



Overview
Quelle: Susannah C. Shissler1, Tonya J. Webb1
1 Institut für Mikrobiologie und Immunologie, University of Maryland, Baltimore, MD 21201
Immunpräzipitation (IP, auch bekannt als "Pull-down"-Assay) ist eine weit verbreitete Technik, die Anwendungen in einer Vielzahl von Bereichen hat. Erstmals 1984 konzipiert, wurde es 1988 verfeinert (1, 2). Das grundlegende Ziel der IP ist die Reinigung und Isolierung eines bestimmten Proteins mit einem Antikörper gegen dieses Protein. Das Wort "Immuno" bezieht sich auf die Verwendung eines Antikörpers, während das Wort "Ausfällung" sich auf das Abziehen einer bestimmten Substanz aus einer Lösung bezieht. Das Zielprotein kann endogene oder rekombinant ewanant sein. Die meisten rekombinanten Proteine haben ein Epitop-Tag (d. h. myc oder Flag), das an ihnen angebracht ist, um die nachfolgende Reinigung zu vereinfachen. In der Regel ist es einfacher, rekombinante Protein-IP zu optimieren, da die Antikörper gegen rekombinante Epitop-Tags sehr stark und wirksam sind. Antikörper gegen endogene Proteine haben eine extrem variable Wirksamkeit , was es viel schwieriger macht, diese IPs zu optimieren. Ein notwendiger Schritt nach der Immunpräzipitation ist die Überprüfung der Reinigung. Das isolierte Protein wird mit SDS-PAGE aufgelöst und anschließend durch westliche Flecken auf Reinheit untersucht (Abbildung 1). Eine wichtige Kontrolle ist die Verwendung eines anderen Antikörpers während des Western Blot, um den Abziehdes des richtigen Proteins zu überprüfen. Die Kombination von IP mit nachfolgenden Techniken ist ein leistungsstarkes Analysetool. Das Ziel nach der Reinigung kann die Charakterisierung des Proteins selbst durch NMR, Massenspektrometrie und In-vitro-Assays oder die Analyse der interagierenden Partner des Proteins (z. B. Protein, DNA, RNA) (3, 4, 5) sein.
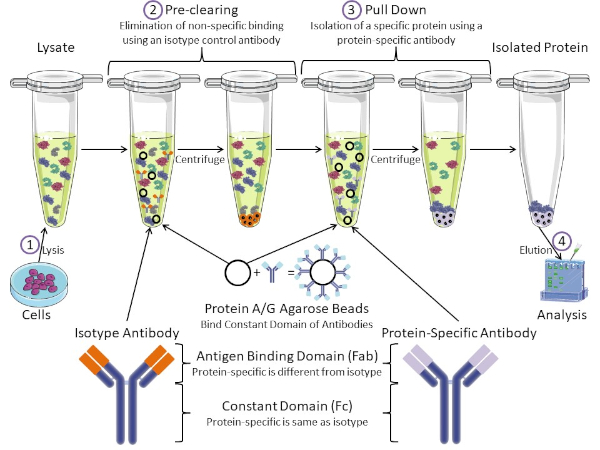
Abbildung 1: Überblick über das Immunpräzipitungsverfahren. Immunpräzipitation ist die Isolierung eines bestimmten Proteins mit einem Antikörper. Nach der Produktion von Lysat aus Zellen, gibt es zwei wichtige Schritte - Vor-Clearing und ziehen nach unten. Während des Vorklärungsschritts werden die Zelllysate von Proteinen vorgereinigt, die sich nicht spezifisch mit einem Isotyp-Kontrollantikörper an Antikörper binden. Im Pull-Down-Schritt wird das Zielprotein mit einem proteinspezifischen Antikörper nach unten gezogen. Das isolierte Protein wird dann durch Western Blot analysiert. Isotyp-Antikörper und proteinspezifische Antikörper haben die gleiche konstante Domäne, aber unterschiedliche Antigen-Bindungsdomänen. Ein wichtiger Bestandteil dieses Protokolls sind Protein-A/G-Agarose-Perlen, die die konstante Domäne von Antikörpern binden und eine Immunpräzipation des Zielproteins ermöglichen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Antikörper sind der Schlüsselbestandteil einer Immunpräzipitation, die sie von anderen Formen der Proteinreinigung (d. h. Nickelaffinitätssäulenreinigung) unterscheidet. Antikörper sind Moleküle von B-Zellen, die bestimmte Proteinepitope erkennen können. Antikörper haben zwei Domänen: Konstante (Fc) und Antigenbindung (Fab) (Abbildung 1). Die konstante Domäne identifiziert den Typ des Antikörpers und diktiert die Funktion in vivo. In der Regel sind die konstanten Domänen von Antikörpern, die für IP verwendet werden, Maus, Ratte oder Kaninchen IgG. Der Antigenbindungsanteil des Antikörpers erkennt ein spezifisches Epitop eines bestimmten Proteins. Antikörper können Epitope an gefalteten Proteinen erkennen, die möglicherweise nicht existieren, wenn das Protein denaturiert ist und umgekehrt. Daher hängt die Verfügbarkeit des Epitops von der Proteinfaltung ab - die Identifizierung eines wichtigen Faktors, der bei der Wahl von Antikörpern und Bedingungen für IP zu berücksichtigen ist.
