文化を遠心ファージ粒子を含有する上清を回収し、破片のペレットを捨てる。目に見える破片を取り除くことを確認するために、第2の遠心分離を行います。
- ファージ濃度を測定するために標準的なファージの滴定プロトコル8を使用してください。ファージの力価は、5〜10×10 9 pfu / mlである必要があります。そのようなので、蛍光ファージの遺伝子型でSAM7変異の指標株としてLE392としてsupF株を使用し、(FI大きなプラークを取得するために豊富なNZYMで作られたトップアガーと寒天プレートを使用グレ2)。
2。ファージの精製(図1)
- 溶解された細菌から遊離核酸を消化するために、溶解液にDNase IをおよびRNase(1μg/ mlのそれぞれ)を追加し、大規模な(例えば2リットル)をフラスコに溶解液を注ぎ、室温で1時間インキュベートする。
- ライセートに1M NaClを追加し、250 mlの遠心ボトルにライセートを、氷上で3時間インキュベートする。 4℃で15分間10,000 rpmでソーバルGSAでライセートを遠心℃、上清を回収。ファージの力価は、5〜10×10 9 pfu / mlである粗溶解物のそれと類似しているはずです。 NaClの添加は、細菌破片からファージ粒子の解離を促進するとPEG 8でファージ粒子を効率的に降水量が必要です。
- 大型フラスコ、例えば2リットルのフラスコにライセートを注ぎ、室温でゆっくりPEG8000を溶解するためにかき混ぜるか、振って、ライセートに10%(w / v)のPEG8000を追加します。 250ミリリットルセントにライセートをrifugeボトルは、その後4℃で(〜16時間)で一晩インキュベートする4℃で15分間10,000 rpmでソーバルGSAローターでライセートを遠心℃、上清を捨てる。
- ファージSM緩衝液(初期ファージ溶菌液を250mlあたり4 mlのSM緩衝液)でペレットを(ファージ粒子はPEG8000で沈殿)に浸します。非常に穏やかな揺れない、または4℃で16時間振とう培養℃、
- 穏やかに50ミリリットルエッペンドルフ遠心管にライセート(ファージ粒子をSMバッファー)を取り出して、その後0.5と残りのペレットを洗浄 - SM緩衝液の1ミリリットル。
- ライセートに等量のクロロホルムを追加します。穏やかに数回反転上下によってクロロホルムで溶解液を混ぜる。 15エッペンドルフ5804Rで4℃minまたは類似のベンチトップ遠心分離機4,000 rpmで遠心分離します。
- 明確ライセートを取得する手順2.6を繰り返します。ファージ力価は約1〜2×10 11 pfu / mlである必要があります。
- 1.3グラム/ミリリットル、1の3つの異なる密度(ρ)とSM / CsClの溶液を調製0.5グラム/ mlおよび1.7グラム/ミリリットル。より正確な密度測定値を得るために屈折率(η)を測定します。 25℃13.4974℃ -密度変換9は 、ρ= 10.8601ηです詳細については、 表3を参照してください。
- 14ミリリットル超クリアベックマン40Ti超遠心チューブに溶液をロードするのに長い針で注射器を使用しています。混ぜないようにすると良い密度勾配を形成するために、ソリューションを使用する必要があります(もう一つ下の密度の増加、すなわちレイヤリングソリューション)は、すなわち、静かに1.3グラム/ミリリットル、1.5の順でSM / CsClのソリューションの2ミリリットルをロード下地g / mlのチューブの底に3mlの注射器で針を挿入して1.7グラム/ミリリットル。
- 優しく14mlの遠心管の上部から重ねることによってファージ溶菌液8mlをロードします。バランスチューブを準備します。 4℃で4時間、24,000 rpmでベックマンSW40Tiローターで遠心する℃、
- 優しくAFを使用して暗い部屋の中でチューブを取り出して、黒い背景にチューブの上部から照らすlashlight。ファージバンドは1.3グラム/ mlおよび1.5グラム/ mlのSM / CsClの層( 図3A)との間のインタフェースの位置ではっきりと見えるはずです。わずかに3 mlのシリンジで21.5ゲージの針を持つバンド下管の側面を通して穿刺。優しく収集〜ファージ懸濁液500μlを。ファージの力価は、5〜10×10 11 pfu / mlである必要があります。
- 4ミリリットル超クリアベックマン超遠心機SW60Tiローターチューブにファージ懸濁液を置きます。 1.5グラム/ mlのSM / CsClの溶液を用いて管を埋める。バランスチューブを準備します。 4℃で24時間、35,000 rpmでベックマンSW60Tiローターで遠心する℃、
- 可視バンドからファージを回収するには、手順2.11と同様の手順を繰り返します。 図3Bに示すようにバンドが見えるはずです。
