



Overview
ソース: マイケル S. リー1とトーニャ J. ウェッブ1
1メリーランド大学医学部微生物学・免疫学科、マーリーン・スチュワート・グリーンバウム総合癌センター、ボルチモア、メリーランド州 21201
免疫組織化学(IHC)および免疫細胞化学(ICC)は、抗体を用いた特定抗原の発現および局在化を可視化するために用いられる技術である。IHCの最初の使用は、アルバート・クーンズが肺炎球菌に感染したマウスからの組織切片における肺炎球菌抗原の存在を視覚化する技術を使用した1941年でした(1)。免疫組織化学という名前は、IHCで使用される組織切片を参照して、抗体を参照して「免疫-」、および「histo-」の根に由来する。免疫細胞化学の根元「サイト-」は、ICCとIHCの主な違いを強調しています。IHCは組織全体の切片を使用しますが、ICCは組織から単離された細胞や培養中に増殖した細胞を使用します。使用されるサンプルの違いは、サンプル調製が技術的にIHCとICCの間で異なることを意味しますが、そうでなければICCとIHCのプロトコルは同一であり、用語は頻繁に同じ使用されることがわかります。
IHCおよびICCの両方において、ペルオキシダーゼまたはロダミンなどの化学的または蛍光タグを有する抗体は、タグ付けされた抗体を抗原に対する特異的結合を通じて目的の任意の抗原の分布を可視化するために使用される。IHCの場合、組織の薄いスライスは、染色される前に組織の構造を維持するためにスライド上に固定され、組織全体のコンテキストで抗原の視覚化を可能にする(図1)。ICCの場合、細胞は染色される前にスライド上に均等に分布し、個々の細胞内の抗原分布の可視化を可能にするが、特定の組織の構造内には存在しない。2 つのプロトコル間の類似性により、このプロトコルは IHC に関連するサンプル調製の追加の複雑さに対処するために IHC に焦点を当てます。
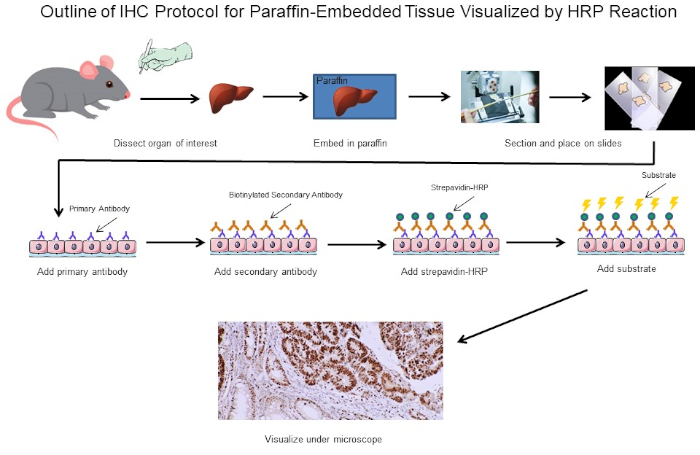
図1:IHCプロトコルの概要マウスから解剖したパラフィン埋め込み組織のIHCプロトコルの視覚的輪郭。このプロトコルは、ビオチン化二次抗体とストレパビジン-HRPを使用して、抗体結合の位置を可視化します。蛍光タグ付き抗体などの他のオプションも可能です。この図のより大きなバージョンを表示するには、ここをクリックしてください。
IHCを行う際の最初の主要な決定は、染色プロセス全体を通して組織の構造を維持するために組織セクションを調作成する方法である。2つの主要な選択は、パラフィン埋め込み組織のホルマリン固定セクションまたは凍結組織の新鮮なセクションである。どの方法を使用するかは、下流の解析を行うかによって異なります。パラフィン埋め込み組織のホルマリン固定は、一般的に、新鮮な組織を凍結しながら、IHCの外の後続のアッセイのためのタンパク質機能を維持しながら、最適なイメージングのための組織形態をより良く保存すると考えられている。さらに、新鮮な凍結組織切片は、遺伝子発現分析(2)により適することが示されている。第3の考慮事項は、一部の抗体は特定のタイプのセクションに対してのみ最適化され、他の抗体では機能しない可能性があるため、目的の抗原に対する抗体が固定組織セクションまたは凍結組織切片に適しているかどうかです。最後に、新鮮な凍結サンプルは-80°Cで保存する必要があり、固定されたセクションは室温ではるかに長く保存することができる間、1年を超えて持続しない可能性があるため、組織切片を保存する必要がある期間を決定する必要があります。これらは、パラフィン埋め込み組織のホルマリン固定切片または凍結組織の新鮮なセクションを使用するかどうかを決定するための主要な考慮事項のいくつかです。最終的には、十分な組織を持っている場合は、両方の一部を持っているだけで良いかもしれません。
本実験では、リンパ腫発症の自発的マウスモデルから拡大脾臓においてサイクリンD1発現が増加したかどうかを調べる。脾臓組織試料は、まず、野生型マウス、リンパ腫を有しないトランスジェニックマウス、または自発的にリンパ腫を発症したトランスジェニックマウスのいずれかから単離した。脾臓組織試料をパラホルムアルデヒドに固定し、パラフィンに埋め込み、切除し、マウス抗サイクリンD1一次抗体を用いて染色し、続いて馬の抗マウス二次抗体を用いて、3,3-ジアミノベンジジン(DAB)を用いて開発した。その後、ハリス・ヘマトキシリン溶液でセクションを逆染色し、その後、セクションを20倍の倍率で画像化した。