Sowohl prokaryotische als auch eukaryotische Systeme haben Antikörper-bindende Proteine. In eukaryotischen Systemen, der Zweck ist Immunschutz vor Bakterien, während in prokaryotischen Systemen, der Zweck ist der Schutz vor dem Immunsystem. Antikörperbindende Proteine beeinflussen die IP-Methodik auf zwei Arten. Erstens gibt es einen notwendigen Vorklärungsschritt (Abbildung 1), um das Lysat von Proteinen zu befreien, die Antikörper binden - wodurch die unspezifische Bindung im Endprodukt reduziert wird. In diesem Schritt wird ein Isotyp-Antikörper verwendet, der die gleiche konstante Domäne wie eine andere Antikörperbindungsdomäne als Ihr proteinspezifischer Antikörper hat. Bakterielle Antikörper-bindende Proteine sind die zweite Schlüsselkomponente dieser Methode. Nachdem der proteinspezifische Antikörper das Zielprotein bindet, muss der Antikörper: Proteinkomplex heruntergezogen werden (Abbildung 1). Proteine A, G und L sind bakterielle Proteine, die die konstante Domäne von Antikörpern binden. Während Bakterien dies verwenden, um das Immunsystem zu untergraben, Forscher haben dieses System für eine einfache Antikörperreinigung kooptiert, und es wird sowohl während der Vor-Clearing-und Pull-down-Schritte verwendet. Diese Proteine haben unterschiedliche Bindungsaffinitäten für verschiedene Arten und verschiedene konstante Domänensubtypen - ein weiterer Faktor, der bei der Auswahl der Bedingungen für IP zu berücksichtigen ist. Viele Unternehmen verkaufen Protein A/G-markierte Agaroseperlen (Abbildung 1), vorgefertigte Spinsäulen oder Harze, um Säulen herzustellen. Im Allgemeinen werden Perlen und Spinsäulen für kleinere Probengrößen verwendet, während Harze für die Massenreinigung verwendet werden.
In dieser Übungsübung zeigen wir, wie das endogene Protein c-myc aus primären murinen Thymosyten mit Protein A/G Plus Agaroseperlen auf basisiger Immunpräzipitationstechnik gereinigt werden kann. Das Protokoll beginnt mit der Zelllysatzubereitung und endet mit der Überprüfung des erfolgreichen Protein-Pull-Downs mittels Western-Blot-Analyse.
Procedure
1. Immunpräzipitation mit Protein A/G PLUS Agarose Perlen
Zelllysat-Vorbereitung
- Zentrifugieren Sie 108 Thymose in einer Mikrozentrifuge bei 13.000 U/min für 3 min und entfernen Sie den Überstand.
Hinweis: Die Zellzahl variiert je nach Expressionsniveau des gewünschten Proteins und dem gewählten Zelltyp. - Setzen Sie die Zellen in 500 L Lysepuffer RIPA mit PMSF wieder aus.
- Unterbrechen Sie Zellen mit ein paar schnellen Impulsen mit einem Wirbel und dann das Lysat ein paar Mal mit einer 25 G Nadel an einer Spritze befestigt aspirieren.
Hinweis: Vermeiden Sie blasen. Verwenden Sie eine größere Nadel, z. B. eine 21G-Nadel für größere Zelltypen. - Die Zelle 10 min auf Eis lysieren.
- Zentrifugieren Sie das Lysat bei 13.000 U/min für 15 min bei 4°C.
- Übertragen Sie den Überstand in ein frisches, beschriftetes Mikrozentrifugenrohr.
Pre-Clearing
- Fügen Sie dem Lysat 20 L Protein A/G PLUS Agarose-Perlen und 1 g eines Isotyp-Kontrollantikörpers (hier verwendet der IgG1-Isotyp-Kontrollantikörper der Maus) hinzu.
Hinweis: Die Wahl des verwendeten Isotyp-Antikörpers hängt von dem proteinspezifischen Antikörper ab, der später im Pull-Down-Schritt verwendet wird. - Die Lysatmischung auf einem Zentrifugenrotator im Kühlraum (4°C) 30 min inkubieren.