- 透析膜カセット( 表2)にファージ懸濁液をロ ードおよび4でSMバッファーの1000倍体積に対して3回透析で3時間の持続期間のためのC、3時間とovernigHT(〜16時間)。透析の目的は、ファージ懸濁液中に存在する塩化セシウムを取り除くことです。最後のファージ力価は、5〜10×10 11 pfu / mlである必要があります。
3。 1アガロースゲルスラブ(図4)を準備
- 70%エタノールで6顕微鏡スライド(75×50ミリメートル、厚さ1mm)を清掃してください。
- 5スライドをアレンジして、図4に示すようにテープで固定します。
- しがみつくラップ(1.5%アガロースが得られる)で覆われた小さなビーカーで6mlの培地に0.09グラムアガロースを混ぜる。溶液が透明に変わるまで、ホットプレート上で加熱する。
- 保護されたスライド上にアガロース溶液を注ぐ。
- 慎重に気泡を避け、上に最後のスライドを配置します。上部に重量を配置し、約30分間放置してください。
- 側に4スライドを外し、しがみつくラップでトップとボトムのスライドと一緒にスラブを包む。スラブは最大3日間まで4℃で保存することができます。
4。精製ファージストックのテスト
- 準備する上記のように、PBS-アガロースゲルスラブ(第3節)。
- DAPIで精製されたファージを染色。 10μgの/ 30 4時分℃または室温で10分間インキュベートミリリットルDAPI(5μg/ mlの最終DAPIの濃度)、10μlのファージ10μlの(〜1×10 10 pfu / mlの)を混ぜます。
- 場所1番24×50mmのカバーグラスの中心にファージ/ DAPI混合物の添加、オーバーレイ、あらかじめ用意し、PBS-アガローススラブの小片(約10×10mm)で。サンドイッチゲル上の一番上のスライドが削除された後、アガローススラブの小片をカミソリの刃でカットされます。 YFPおよびDAPIのチャネルを介して落射蛍光顕微鏡下の画像サンプルを。個々のファージは、両方のチャネルで回折限界の蛍光灯"スポット"( 図5)のように見えるはずです。下記のステップ6.2と同じ顕微鏡とカメラのセットアップを使用します。
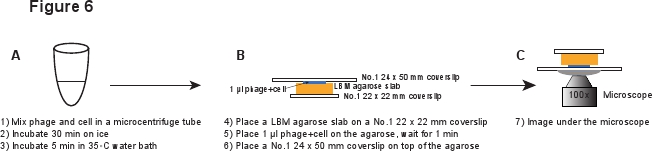
5。感染症(図6)
- 14ミリリットルファルコンチューブに、LE392 [PP RE-MCHの新鮮なコロニーを接種erry](詳細については表1を参照)100μg/ mlのアンピシリン、10mMのMgSO 4及び0.2%マルトースを添加したLB培地2mlに。適度な振とう(265 rpm)しながら37℃で一晩培養します。
- LBMM(LBは、10mM MgSO 4及び0.2%マルトース添加)に文化1:1000に希釈、すなわち50ミリリットルのフラスコ内の5ミリリットルLBMM培地に5μlを一晩培養した培養液を追加します。 ℃の適度な振盪(265 rpm)しながら37℃でOD 600が ≈0.4に育つ。
- 上記のセクション3に記載したようにLBM-アガロースゲルスラブを準備するLBM媒体を使用しています。
- 室温で2分間ベンチトップ遠心機で2,000 gで細胞を1 mlを遠心分離します。上清を取り除き、ゆっくりと外径600&20に到達する20μlの氷のように冷たいLBMMに細胞を再懸濁します。
- 精製ファージストックを操作する際には、広いピペットチップを使用するか、またはファージ粒子3を断片化を避けるために、より広い開放先端を作るために定期的なピペットの先端をカットします。優しく男5から0.1の範囲内の平均ファージと細胞の比率に到達するファージを精製し、20μlの持つ細胞のix20μlの。ファージの吸着を可能にするために、氷上で30分間インキュベートした後、ファージDNA注入6をトリガするために5分間35℃の水浴中でインキュベートする。
- 任意の細胞凝集体を分離するために数回ピペッティングします。再びファージを断片化を避けるために広いピペットチップを使用しています。 LBMM、45μlのLBMMに例えば5μlを混合物に混合1:10に希釈します。
- No.1の22×22mmのカバースリップ上のLBM-アガローススラブ(〜10×10mm)のピースを置きます。アガローススラブが室温に達したことを確認するために使用する前に事前に準備LBM-アガローススラブは、少なくとも1時間、室温で配置する必要があります。アガローススラブ上ファージ/細胞混合物の1μlを置き、混合物をアガローススラブに吸収できるようにするために1分間待つ。優しくアガローススラブの上に第1号24×50mmのカバーグラスを置く。この手順は、INFからファージを断片化を避けるためのものですected細胞( 図6)。
6。顕微鏡下で、次の細胞の運命
- 注意深く顕微鏡のステージ上にカバースリップをマウントします。イメージングのために、高倍率(例えば100倍)対物レンズを( 下の説明では顕微鏡システムを参照) を使用します 。
