



Overview
Source: Jonathan F. Blaize1, Elizabeth Suter1, and Christopher P. Corbo1
1 Department of Biological Sciences, Wagner College, 1 Campus Road, Staten Island NY, 10301
Quantitative assessment of prokaryotes can be onerous given their abundance, propensity for exponential proliferation, species diversity within a population, and specific physiological needs. Compounding this challenge, is the four-phase nature in which bacteria replicate (lag, log, stationary and death). The ability to accurately estimate microorganism concentration is necessary for successful identification, isolation, cultivation, and characterization (6). As such, microbiologists have employed serial dilution and various plating techniques for over a century to reliably quantify bacterial and viral load in clinical, industrial, pharmaceutical, and academic laboratory environments (2,4,6). Descriptions of this methodology first appeared in 1883 when the German scientist and physician Robert Koch published his work on infectious disease-causing agents (2). Often referred to as the father of modern bacteriology, Koch's forenamed techniques have become the gold standard for enumeration of microorganisms, culturable or otherwise, throughout the world.
Serial dilution is a systematic reduction of a known or unknown entity (a solute, organism, etc.) through successive re-suspension of an initial solution (solution0) into fixed volumes of a liquid diluent (blanks). These blanks usually consist of 0.45% saline, though the composition can be varied (7). While an experimenter can choose any volume for each diluent, it is most often a multiple of 10, facilitating logarithmic reduction of the sample. For example, solution0 contains a total of 100 E. coli cells suspended in 10 mL of nutrient broth. If 1 mL of solution0 is removed and added to 9 mL of saline (diluent1), the new solution (solution1) would contain 1/10th of the initial concentration of E. coli. In this example, the new solution (solution1) would contain 10 E. coli cells. Repeating this process by removing 1 mL of solution1 and adding it to another 9 mL of saline (diluent2) would yield solution2, containing only a single E. coli cell. Since each new solution (9 mL of diluent + 1 mL of solution) contains a total of 10mL, we can conclude that the dilution factor for this reduction is 10 or that this was a 10-fold serial dilution (Figure 1). Since we only began with 100 cells in this example and we are diluting by a factor of 10, only two steps are required to reach the absolute minimum concentration of 1 cell.
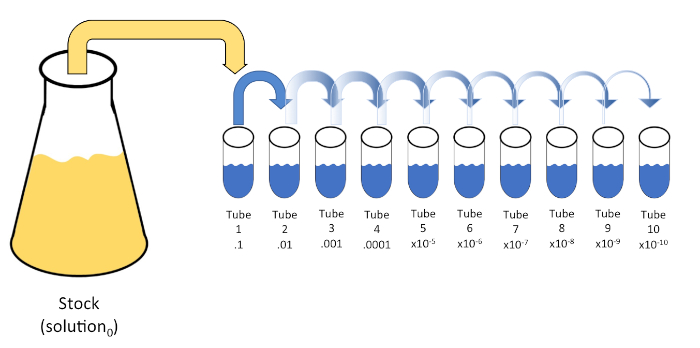
Figure 1: Serial dilution of a stock solution. A 1 mL aliquot of the stock solution (solution0) is added to tube 1 which contains 9 mL of 0.45% saline (dilent1); the product of this mixture is solution1. Repeat by aliquoting 1 mL of the newly created solution1 and adding it to tube 2. Aliquoting and resuspension continues in this fashion until the final tube is reached, diluting the stock concentration by a factor of 10 each with each step. Please click here to view a larger version of this figure.