- Zentrifugieren Sie die Probe bei 3200 U/min für 30 s bei 4°C.
- Übertragen Sie den vorgereinigten Überstand in ein frisches, beschriftetes, 1,5 ml Mikrozentrifugenrohr. Entsorgen Sie das Pellet.
Proteinkonzentrationsbestimmung
- Bestimmen Sie die Proteinkonzentration der Zelle lysiert, indem Sie einen Bradford Assay durchführen.
- Aliquot 1000 L Bradford Reagenz in 7 Mikrozentrifugenröhren.
- Fügen Sie die folgenden Mengen des BSA-Proteinstandards (2 mg/ml) in 6 der Röhrchen ein (Tabelle 1).
| Rohrnummer | BSA-Volumen (l) (2 mg/ml) | Proteinkonzentration (g/l) |
| 1 | 0 | 0 |
| 2 | 1 | 2 |
| 3 | 2 | 4 |
| 4 | 3 | 6 |
| 5 | 4 | 8 |
| 6 | 5 | 10 |
Tabelle 1: BSA-Proteinstandardmengen
- In der7. Röhre 1 l des vorgereinigten Lysats hinzufügen.
Hinweis: Um sicherzustellen, dass die Probenkonzentration im Assay-Detektionsbereich liegt, bereiten und analysieren Sie auch eine 1:2- oder 1:5-Lysatverdünnung. - Legen Sie 200 l aus jedem der 7 Rohre in einzelne Brunnen einer flachen, 96-Well-Platte, die jede Probe in dreifacher Ausfertigung wiederholt.
- Leseplatte auf einem Plattenleser bei 595 nm.
- Generieren Sie die Standardkurve in Excel und berechnen Sie die Proteinkonzentration des vorgereinigten Lysats.
herunterziehen
- Beschriften Sie zwei frische 1,5 ml Mikrozentrifugenrohre - eine als "Kontrolle" und andere als "Test", die in diesem Beispiel c-myc ist.
- Legen Sie 500 g vorgereinigtes Lysat in jedes dieser Rohre.
Hinweis: Die hier verwendete Proteinmenge hängt von der Menge des zu reinigenden Proteins ab. - Erhöhen Sie das Gesamtvolumen für jedes Rohr mit einem Lysepuffer bis zu 500 l.
- Fügen Sie dem Testgruppenröhrchen 2 g Anti-C-Myk-Antikörper und dem IgG1-Isotyp-Kontrollantikörper mit 2 g der Maus einen Antikörper zur Kontrollgruppe hinzu.
Hinweis: Die Menge des Antikörpers hängt von der Wirksamkeit des Antikörpers und der Menge des Zielproteins ab. - Inkubieren Sie die Rohre auf einem Rotator im Kühlraum (4°C) für 2 h.
- Fügen Sie 20 L Protein A/G PLUS Agarose Perlen zu jeder Tube hinzu.
Hinweis: Es ist ratsam, Pipettenspitzen mit abgeschnittenem Ende zu verwenden, um Schäden an den Perlen zu vermeiden. - Über Nacht auf einem Rotator im Kühlraum (4°C) inkubieren.
Hinweis: Je nach Zielprotein und Antikörperwirksamkeit kann dieser Schritt von 1 h bis über Nacht variieren. - Zentrifugieren Sie die Rohre bei 3200 U/min, damit bei 30 s 4°C die Perlen heruntergezogen werden.
- Aspirieren Sie den Überstand aus jeder Röhre.
Hinweis: Das Zielprotein ist nun an die Perlen gebunden. - Waschen Sie die Perlen zweimal mit 500 l 1X Dulbecco s PBS.
- Zentrifugieren Sie die Rohre bei 3200 U/min für bei 30 s 4°C.
Hinweis: Für strengeres Waschen verwenden Sie strengere Puffer wie RIPA. - Aspirieren Sie den Puffer aus jedem Rohr. Entfernen Sie mit Gel-Ladespitzen den Restpuffer von den Perlen und halten Sie die Perlen auf Eis, um das Protein zu entschärfen.
Hinweis: In diesem Beispiel wird das Protein in SDS-PAGE-Laufpuffer eluiert, indem die Perlen für die Western-Blot-Analyse gekocht werden. Dieser Ansatz eignet sich zur Überprüfung von IP-Ergebnissen oder zur Untersuchung von Protein-Protein-Wechselwirkungen. Für andere nachgelagerte Anwendungen, wie die Reinigung von Proteinen für die strukturelle oder enzymatische Analyse, werden anspruchsvollere Systeme wie Epitop-Tags (Flag-Tag oder myc-tag) verwendet, um die Elution des Antikörpers mit dem Protein von Interesse zu vermeiden.