- 初期のタイムフレームで設定した画像を取得する。この画像のセットは、すべての感染したファージの最初の番号と位置を特徴付けるために使用されます。 200nmのz軸(垂直方向)の間隔で15枚のシリーズを取る。 YFPのチャネルを介してイメージ。また、位相コントラストとmCherryチャネルを介して単一の合焦画像を撮影する。 ( 下の説明ではイメージの取得を参照してください)漂白と細胞の損傷を最小限に抑えながら、十分な信号を得るために、光強度、露光時間を最適化します。
- 感染後の細胞の運命のタイムラプスムービーを取得する。画像位相コントラスト、YFPおよびmCherry内のサンプル周りに4時間10分の時間間隔でチャネル。タイムラプスムービーの途中で、漂白、光毒性につながる可能性があり、試料の不必要な被ばくを避けるために、時点ごとのチャネルごとに1つのz位置イメージを使用します。
7。画像解析
- 手動ファージ及び初期時間枠内のレコード·ファージの場所と細胞の長さの数を数えます。これは、MetaMorphまたはImageJのようなソフトウェアを使用して行うことができます。細胞運命(、溶菌溶原または、感染していない)、溶解時間、タイムラプスムービーを再生することにより、任意の他の所望の情報を記録します。異なる細胞運命を識別するには、以下の代表的な結果セクションにタイムラプスムービーを参照してください。
- 上記の手動分析に加えて、より定量的な情報(個々の細胞の経時例えば蛍光レベル)は自動細胞認識と系統トレーシングのアルゴリズムを使用して抽出することができます。我々は、TRの自宅MATLAB組み込みプログラムを使用セルセグメンテーションのSchnitzcell Matlabコード(カリフォルニア工科大学Elowitzグループによって書かれた)と一緒に、細胞系譜および蛍光レベルをacing。
8。代表の結果:
ファージメッキ:
蛍光標識したファージのプラークは(ステップ1.6および第2節の)野生型( 図2)に比べて有意に小さくなります。そこで我々は、プラークの℃のインキュベーターが見えるように37にプレートを少なくとも12時間インキュベートする。
ファージ遠心法:
塩化セシウムステップ勾配(ステップ2.10)を有するファージ試料の遠心分離の後、2つのバンドは( 図3A)見えるはずです。トップバンドは、ファージ懸濁液とSM / CsClを1.3グラム/ mlの層との界面に、細胞の残骸と空のファージキャプシドを含んでいます。下部バンドは、SM / CsClを1.3グラム/ mlおよび1.5グラム/ mlの層間の界面で、ファージバンドです。チーバイSバンドは蛍光λファージLZ2ための緑がかった表示されます。野生型ファージλIG2903のためのバンドは、5青っぽく表示されます。ステップ2.12のCsCl平衡勾配超遠心分離の後、1ファージバンドは管の中央部分( 図3B)に表示する必要がある。蛍光λファージLZ2は GPD-EYFPおよびGPDキャプシドの混合物を含んでいるので、タンパク質対DNAの比は野生型のそれよりも高くなっています。したがって、蛍光λファージLZ2のバンドが野生型λIG290310のそれより(チューブ内の高い位置にあるように表示されます)少し軽いです。
DAPI染色:
図5は、DAPI(4節)とファージを標識した後に得られた典型的なイメージを示しています。精製に成功したファージのYFPおよびDAPI信号は100%に近いので対応している必要があります。我々は一般的に観察することのYFPスポットの1%未満sはDAPIを(ウイルスゲノムなしキャプシドを表す)が含まれていません。 DAPIのスポットの1%未満はYFP(非蛍光ファージに対応)5が含まれていません。
タイムラプスムービー:
溶解細胞を、細胞溶解、続いて細胞内のYFPの蛍光(緑チャンネル)の蓄積によって認識されます。溶原性細胞は、細胞と正常細胞の増殖と分裂の再開内部均一mCherry蛍光(赤色)の蓄積によって認識されます。非感染細胞(または感染が失敗した細胞)上記の表現型のいずれかが表示されませんし、成長し、正常に分割されます。 図7は、いくつかの位相コントラストの画像セット、YFPおよびmCherryチャネル、およびこれらの対応するオーバーレイ画像を示しています典型的なタイムラプスムービー(第6節)からの3つのチャンネルが含まれています。個々のファージ(グリーンスポット)は、初期のタイムフレーム( 図7A)ではっきりと見える。典型的には、数他のファージを吸着されている間、 図7B(左パネル)に示すように、ファージの、(おそらくそれらの細胞を感染させること)は、細胞表面上に見られる。感染の結果は時間の経過区別になります。溶菌サイクルは、細胞溶解(解放緑ファージ、 図7Dと爆発したセル)に続いて新たなファージの細胞内産生(緑、 図7C)によって示されます。溶原性はP REのプロモーター(赤、 図7C)と細胞の増殖と分裂(赤、 図7D)の再開からmCherryの生産によって示されます。