試薬
パラフィン埋め込みセクション
- 4% パラホルムアルデヒド(PFA)
- エタノール(無水変性、組織学的等級100%、95%、80%、75%、50%)。二重蒸留水(ddH2O)を使用して100%のストックから希釈することができます
- キシレン
- IHC互換ガラススライドは、組織セクションが手順全体を通して取り付けられたままであることを確認します。IHC互換ガラススライドは、特殊なコーティングを持っており、複数の小売業者から容易に入手可能です。ICCを実行する場合は、チャンバスライドを使用してください。チャンバースライドは、細胞がチャンバーに播種され、細胞がスライドに付着し、適切な合流に達するまでインキュベーターに置くことができ、その時点でチャンバーを除去し、染色をIHCと同様に進めることができます。
- パラフィン
- 0.3% 過酸化水素 (H2O2)/メタノール: 調味するには、1 mL 30% H2O2 ~ 99 mL メタノールを添加します。-20°Cでの保管
- 抗原検索バッファー: IHC クレートバッファー pH 6.0
新鮮な冷凍セクション
- 最適な切断温度(OCT)埋め込み化合物
- 最適な固定性:-20°Cに冷却された4%のPFAまたはアセトン
染色
- ブロック バッファ: ユーザーが決定する必要があります。一例は、1X PBSで希釈された馬血清です
- 希釈された一次抗体:メーカー仕様を参照
- 希釈ビオチン化二次抗体:メーカー仕様参照
- 希釈されたアビジン-ホースラディッシュペルオキシダーゼ(HRP):ペルオキシダーゼ可視化のみ。製造元の仕様を参照してください。
- DABまたは他の互換性のある基板
- カウンターステイン(オプション)
- エタノール(無水変性、組織学的グレード100%および95%)
- キシレン
- オルガノ/リモネン山
Procedure
1. 免疫細胞化学用細胞の調製
- チャンバ付きスライドまたはチャンバ付きカバーに関心のある種子細胞は、24ウェル培養プレートのウェルに0.5 mLの細胞懸濁液を加えることによってスリップする。
注:一部の細胞は、ポリリジンで処理されたカバースリップなど、治療されたカバースリップに成長を必要とする場合があります。最適な治療条件は、使用する細胞の種類に応じてユーザーによって決定されるべきである。 - プレートを加湿したCO2インキュベーターに入れ、細胞を37°Cで50~70%コンフルエントになるまで成長させます。
- 細胞が最適な合流に達したら、各ウェルから培養培地を取り出し、0.5mLの4%PFA(1X PBSで希釈)で細胞を固定し、室温で20分間インキュベートします。
- 固定液を取り外し、1X PBSの1 mLでウェルを3回洗います。
- 次に、各ウェルに0.1%トリトンX-100の0.5mLを1X PBSに加えて細胞を透過化し、室温で15分間インキュベートする。
- 透過性バッファーを吸引し、1X PBSの1 mLでウェルを3回洗浄します。
- カバースリップ上の細胞は固定され、透過化されるようになりました。次の免疫組織化学の例について実証された染色手順に進みます- インキュベーションは、組織セクションスライド上で直接ではなく、24ウェルプレートのウェル内で行われるべきである場合を除きます。
2. 染色のためのホルマリン固定、パラフィン埋め込みセクションの調製
- ホルマリン固定、パラフィン埋め込み組織切片を得る。
デパラフィン化
- スライドを100%キシレンに2回、それぞれ5分間浸します。
水分 補給
- スライドを100%エタノールに2回ずつ3分間浸します。
- スライドを95%エタノールで3分間浸します。
- スライドを70%エタノールで3分間浸します。
- スライドを50%エタノールに3分間浸します。
内因性ペルオキシダーゼ活性の遮断
- 室温で30分間、0.3%H2O2の100mLでスライドをインキュベートします。
- スライドを1X PBSで2回5分間洗います。
抗原検索
- スライドをIHCク硝酸バッファー(pH6)に浸し、20分間沸騰させます。
- ティッシュスライドは今染色の準備ができている。
3. 染色のための新鮮な冷凍、OTC埋め込みセクションの準備
- 5mmの新鮮な分離された組織を金型に入れ、セクションが完全に覆われるまでOCTを加えます。