2. IP-Verifizierung durch Western Blot-Analyse
SDS-PAGE Elektrophorese:
- Re-suspend Perlen in 20 L SDS-PAGE-Ladefarbstoff mit dem -Mercapto-Ethonol.
- Die Proben bei 95 °C 5 min kochen.
- Zentrifugieren Sie die Perlen bei 13.000 U/min für 10 s bei Raumtemperatur.
- Mit Gel-Ladespitzen die aus den Perlen gewonnenen Proben sorgfältig pfeifen und in Brunnen mit 4-15% Gradienten-SDS-PAGE-Gel laden.
- Laden Sie zusätzlich zu den Proben eine Fahrspur mit einer Proteinleiter sowie eine Fahrspur mit dem vorgereinigten Lysat als Ladekontrolle.
- Laufen Sie bei 100 V, bis die Farbe vorne den Boden des Gels erreicht (ca. 1h).
Western Blot Analyse:
- Machen Sie Western Blot Sandwich, um sicherzustellen, dass PVDF Membran zwischen Gel und roter Kathode ist.
- Transfer für 1 h bei 100 V.
- Legen Sie die Membran in 5 ml Sperrpuffer bei Raumtemperatur für 1 h auf eine Wippe bei niedriger Einstellung, um die unspezifischen Proteinbindungsstellen zu blockieren.
Hinweis: Die Mengen an Blockierpuffern, primärer Antikörper, sekundärer Antikörper und Waschungen müssen möglicherweise für größere Blots erhöht werden. - Inkubieren Sie den Blot mit 5 ml Anti-C-Myc-Antikörper im Sperrpuffer über Nacht bei 4°C auf Rocker bei niedriger Einstellung.
Hinweis: Der hier verwendete Antikörper sollte sich von dem im Pull-Down-Schritt verwendeten unterscheiden. - Waschen Sie den Fleck 3-6 mal mit 5 ml TBST mit jeder Wäsche von 5 min bei Raumtemperatur auf einer Wippe bei niedriger Einstellung.
- Inkubieren Sie den Blot mit HRP-getaggten Anti-Kaninchen-Lichtketten-Sekundärantikörper im Sperrpuffer, für 1 h bei Raumtemperatur auf Derawippe bei niedriger Einstellung.
Hinweis: Die Wahl des sekundären Antikörpers hängt von primären Antikörpern ab, die für den Western Blot verwendet werden. Zusätzlich wird im Protokoll eine leichte Kettenspezifische Sekundäre verwendet, da das Zielprotein im Molekulargewicht nahe an der schweren Kette des Antikörpers liegt. Wenn das Zielprotein nahe 50kDa ist, verwenden Sie eine leichte Kette spezifische sekundäre. Wenn das Zielprotein nahe 25kDa ist, verwenden und schwere Kette spezifische sekundäre. - Waschen Sie den Fleck 3-6 mal mit 5 ml TBST mit jeder Wäsche von 5 min bei Raumtemperatur auf einer Wippe bei niedriger Einstellung.
- Entfernen Sie Flüssigkeit aus Fleck und Tupfer Rand von Fleck auf Labortücher, um überschüssige Flüssigkeit zu entfernen.
- Bedecken Sie Blot mit 1x chemiluminescent Detektionreagenz' und inkubieren für 1 min.
Hinweis: Die folgenden Schritte sollten in schneller Folge durchgeführt werden, da das Detektionsreagenz leicht und zeitempfindlich ist. - Dab Rand des Flecks auf Labortücher, um überschüssige detektionreagenz zu entfernen.
- Platzieren Sie den Fleck auf der Bildfläche des Imager-Fachs.
Hinweis: Chemilumineszierende Flecken können auch mit Film visualisiert werden. - Bild mit dem 'Chemiluminescent Program', um mehrere Zeitpunkte von 10 s bis 5 min zu erfassen.
Hinweis: Die optimale Zeit kann sich je nach Proteinmenge und Der Qualität des Antikörpers ändern. - Wählen Sie ein Bild mit optimaler Bandsichtbarkeit aus, und exportieren Sie es dann.
- Bevor Sie den Fleck bewegen, machen Sie ein Bild des Flecks mit dem Imager, um die Position der Leiter zu erfassen. Exportieren Sie dann auch dieses Bild.
- Mit einer Folienvorbereitungssoftware (z. B. PowerPoint) richten Sie die Bänder und Leiterbilder so aus, dass sie ein einzelnes Bild bilden.
Immunpräzipitation, oder IP, ist eine weit verbreitete Technik, um ein Protein von Interesse aus einer Zelle oder Gewebelysat oder einer Körperflüssigkeit zur Proteincharakterisierung zu isolieren oder Protein-Protein-Wechselwirkungen zu untersuchen.