図1。 AB)をかくまって、GPD-EYFPファージの溶原菌を誘導することによって得られる。ファージはステップ(パネルCL)のシリーズを通して精製されています。

図2ファージプラーク。蛍光ファージのプラーク(左)が37℃で12時間プレートをインキュベートした後、野生型(右)に比べて小さく℃である

図3遠心分離した後にバンドをファージ。)2つのバンドはCsCl段階勾配で超遠心分離後に表示されます。トップ1は、細胞の残骸と空のファージキャプシドに相当し、ボトムバンドが目的のファージを含んでいます。左:蛍光ファージ、右:野生型B。 )は、単一のファージバンドがCsCl平衡勾配で超遠心分離後に表示されます。蛍光ファージバンド(左)野生型ファージ(右)青みがかったバンドに比べて、緑がかっています。

図4アガロースゲルスラブを準備する手順。

図5:DAPI染色後のファージの蛍光画像。個々のファージは容易に区別できます、とYFPおよびDAPI信号は非常によく共局在。

図6ファージ感染およびイメージングセットアップの概略説明が。 この画像のフルサイズバージョンを表示するには、ここをクリックしてください。
グレ7 "SRC =" / files/ftp_upload/3363/3363fig7.jpg "/>
図7ファージ感染のタイムラプスムービーからの典型的なイメージ。示す位相コントラスト、YFPおよびmCherryチャネルだけでなく、3つのチャネルのオーバーレイです。最初の時間枠から、(A)YFPチャンネルの画像。左、異なるz位置におけるYFP画像の合計。 3右の画像は、異なるz位置、細胞表面のさまざまな領域に対応する時の試料のYFPイメージです。 (B)、(C)及び(D)の位相差(中央左)、異なる時間枠でYFP(中央右) とmCherry(右)チャンネルのオーバーレイイメージ(左)。 (B)は、T = 0で、2つのセルは、単一のファージ(グリーンスポット)に感染し、それぞれを見てきており、1つのセル3ファージに感染している。また、観測されたいくつかの非吸着ファージである。 (C)T = 80分で、シングルファージに感染した2細胞はそれぞれ溶菌経路に入った、として示す新しいファージの細胞内産生(緑)によるd。 PREプロモーター(赤)からmCherryの生産によって示されるように3ファージに感染した細胞は、溶原経路に入りました。溶原性細胞が分かれているが(D)はt = 2の時間で、溶菌経路は、細胞溶解(セルが爆発)をもたらしました§。
図7の§左パネル(C)と(D)携帯から転載されており、141、曽、サミュエル·O·スキナー、Chenghang宗、ジャン·シッピー、マイケルFeiss、およびIDOゴールディングをLanying、細胞内レベルでの意思決定は、結果を決定エルゼビアの許可を得て、バクテリオファージ感染、682から691まで、著作権(2010)。
| 系統名 | 関連する遺伝子型 | ソース/参照 |
| 菌株 | ||
| LE392 | 商標F | ジョンCronan、イリノイ大学 |
| ファージ株 | ||
| λLZ1 | GPD-EYFP、cI857 SAM7 D-EYFP B :: kanR | 曽ら 5 |
| λLZ2 | λLZ1としてGPD-モザイク、同じ遺伝子型 | 曽ら 5 |
| プラスミド | ||
| PP RE - mCherry | P、RE、 アンプ Rの制御下mCherry | 曽ら 5 |
| pPLate * D | λ後期プロモーターの制御下で、GPD、 アンペアR | 曽ら 5 |
表1菌株、本研究で使用したファージとプラスミド。
| 密度ρ(g / ml)を | CsCl密度(g)を | SM(ミリリットル) | 屈折率η |
| 1.30 | 39 | 86 | 1.3625 |
| 1.50 | 67 | 82 | 1.3815 |
| 1.70 | 95 | 75 | 1.3990 |
ステップ勾配をSMバッファー(100ミリリットル)で調製表3。CsClのソリューションを提供しています。
Discussion
菌株、ファージ及びプラスミド:
ひずみLE392はsupFです。それは(詳細については表1を参照)は、ファージゲノム中SAM7突然変異を抑制するために選ばれました。溶菌経路を選択した感染細胞が意志としてこのように、誘導され溶原は、最終的に、ファージ粒子を溶解し、解放します。溶原性細胞は30℃で栽培されている℃のファージゲノム中857アレルCI温度感受性の存在に起因する。熱誘導後、GPD-EYFPおよび野生型GPDは、λLZ1のゲノムおよびプラスミドpPlate * Dのそれぞれから同時発現される。その結果、新たに作成されたλファージLZ2のキャプシドはGPD-EYFPおよびGPDタンパク質の混合物を含んでいます。このモザイクファージは、構造的に安定しており、個々のファージ5の検出を可能にするために十分に蛍光性である。 PP RE - mCherryは溶原pathwaの選択を検出するために使用されるプラスミドレポーターですyである。プロモーターP REは溶原性1,11の確立時にCIIによって活性化される。 PP RE - mCherry 5は mCherry 12でGFPを置き換えることにより、PE-GFP 11から派生したものです。詳細については、当社の以前の仕事5を参照してください。
成長条件パラメータ:
溶原誘導(第1節)の間に、180 rpmで軽度の揺れは良いウイルス収量13を与える。グルコース代謝があるので避けてください成長培地中のグルコースの使用は酸性代謝産物を生成し、成熟したラムダ粒子が酸性pH 13で不安定である。 MgSO 4の加算はファージカプ3を安定させることを目的としている。野生型CI(代わりにCI 857)を運ぶファージは、溶原菌は、DNA損傷剤のマイトマイシンC 3を使用して誘導することができる。ステップ1.3では、37℃でインキュベーション℃は通常90分を超えてはいけません。それは、USEFです 30分ごとにOD 600により細胞密度を確認してUL。良いライセートは、OD 600が約0.2以下に低下し、残りのOD 600が細胞破片の結果です。新しく作成されたファージは、細胞の破片に自分のDNAを注入するために始めることができるから時間が経ちすぎインキュベートすると、下のファージの収率で発生する可能性があります。ステップ2.11と2.13で見えるファージバンド(少なくとも1×10 11ファージ粒子)を取得するには、ステップ1.2で少なくとも500 mlの培養液を栽培しています。ステップ5.1および5.2の増殖培地中に0.2%マルトースの添加は子羊、ラムダファージの吸着3,14に対する受容体の発現を誘導することを目的としている。 mCherry -ステップ5.2の代わりに、100倍1000倍に希釈し、レポータープラスミドのPP REからmCherryのバックグラウンドレベルを減らすことを目的としています。 、35をトリガファージDNA注入のためのステップ5.5°Cでは温度感受性cI857対立の誘発を避けるために選択されます。
ファージ精製:
jove_content ">ファージの精製工程(2.11〜ステップ2.1)は、他の精製プロトコル5に置き換えられますが、CsCl平衡勾配(ステップ2.12と2.13)を介して最終的に超遠心分離は不可避であることができます。スイングバケットローターはステップ2.10と2.12で必要とされる鋭い目に見えるファージバンドを確保する。純粋ファージ株式の取得には一週間ほどかかる場合がありますので、中間段階の間に何かがうまくいかないことを確認するための方法に沿ってファージ力価を確認する必要があります。ファージの取り扱い:
第2節の全精製手順の間に、それはファージは、ファージ頭部からファージ尾部を剪断避けるために穏やかにライセートを処理するために重要です。第5節で細胞感染(例えば、5.7を通じて5.5ステップ)の間に、それはまた、感染細胞からファージ粒子のせん断を避けることが重要です。のIE、ファージのDNAを注入した後、感染細胞から剪断される場合、結果は "暗い"感染症であることに注意してくださいクション結果が実験で観察されたが、感染ファージはしませんされます。ファージまたはファージ/細胞混合物を取り扱うときは、必ずこのような問題の発生を防ぐために、我々は広いピペットチップを使用しています。
DAPIのテスト:
DAPI(4節)とファージストックを染色すると、ファージストックの純度を確認するには、迅速かつ効率的な方法です。また、時間をかけて既存のファージストックの分解の可能性をテストするために使用することができます。純粋な株式については、蛍光顕微鏡下でYFPおよびDAPI信号の共局在はほぼ100%であるべきである。我々は通常のYFPスポットの1%未満では、これらの粒子が正常にウイルスDNAをパッケージ化しなかったことを示していたり、既に他の場所で彼らのDNAを注入したDAPIを(ウイルスゲノムなしキャプシドを表す)は、含まれていないことを確認します。 DAPIのスポットの1%未満はYFP(非蛍光性のファージに相当)が含まれていません。この条件が満たされない場合、2.14を通じて2.12はoに繰り返されるために必要な手順再び精製するrder。長期的なライブセルイメージングがここに必要としないため、撮像パラメータに関しては、ステップ4.3の顕微鏡のセットアップでは、第5節でほど重要ではありません。 1の願いはただ一つのファージ粒子の蛍光強度を校正する場合は、第5節と同様に顕微鏡の設定を保存しておくことは有用である。 PBS-アガローススラブは非常にきれいで、またはDAPI染料が使用されても過言ではない場合には、ファージDNAに対応するいくつかのDAPIスポットは "ハロ"で囲まれていてもよい。少なすぎるDAPIの染料が使用されている場合は、DAPIのチャネルからの信号は非常に弱いかもしれません。