- 完全に凍結するまで、ゆっくりと組織ブロックを液体窒素に沈めます。サンプルは今1年まで-80°Cで貯えることができる。
- 断面の準備ができたら、凍結組織ブロックをクライオスタットに移し、セットアップ全体を-20°Cにします。
- クライオスタットを使用して5〜10 μmの厚い組織切片を切断し、ブラシを使用してIHC互換ガラススライドに直接セクションを配置します。
- スライドを室温で一晩乾燥させます。スライドはまた-80°Cで貯えることができる。
- 室温で15分間、4%PFAの250 mLでスライドを浸し、染色前にスライドを固定します。最適な固定方法は、ユーザーが決定する必要があります。
- スライドを1X PBSの250 mLで2回、それぞれ5分間浸します。
- 内因性ペルオキシダーゼ活性を遮断するために、室温で30分間、0.3%H2O2の250 mLにスライドを浸します。
- スライドを1X PBSの250 mLで2回、それぞれ5分間浸します。
- これで、スライドを染色する準備が整いました。
4. 染色
- バリアペンを使用して疎水性バリアで組織を円形にします。
ブロック
- ピペットを使用して、100 μLのブロッキングバッファー(1X PBSで希釈された馬血清)を室温で1時間にわたってセクション上に置きます。
- ピペットを使用してブロッキングバッファを削除します。
一次抗体インキュベーション
- 100 μL希釈一次抗体溶液(マウス抗ヒトサイクリンD1希釈1:100をブロッキングバッファーで1:100)で包まれた組織を室温で30分間インキュベートする。
- 各スライドから一次抗体を排出し、1X PBSでスライドを5分間5分間洗浄します。
二次抗体インキュベーション
- 希釈されたビオチン化二次抗体(ビオチン化された馬抗マウスIgG希釈1:200)を室温で30分間100μLでインキュベートします。
- 切片を排出して二次抗体を取り出し、それぞれ5分間1X PBSで2回洗浄します。
色の開発
- HRPを用いた可視化:アビジンビオチン複合体(ABC)試薬の100 μLを追加し、室温で30分間暗闇の中でセクションをインキュベート
注:蛍光タグ付き抗体は、適切な顕微鏡を用いて使用および可視化することもできる。 - スライドを1X PBSで2回5分間洗います。
- DABの100 μLの断面を最大5分間インキュベートしてスライドを開発します。
- 蒸留水(dH2 O)を室温で5分間加えて開発を中止します。
カウンターステイン(必要に応じて)
- ハリス・ヘマトキシリン溶液(または0.1M酢酸ナトリウム(pH 4.2)で0.5%メチルグリーン)に10分間スライドを簡単に浸します。
- dH2Oでスライドを2回5分間洗い流し、カウンターステインを洗い流します。
脱水
- スライドを95%エタノールに2回ずつ5分間浸します。
- スライドを100%エタノールに2回ずつ5分間浸します。
- スライドを100%キシレンに2回、それぞれ5分間浸します。
- スライドをペーパータオルで拭きます。
取り付けおよびカバースリップアプリケーション
- オルガノ・リモネン・マウントなどの取り付けメディアをスライドに1滴追加し、セクションの上にカバースリップを配置します。
顕微鏡分析
- 分析のために適切な顕微鏡下で染色されたセクションを観察します。ここでは、観察に標準的な光顕微鏡を使用し、イメージングには取り付けられたデジタルカメラを使用しました。
免疫細胞化学と免疫組織化学は、培養細胞および組織に関心のあるタンパク質の染色方法をそれぞれ有する。両方の関連技術の基本原理は、検出システムでタグ付けされた特定の抗体を使用してタンパク質を同定および視覚化し、細胞および組織内の位置、ならびに相対レベルを決定することを含む。いずれの実験においても、プロセスはサンプル調製から始まる。
細胞内のタンパク質または抗原の位置を特異的に可視化する免疫細胞化学の場合、これには3つのステップが含まれます。最初のステップはめっきであり、これは、カバースリップまたはスライド上の成長培地中の細胞を培養することを伴う、典型的には、培養プレートのウェル内にある。続いて固定が続き、パラホルムアルデヒドのような沈殿剤や架橋剤を細胞に加えて、タンパク質の構造的完全性を維持し、酵素活性が分解するのを防ぎます。最後のステップは透過性であり、染色のために細胞膜を透過性にするために洗剤を添加することを含む。