Der Prozess beginnt mit einem Antikörper, der eine hohe Affinität und Spezifität für das Zielprotein hat. Dieser Antikörper wird mit der Probe gemischt, so dass sich Antikörper-Ziel-Komplexe bilden können. Jedes Protein, das an das Zielprotein gebunden ist, wird dabei auch indirekt an den Antikörper gebunden. Als nächstes wird die Lösung mit Agaroseperlen inkubiert, die zu einem bakteriellen Protein konjugiert sind, das eine starke Affinität zur konstanten Region der Antikörper hat. Das bakterielle Protein bindet an den Antikörper und verbindet die Antikörper-Ziel-Komplexe mit den Perlen. Dann wird die Lösung zentrifugiert, um die Perlen auszufällen, wodurch der gesamte Komplex extrahiert wird, der den bindungden Antikörper, das Zielprotein und alle interagierenden Proteine enthält. Schließlich werden die gebundenen Proteine aus den Perlen extrahiert und voneinander freigesetzt und für die weitere Analyse durch Techniken wie Western Blotting verwendet.
Mehrere Variationen verschiedener Teile dieser Technik werden häufig verwendet, wie Z. B. Pre-Clearing, mit Peptid-Tags oder magnetischen Perlen oder die Analyse anderer Nicht-Protein-Bindungspartner. IP kann durch einen Pre-Clearing-Schritt vorangegeben werden, um unspezifische Antikörper-bindende Proteine in der Probe zu entfernen und den Hintergrund zu minimieren. Dabei wird die Probe zunächst mit Isotypkontrollantikörpern inkubiert, so dass sie sich an diese Proteine binden können, und dann mit Agaroseperlen die Komplexe ausgefällt. Das Beispiel ist dann bereit, mit der tatsächlichen IP fortzufahren.
Peptid-Tags sind nützlich, wenn ein bestimmter Antikörper für IP nicht verfügbar ist. Hier kann das Zielprotein genetisch verändert werden, um ein Peptid-Epitop-Tag zu enthalten, und ein Antikörper gegen das Tag ist in der Lage, das Protein von Interesse herauszuziehen. Magnetperlen werden oft anstelle von Agarose verwendet, um das Ziel zu fällen. Nach der Bindung an den Antikörper-Ziel-Komplex wird das Probenröhrchen in ein starkes Magnetfeld gelegt, das die Perlen aus der Lösung extrahiert. Dies eliminiert die Notwendigkeit der Zentrifugation und verbessert Geschwindigkeit und Komfort.
Immunpräzipitation wird auch zur Untersuchung von DNA- oder RNA-bindenden Proteinen verwendet und sind als Chromatin-Immunpräzipitation bzw. RNA-Immunpräzipitation bekannt. Diese Variationen sind nützlich für die Fehlerbehebung und Anpassung der Methode für verschiedene experimentelle Anwendungen. In diesem Video werden Sie beobachten, wie man eine Zelle lysiert und Immunpräzipitation durchführt, um ein Protein von Interesse zu extrahieren, gefolgt von einer Western-Blot-Analyse, um das Experiment zu validieren.
Um zu beginnen, legen Sie die vorgesammelten Zellen in eine Mikrozentrifuge und drehen Sie bei 13 Tausend U/min für drei Minuten. Entfernen Sie nach dem Spin den Überstand und setzen Sie die Zellen in 500 MikroliterLysis-Puffer RIPA mit PMSF wieder aus. Nun, stören Sie die Zellen mit ein paar schnellen Impulsen mit einem Wirbel und dann das Lysat ein paar Mal mit einer 25-Meter-Nadel an einer Spritze befestigt, wobei darauf zu achten, dass Blasen zu vermeiden. Legen Sie die Zellen für 15 Minuten auf Eis. Nach dem Inkubieren der Proben auf Eis zentrieren Sie das Lysat für 15 Minuten bei vier Grad Celsius.
Beschriften Sie ein neues 1,5 Milliliter Mikrozentrifugenrohr. Nach der Drehung den Überstand auf das frisch beschriftete Rohr übertragen und das Pellet entsorgen. Als nächstes vorab das Lysat von Verunreinigungen, die nicht spezifisch an die Agaroseperlen oder den primärantikörper binden, durch Zugabe von 20 Mikrolitern der Protein A/G PLUS-Agarose-Perlen und ein Mikrogramm eines Isotyp-Kontrollantikörpers an das Lysat, das in diesem Beispiel ist eine Maus-IgG1-Isotypsteuerung. Inkubieren Sie das Rohr auf einem Rotator in einem kalten Raum für 30 Minuten. Nachdem Sie das Lysat im Kalten Raum 30 Minuten lang rotiert haben, zentrieren Sie die Probe 30 Sekunden lang bei 4 Grad Celsius bei 3200 U/min. Entfernen Sie das Rohr aus der Zentrifuge und übertragen Sie den vorgereinigten Überstand in ein frisch beschriftetes 1,5 Milliliter Mikrozentrifugenrohr. Entsorgen Sie das Pellet.