顕微鏡システム:
第6節のイメージングのために、私たちは100倍対物レンズ(プランのFluo、開口数1.40、油浸)と標準フィルターセット(ニコン)と、商業倒立落射蛍光顕微鏡(エクリプスTE2000-E、ニコン)を使用します。蛍光光源は、光強度の制御とアーク灯である。次の機能は、コンピュータ制御されています:x、y、zのPOsition、明視野と蛍光シャッター、及び蛍光フィルター選択。オートフォーカス機能が必要です。そうしないと、フォーカスが容易にタイムラプスムービー(通常は4時間長い)の間に流れ出すことがあります。それが並列で複数の感染イベントを追跡することが可能として、各時点で(x、y)は、複数のポジションを獲得する能力は、便利です。我々は一般的に100感染イベントに追従し、それぞれの映画の中で8段階のポジションを獲得。我々が使うカメラは16ビット(Cascade512、Photometrics)のダイナミックレンジを持つ16×16μmのピクセルのカメラと512×512冷却CCDである。買収は、MetaMorphソフトウェア(Molecular Devices)を使用して実行されます。顕微鏡は、恒温室に置かれるべきである、或いは、顕微鏡ステージは、温度制御室で囲む必要があります。
画像取り込み:
ライブセルイメージングのために、それは漂白とフォーにつながる可能性があり、試料の不必要な被ばくを避けるために重要ですtotoxicity。したがって、それは最初に漂白過大または細胞増殖を阻害するに至らない間に蛍光を検出することができ、最適な露光量を見つけるためにあなたのシステムを特徴づけるために最善の方法です。良い蛍光画像を得るためには、励起光強度、露光時間とカメラゲインと遊ぶ。ステップ6.2から6.3で、10分のフレーム間隔は、光の露出を最小限に抑えることを目的として選択されます。各フレームでは、単一の合焦画像は、位相コントラスト(細胞認識のために)および蛍光チャネル(細胞の運命を決定するため)が必要である。最初の時点では、しかし、YFPのチャネルを介して、複数のz位置の画像は、細胞表面上のすべての感染したファージをキャプチャする必要があります。初期フレームにおけるYFPの露光時間はまた後で時間枠でタイムラプスムービーで使用しているよりも高くする必要があるかもしれません。
画像解析:
非常に慎重にステップ7.1における細胞表面の周りファージ粒子を数える。として上で述べたように、我々は、ステップ6.2におけるYFPのチャネルを介してZ-スタックのシリーズを取る。しかし、これはまだカウントに挑戦一部の蛍光ファージ粒子アウトフォーカスを離れることができる。最初の時間枠内のセルの長さがMetamorphソフトウェアを使用して測定されています。セル長はまた、ImageJのか、他のソフトウェアツールによって測定することができる。さらに、自動化された家庭MATLAB組み込みプログラムは、そのような細胞系譜に沿って時間をかけて蛍光変化などの情報を取得する際に非常に役立つことがあります。
Disclosures
特別な利害関係は宣言されません。
Acknowledgments
我々はマイケルFeissとファージの作成と精製に関するガイダンスジャンシッピーに感謝しています。我々は、細胞認識ソフトウェア、Schnitzcellを提供するためのマイケルElowitzに感謝します。 、ウェルチ財団(助成Q-1759)とヒューマン·フロンティア·サイエンス:ゴールディングラボの仕事は国立衛生研究所(R01GM082837)、国立科学財団(生きている細胞の物理学研究センター082265、PFC)からの補助金によってサポートされていますプログラム(RGY 2008分の70)。
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform | Fisher Scientific | C298-500 | |
| NaCl | Fisher Scientific | S271-3 | |
| DNase I | Sigma-Aldrich | D4527-10KU | |
| RNase | Sigma-Aldrich | R4642-10MG | |
| PEG8000 | Fisher Scientific | BP233-1 | |
| SM buffer | TEKnova, Inc. | S0249 | |