対応する方法では、免疫組織化学、タンパク質または抗原は組織内で可視化され、試料調製には5つのステップがある。まず、組織全体を固定し、通常はパラホルムアルデヒドを投与する。これは、パラフィンのブロックに組織を埋め込み、次に、マイクロトームと呼ばれるマシンを使用して、スライドに配置することができる薄いスライスに組織を切断するために、このブロックのセクション化が続きます。次に、スライドは、デパラフィン化、または組織スライスの周りからパラフィンの除去を行う。次に、任意抗原検索工程を行うことができる。これは、固定中に架橋されたエピトープのマスクを解除するために熱または酵素を使用して行うことができ、抗体結合のためにそれらを利用できます。適切なサンプル調製後、標的特異的一次抗体を細胞または組織試料に添加する。この一次抗体は、目的のタンパク質に結合する必要があります。次に、二次抗体を添加し、一次抗体を検出して結合する。この二次抗体は、HRPと呼ばれる酵素に結合するか、または結合することができる。その特定の基板DABが添加されると、HRPはこれを不溶性の褐色沈殿物に変換します。この茶色の染色は、標的タンパク質の位置を示す。スライドはまた、青色で核を標識し、細胞内局在を決定するための空間基準点を提供するヘマトキシリンで染色されています。その後、取り付け媒体をスライドに追加し、続いて染色されたサンプルをシールして保存するためにカバースリップを行います。最後に、スライドは軽い顕微鏡でイメージすることができる。
このビデオでは、めっきされた細胞および組織セクションのサンプル調製技術を観察し、続いて組織セクションの免疫染色を行う。
まず、目的の細胞は、カバースリップに座っている必要があります。これを行うには、組織培養フードで作業し、個々のカバースリップを24ウェルプレートのウェルに配置します。その後、サッシを閉じ、UVライトをオンにして、カバースリップを少なくとも15分間殺菌します。次に、UV ライトをオフにします。コンフルエントな10センチメートルの皿から目的の細胞を持ち上げるには、メディアを吸引し、PBSで簡単に洗浄し、2分間細胞にトリプシンを追加します。次に、プレートの側面をタップして、細胞が切り離されていることを確認し、メディアでトリプシンを中和します。次に、0 を追加します。各ウェルにセル懸濁液の5mLは、カバーリップをカバーすることを確認してください。加湿されたCO2インキュベーターにプレートを入れ、細胞が50-70%のコンフルエントになるまで37°Cで成長させることができます。
細胞が最適な合流に達したら、各ウェルから培養培地を吸引し、でそれらをインキュベートして細胞を固定します。室温で20分間1X PBSで希釈した4%パラホルムアルデヒドの5mL。固定剤を取り除き、各カバースリップの上に1X PBSの1 mLを加える細胞をすすいで下します。直ちにPBSを吸引し、その後、合計3回の洗い流しのために2回以上すすり続けた。
さて、各ウェルに1X PBSに0.1%トリトンX-100の0.5mLを加えて細胞を透過化する。プレートを室温のままに15分間放置します。透過性バッファーを吸引し、各ウェルに1x PBSの1 mLを加えて細胞をすすいで下します。直ちにPBSを吸引し、合計3回の洗い流しのためにさらに2回すすり続けてください。カバースリップ上の細胞が固定され、透過化されたので、インキュベーションが24ウェルプレートのウェル内で行われるべきであるという例外を除き、次の免疫組織化学の例で実証された染色手順に進みます。組織セクションスライドに直接ではなく。
開始するには、調製された、ホルマリン固定、パラフィン埋め込み組織セクションを得る。スライドラックに入れ、100%キシレンの250 mLに完全に浸漬することにより、スライドを脱パラフィン化します。スライドがキシレンで5分間インキュベートできるようにします。その後、容器からスライドを取り出し、ペーパータオルで拭き取り、さらに5分間新鮮な容器に入れ、新しいキシレン浴に入れます。
次に、100%エタノールから始まる一連のグレードのエタノール溶液でセクションを3分間水分補給する。ペーパータオルでスライドラックを拭き取り、スライドを100%エタノールの新しい容器に移し、さらに3分間使用します。この洗浄、ペーパータオルで乾燥し、指定された時間のエタノール濃度に従ってスライドを新しい浴場に移します。最終的なエタノール洗浄後、ペーパータオルでラックを拭き取り、内因性ペルオキシダーゼ活性を遮断するために、室温で30分間、100mLの過酸化水素を30分間インキュベートします。スライドを1X PBSの250mLで5分間洗います。新鮮な1X PBSの容器でこの洗浄をさらに5分間繰り返します。