Bestimmen Sie nun die Proteinkonzentration der Zelle lysieren, indem Sie einen Bradford-Assay durchführen. Etikett sieben 1. 5-Milliliter-Mikrozentrifugenröhrchen eins bis sechs und Proben und Aliquot 1000 Mikroliter des Bradford-Reagenzes in jede Röhre. Sechs der Rohre werden verwendet, um eine Standardkurve zu machen, indem verschiedene Mengen bekannter Mengen von BSA zu jedem Rohr hinzugefügt werden. Die hinzuzufügenden Beträge sind in dieser Tabelle aufgeführt. Im siebten Probenröhrchen einen Mikroliter des vorgereinigten Lysats hinzufügen. Legen Sie 200 Mikroliter aus jedem der sieben Röhren in einzelne Brunnen einer flachen 96-Well-Platte und wiederholen Sie jede Probe in Dreifacharbeit, so dass es drei Spalten mit sieben Proben gibt. Lesen Sie die Platte auf einem Plattenleser mit einer Wellenlänge von 595 Nanometern. Nach dem Erstellen einer Standardkurve in Excel berechnen Sie die Proteinkonzentration des vorgereinigten Lysats.
Als nächstes beschriften Sie zwei 1,5-Milliliter-Mikrozentrifugen-Röhren - eine als Kontrolle und die andere als Test, die in diesem Beispiel der c-myc-Antikörper sein wird. Legen Sie 500 Mikrogramm des vorgereinigten Lysats in jedes dieser Rohre und bringen Sie dann das Gesamtvolumen für jedes Rohr mit Lysepuffer auf bis zu 500 Mikroliter. Als nächstes fügen Sie dem Testgruppenröhrchen zwei Mikrogramm des Anti-C-Myc-Antikörpers hinzu. Fügen Sie für die Steuerung zwei Mikrogramm des IgG1-Isotyp-Kontrollantikörpers der Maus hinzu. Sobald die Antikörper in die Rohre gegeben sind, legen Sie die Proben auf einen Rotator in einem kalten Raum und inkubieren Sie für zwei Stunden. Nun fügen Sie die Agarose Perlen. Dazu wird empfohlen, das Ende einer Pipettenspitze abzuschneiden und dann mit dieser modifizierten Spitze 200 Mikroliter der Protein A/G PLUS-Agarose-Perlen in jedes Rohr einzutragen. Inkubieren Sie die Rohre auf einem Rotator im Kühlraum über Nacht.
Nach der Inkubation entfernen Sie die Rohre aus dem Rotator und drehen Sie die Lysate in der Mikrozentrifuge, um die Perlen herunterzuziehen. Nachdem die Drehung abgeschlossen ist, entfernen Sie die Rohre aus der Zentrifuge und aspirieren Sie den Überstand aus jedem Rohr. Als nächstes waschen Sie die Perlen mit 500 Mikrolitern von 1X Dulbecco PBS. Die Rohre in eine Mikrozentrifuge geben und 30 Sekunden bei vier Grad Celsius nach unten drehen. Entfernen Sie anschließend den Überstand. Wiederholen Sie die Wasch- und Zentrifugenschritte noch einmal für insgesamt zwei Mal. Entfernen Sie die Rohre aus der Mikrozentrifuge und aspirieren Sie den Puffer aus jedem Rohr. Entfernen Sie mit Gel-Ladespitzen den restverlassenen Puffer von den Perlen und halten Sie die Perlen auf Eis, um das gebundene Protein zu vereitten.
In diesem Beispiel wird das Protein in SDS-PAGE-Laufpuffer eluiert, indem es für die Western Blot-Analyse kocht. Um dies zu tun, setzen Sie die Perlen in 20 Mikroliter SDS-PAGE Ladefarbstoff mit Beta-Mercaptoethanol, oder BME. Kochen Sie die Proben bei 95 Grad Celsius für fünf Minuten, um die Immunokomplexe von den Perlen zu trennen. Dann zentrifugieren Sie die Perlen mit maximaler Geschwindigkeit für 10 Sekunden bei Raumtemperatur. Entfernen Sie die Rohre aus der Mikrozentrifuge und halten Sie sie bei Raumtemperatur in einem Rack. Mit Gel-Ladespitzen die Proben sorgfältig aus den Perlen pipetten und in Brunnen eines 4 bis 15% Gradienten SDS-PAGE Gel sladen. Laden Sie zusätzlich zu den Proben eine Fahrspur mit einer Proteinleiter sowie eine Fahrspur mit dem vorgereinigten Lysat als Ladekontrolle. Sobald das Gel geladen ist, führen Sie das Gel bei 100 Volt aus.