| NZYM | TEKnova, Inc. | N2062 | |
| CsCl | Sigma-Aldrich | C3011-250G | |
| Syringe | BD Biosciences | 309585 | |
| Needle | BD Biosciences | 305176 | |
| Dialysis cassette | Thermo Fisher Scientific, Inc. | 66333 | |
| Microscope slide | Corning | 2947-75x50 | |
| Agarose | Fisher Scientific | BP160-100 | |
| SW40Ti ultra-clear tube | Beckman Coulter Inc. | 344060 | |
| SW60Ti ultra-clear tube | Beckman Coulter Inc. | 344062 | |
| SW40Ti rotor | Beckman Coulter Inc. | 331302 | |
| SW60Ti rotor | Beckman Coulter Inc. | 335649 | |
| Refractometer | Fisher Scientific | 13-947 | |
| Epifluorescence microscope | Nikon Instruments | Eclipse TE2000-E | |
| Table 2. Reagents and equipment. | |||
References
- Oppenheim, A. B., Kobiler, O., Stavans, J., Court, D. L., Adhya, S.
- Ptashne, M. A genetic switch : phage lambda revisited. , 3rd edn, Cold Spring Harbor Laboratory Press. (2004).
- Hendrix, R. W. Lambda II. , Cold Spring Harbor Laboratory. (1983).
- Hershey, A. D. The Bacteriophage lambda. , Cold Spring Harbor Laboratory. (1971).
- Zeng, L. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell. 141, 682-691 (2010).
- Edgar, R. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. , (2008).
- Ausubel, F. M. Current protocols in molecular biology. , John Wiley & Sons. (1994).
- Sambrook, J., Russell, D. W. Molecular cloning : a laboratory manual. , 3rd edn, Cold Spring Harbor Laboratory Press. (2001).
- Fasman, G. D. Practical handbook of biochemistry and molecular biology. , CRC press. (1989).
- Kaiser, A. D. On the internal structure of bacteriophage lambda. J. Gen. Physiol. 49, 171-178 (1966).
- Kobiler, O. Quantitative kinetic analysis of the bacteriophage lambda genetic network. Proc Natl Acad Sci. 102, 4470-4475 (2005).
- Shaner, N. C. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572 (2004).
- Personal communication with M. Feiss. , Forthcoming.
- Schwartz, M. The adsorption of coliphage lambda to its host: effect of variations in the surface density of receptor and in phage-receptor affinity. J. Mol. Biol. 103, 521-536 (1976).

{kind=link}