次に、pH 6.0でIHCクレートバッファーの250 mLにスライドを浸漬し、それらを20分間沸騰させて抗原検索を行います。次に、染色プロトコルに進みます。
IHCの染色プロセスを開始するには、疎水性ペンでセクションを丸で囲み、バッファがカバーする必要がある最小限の領域を特定します。次いで、ピペットを使用して100マイクロリットルのブロッキングバッファーを配置し、この実験では1X PBSで希釈された馬血清をセクション上に置く。室温で1時間スライドをインキュベートします。この後、ピペットを使用してブロッキングバッファを削除します。
次に、1X PBSで希釈した990マイクロリットルの馬血清を1X PBSに添加して1:100希釈で一次抗体および遮断バッファーを希釈する。5 mLエッペンドルフチューブ、続いて一次抗体の10マイクロリットルが続く。希釈した一次抗体を各セクションに100マイクロリットルを加え、室温で30分間スライドをインキュベートする。タイマーが鳴ったら、各スライドから一次抗体を排出し、1X PBSの250 mLで5分間洗浄します。新鮮な1X PBSを使用して、もう一度この洗浄を繰り返します。
スライドが1X PBSで洗浄されている間、1.5 mLチューブに995マイクロリットルのブロッキングバッファーを加え、続いて5マイクロリットルの二次抗体(この場合はバイオチリン化された馬の抗マウスIGG)を加えることによって、二次抗体を1:200希釈に希釈する。希釈した二次抗体を各セクションに100マイクロリットルを加え、室温で30分間スライドをインキュベートする。30分後、セクションから排出して二次抗体を取り出し、1X PBSの250 mLで5分間洗浄します。新鮮な1X PBSを使用して、この洗浄を繰り返します。
さて、アビジンビオチン複合試薬の100マイクロリットルを追加し、室温で30分間暗闇の中でセクションをインキュベートします。次に、スライドを1X PBSの250 mLに5分間浸して洗います。以前の洗浄工程と同様に、新鮮な1X PBSを使用して、この洗浄をもう一度繰り返します。次に、DABの100マイクロリットルのセクションを最大5分間インキュベートしてスライドを開発します。250 mLの蒸留水に5分間浸漬して開発を中止します。
これで、必要に応じてスライドを逆染色できます。これを行うには、ハリスヘマトキシリン溶液の250 mLでスライドを簡単に浸します。250mLの蒸留水で5分間洗い流してカウンターステインを洗い流します。新鮮な蒸留水を使用して、この洗浄をもう1回繰り返します。次に、セクションを脱水します。これを行うには、まず95%エタノールでスライドを5分間インキュベートします。ペーパータオルの上にスライドをブロットし、さらに5分間新鮮な95%エタノールの新しい容器に移します。洗濯、ペーパータオルでブロッティング、スライドを新しいお風呂に移すサイクルを続け、それぞれ5分間の溶液に従います。
最終的なインキュベーションの後、ペーパータオルでスライドをブロットし、オルガノリモネンマウントなどの取り付けメディアのドロップをスライドに追加します。今、空気の泡をトラップしないように注意して、セクションの上にカバースリップを配置します。スライドは今分析のための顕微鏡の下で観察される準備ができている。
染色された部分を観察するには、標準的な光顕微鏡を使用して汚れを視覚化し、デジタルカメラを使用して画像をキャプチャします。IHCのこの特定の例では、野生型および自発性、二重トランスジェニック、またはDTGマウスからの脾臓組織が、リンパ腫におけるDyclin D1発現を研究するために比較される。組織をパラフィンを埋め込み、切除し、抗サイクリンD1抗体で染色し、20倍の倍率で画像化した。細胞を発現するサイクリンD1は、青色組織背景に対して赤褐色で示される。種々のマウスからの画像間の染色強度を比較すると、非拡大脾臓は、マウスの遺伝子型に関係なく、比較的低い量のCyclin D1発現を有する。これに対して、DTGマウスからの脾臓の肥大は、このマウスモデルにおける癌発症とCyclin D1発現との相関を示す赤褐色染色の増加を示す。
Subscription Required. Please recommend JoVE to your librarian.