Nachdem die Färbefront den Boden des Gels erreicht hat, was etwa eine Stunde dauern sollte, stoppen Sie das Gel und machen Sie ein Western Blot Sandwich, um sicherzustellen, dass die PVDF-Membran zwischen dem Gel und der Kathode liegt. Legen Sie das Western Blot Sandwich in das Transfergerät und übertragen Sie die Proteine auf dem Gel für eine Stunde bei 100 Volt auf die Membran. Nachdem die Übertragung abgeschlossen ist, legen Sie die Membran in fünf Milliliter Block, um zu verhindern, dass die Antikörper nicht spezifisch an die Membran binden. Gestein bei niedriger Einstellung für eine Stunde bei Raumtemperatur. Wenn der Timer ertönt, entfernen Sie den Blockierenden Puffer. Fügen Sie fünf Milliliter des Sperrpuffers mit dem Detektionsantikörper in die Membran ein. Hier wird ein Anti-c-Myk-Antikörper verwendet, der sich von dem für den Pull down verwendeten unterscheidet.
Inkubieren Sie den Fleck über Nacht, bei vier Grad Celsius auf einer Wippe in einer niedrigen Umgebung. Entfernen Sie nach der Inkubation den Antikörper und den Blockierpuffer. Waschen Sie den Fleck, mit fünf Milliliter TBST für fünf Minuten bei Raumtemperatur, auf einer Wippe bei einer niedrigen Einstellung. Dieser Waschschritt sollte zwei- bis fünfmal für insgesamt drei bis sechs Waschungen wiederholt werden, wobei für jede Wäsche frisches TBST verwendet wird. Fügen Sie dem Blot fünf Milliliter eines bis 1000 sekundären Antikörpers und einen Blockierpuffer hinzu. In diesem Fall ist der sekundäre Antikörper HRP-markierte Anti-Kaninchen-Lichtkette. Inkubieren Sie den Fleck auf einer Wippe in einer niedrigen Einstellung für eine unsere bei Raumtemperatur. Als nächstes entfernen Sie den Puffer und waschen Sie den Fleck mit fünf Millilitern TBST. Inkubieren Sie diese Wäsche auf einer Wippe bei niedriger Einstellung für fünf Minuten bei Raumtemperatur. Wiederholen Sie diese Wäsche für insgesamt sechs bis zwölf Wäschen, jede mit frischen fünf Millilitern TBST. Entfernen Sie die endende Wäsche, indem Sie zuerst die Flüssigkeit aus dem Fleck gießen. Dann, mit Pinzette, tab die Kante des Flecks auf einem Labortuch, um überschüssige Flüssigkeit zu entfernen und dann legen Sie den Fleck in einem frischen Behälter. Als nächstes den Fleck mit 1X Chemiluminescent Detection Reagenz bedecken und eine Minute lang inkubieren.
Arbeiten Sie schnell, tupmpfen Sie den Rand des Flecks auf einem Labortuch, um überschüssiges Detektionsreagenz zu entfernen, und legen Sie den Fleck dann auf die bildgebende Oberfläche des Imager-Trays. Bild mit dem Chemiluminescent-Programm, um mehrere Zeitpunkte von 10 bis 30 Sekunden zu erfassen. Nachdem der Blot abgebildet wurde, wählen Sie ein Bild mit optimaler Bandsichtbarkeit aus, und exportieren Sie dann dieses Bild. Verwenden Sie vor dem Verschieben des Blots den Imager, um ein Bild des Blots zu machen, um die Position der Leiter zu erfassen. Exportieren Sie dann auch dieses Bild. Schließlich richten Sie mithilfe einer Folienvorbereitungssoftware, z. B. PowerPoint, die Bänder und Leiterbilder zu einem Bild aus.
Dieses Bild zeigt das Western Blot-Ergebnis für die Immunpräzipitation des Proteins c-myc aus Thymosexzellen. Von links nach rechts stellen die Fahrspuren die Isotype-Steuerung, die c-myc IP und die vorgeräumte Lysat-Eingabe dar. Die Spur auf der extremen Rechten ist ein zusammengeführtes Bild der Molekulargewichtsleiter. Das starke Band, bei etwa 25 Kilodaltonen, stammt aus der Leichten Kette und das bei 50 Kilodaltonen aus der schweren Kette des Bindungsantikörpers und ist unspezifisch für die IP oder die Proben. C-myc läuft um 67 Kilodalton auf Western Blots und ist in der Regel knapp unterhalb des 75 Kilodalton Leiterbandes sichtbar. In diesem Blot ist das c-myc-Band in der zweiten Spur sichtbar, fehlt aber in der ersten Spur, was darauf hinweist, dass der IP-Antikörper c-myc erfolgreich heruntergezogen hat. Es gibt kein sichtbares Band in der vorgereinigten Lysat-Spur, was darauf hindeutet, dass dieses Protein niedrige endogene Expressionswerte hat.