Results
IHCとICCは幅広いアプリケーションを持っています。例えば、IHCの1つの使用は、腫瘍発達の自発的マウスモデルにおける腫瘍遺伝子の発現を調べることである。図2では、リンパ腫発症の自発的マウスモデルにおいて、拡大脾臓においてサイクリンD1発現が増加したかどうかを調べる。脾臓組織試料をパラホルムアルデヒドに固定し、パラフィンに埋め込み、切片し、抗サイクリンD1抗体(ブロッキングバッファーで1:200を希釈)を用いて染色し、次いで20倍の倍率で切片を画像化した。細胞を発現するサイクリンD1は、青色組織背景に対して赤褐色で示される。これらの結果は、拡大脾臓においてサイクリンD1発現が増加したことを示唆し、このモデルにおける癌発症とサイクリンD1発現との相関を示す。

図2:リンパ腫の自発的二重トランスジェニック(DTG)マウスモデルにおける脾臓サイクリンD1発現抗サイクリンD1一次抗体で染色された脾臓組織の画像を、メチルグリーンで対照染色し、DAB基板で活性化したビオチン化二次抗体およびABC試薬を用いて可視化した。赤褐色は、抗体が結合した位置を表し、青色に染色された脾臓組織の構造内で腫瘍細胞を発現するCyclin D1の存在を示す。この図のより大きなバージョンを表示するには、ここをクリックしてください。
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
免疫組織化学(IHC)および免疫細胞化学(ICC)は、抗体を用いた特定抗原の発現および局在化を可視化するために用いられる技術である。組織は、最初に組織形態を維持する薄いセクションに切断され、スライド上に配置されます。その後、抗体が追加され、目的の抗原に結合し、顕微鏡下で可視化することを可能にする特定のタグが装備されています。したがって、この基本的な概念を通じて、組織構造の文脈における抗原の分布を可視化し、研究することができる。しかし、包括的な概念は基本的なものですが、これらの手法の複雑さと有用性の両方を高める複数の異なるアプローチとバリエーションが開発されています。本稿では、IHCとICCの基本的な概念、これらの技術を使用する際に考慮する必要がある主な決定事項、および詳細なステップバイステッププロトコルについて説明した。IHCとICCによって生成された画像は、一般的に最終製品であり、異なる条件間の染色の量や分布の明らかな違いを強調するためにそのまま公開することができます。
Subscription Required. Please recommend JoVE to your librarian.
References
- Coons, A. H. Creech, H. J., Jones, N. and Berliner, E. The Demonstration of Pneumococcal Antigen in Tissues by the Use of Fluorescent Antibody, The Journal of Immunology, 45 (3), 159-170 (1942).
- Ripoli, F. L., Mohr, A., Hammer, S. C., Willenbrock, S., Hewicker-Trautwein, M., Hennecke, S., Escobar, H. M. and Nolte, I. A comparison of fresh frozen vs. Formalin-fixed, paraffin-embedded specimens of canine mammary tumors via branched-DNA assay. International Journal of Molecular Sciences, 17 (5) (2016).