Subscription Required. Please recommend JoVE to your librarian.
Results
Die Ergebnisse des oben beschriebenen Verfahrens sind in Abbildung 2 dargestellt. Von links nach rechts enthalten die Bahnen die Kontrollgruppe (Isotyp), die Testgruppe (c-myc), das vorgereinigte Lysat (Lysat) und die Molekulargewichtsleiter (Leiter). Die 25 und 75 kDa Leiterbänder sind markiert. Die beiden markanten Bänder bei 25 kDa und 50 kDa sind die leichte und schwere Kette des Bindungsantikörpers bzw. sind unspezifisch für die IP oder die Proben. c-myc Protein, das um 67kDa auf Western Blots läuft und in der Regel direkt unterhalb des 75 kDa Leiterbandes sichtbar ist. In diesem Blot ist das c-myc-Band in der zweiten Spur sichtbar, fehlt aber in der ersten Spur, was darauf hinweist, dass der IP-Antikörper c-myc erfolgreich heruntergezogen hat. Es gibt kein sichtbares Band in der vorgereinigten Lysat-Spur, was darauf hindeutet, dass dieses Protein niedrige endogene Expressionswerte hat.

Abbildung 2: Ergebnisse einer Western Blot-Analyse, die zur Beurteilung der Reinigung von c-myc durch Immunpräzipitation verwendet wird. Ein Band bei 67 kDa, entsprechend c-myc, ist in der Anti-c-myc-Spur sichtbar, nicht aber in der Isotype-Steuerspur. Beachten Sie, dass die c-myc-Ebenen nicht hoch genug waren, um in der Lysat-Spur visualisiert zu werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
Kurz gesagt, Immunpräzipitation ist die Isolierung eines bestimmten Proteins mit einem Antikörper. In diesem Beispiel wurden die Ergebnisse der Immunpräzipation von Western Blot analysiert, um die Reinheit zu bewerten. Das isolierte Protein könnte in einer Reihe von Anwendungen danach verwendet werden, einschließlich: NMR für die Proteinstruktur, Massenspektrometrie für die Aminosäuresequenz oder In-vitro-Assays zur enzymatischen Charakterisierung. IPs können auch die interagierenden Partner von Proteinen charakterisieren. Zum Beispiel könnten dna oder RNA nach Isolation zur Sequenzierung isoliert werden. Co-Immunpräzipitationen bewerten Protein-Protein-Wechselwirkungen. Wenn das Zielprotein während einer IP nach unten gezogen wird, können auch interagierende Proteine nach unten gezogen werden. Diese interagierenden Partner können durch Massenspektrometrie und Western Blot beurteilt werden. Immunpräzipitation ist eine leistungsstarke Technik zum Studium der Proteinbiologie.
Subscription Required. Please recommend JoVE to your librarian.
References
- Olliver, C. L. and Boyd, C. D. (1984). Immunoprecipitation of In Vitro Translation Products with Protein A Bound to Sepharose. In J. M. Walker (eds), Nucleic Acids. Methods in Molecular Biology (pp. 157-160). New Jersey: Humana Press.
- Thurston, C. F. and Henley, L. F. (1988). Direct Immunoprecipitation of Protein. In J. M. Walker (eds), New Protein Techniques. Methods in Molecular Biology (pp. 149-158). New Jersey: Humana Press.
- Anderson, N. G. (1998). Co-immunoprecipitation: Identification of Interacting Proteins. In R. A. Clegg (eds), Protein Targeting Protocols.Methods in Molecular Biology (pp. 35-45). New Jersey: Humana Press.
- Jackson, D. I. and Dickson, C. (1999). Protein Techniques: Immunoprecipitation, In Vitro Kinase Assays, and Western Blotting. In P.T. Sharpe and I. Mason (eds), Molecular Embryology. Methods in Molecular Biology (pp. 699-708). New Jersey: Humana Press.
- Trieu, E. P. and Targoff, I. N. (2015). Immunoprecipitation: Western Blot for Proteins of Low Abundance. In B. Kurien and R. Scofield (eds), Western Blotting. Methods in Molecular Biology (pp. 327-342). New York, NY: Humana Press.