Serial dilution is the simplest technique for obtaining manageable concentrations of a desired organism and it is complemented by petri dish streaking and spreading, just two of many plating techniques used by microbiologists. This benefit of this approach is that the experimenter can harvest pure strains of a single species or separate strains from a mixed population (7). Streaking is accomplished by introducing an organism to a solid medium (generally consisting of agarose) that it will grow upon if the appropriate nutrients are available. Gently sweeping a sterile inoculating loop across the medium (so that a subtle streak remains) in a rigid sinusoidal pattern will distribute the organism proportionally to the frequency of the experimenter's waveform. Dividing the Petri dish into thirds or fourths (quadrant streak) and decreasing the frequency of each streak as a new region of the dish is entered will gradually reduce the number of microorganisms that can occupy that region, producing single colonies instead of an unquantifiable bacterial lawn. Spread plating does not additionally dilute samples; a sterile glass spreader is used to distribute an aliquot of suspension media across an entire petri dish (Figure 2). The colonies that grow on the spread plate arise from a single cell and each colony on the dish can be counted to estimate the number of colony forming units per milliliter (CFU) in a given suspension, represented as CFU/mL (6) (Figure 3) Soft agar and replica plating are variations of the aforementioned techniques and allow for isolation of bacteriophage and mutant screening, respectively (1,7).
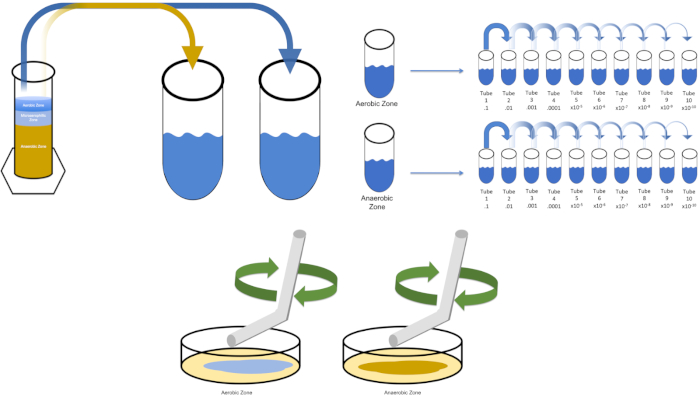
Figure 2: Plate streaking for bacterial enumeration and strain isolation. Label the bottom of a petri dish with identification information (bacteria name, date, media) and divide into thirds. After selecting an appropriate dilution of the stock sample, take a sterile (disposable or flamed) inoculating loop and submerge it the test tube (here, T3). Slightly raise the petri dish cover on one side so that only the inoculating loop can access the agar. Glide the inoculating loop across the top of the media in a zig-zag fashion being careful not to compromise the agar. Rotate the plate by roughly 1/3rd (~118°) and reduce the frequency of zig-zag motion. Rotate a final time and reduce zig-zag frequency once more. Please click here to view a larger version of this figure.
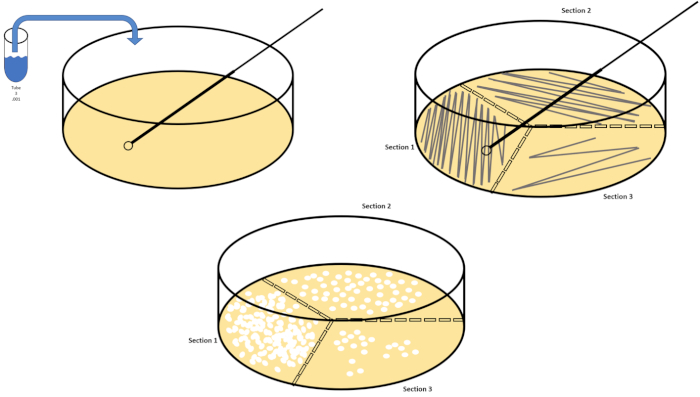
Figure 3: Spread plating. 1 g of the aerobic zone was resuspended in T1 and then serially diluted. A sterile glass or plastic disposable spreading rod is used to distribute inoculum throughout each dish. This was repeated with 1 g of the anaerobic zone. Please click here to view a larger version of this figure.
As with serial dilutions, a logarithmic scale is employed to express organismal concentration. The number of colonies grown in standard petri dishes measuring 100mm x15mm can be enumerated manually (or automated with the aid of computational processing) by identifying isolated clusters of growth. Counts that total fewer than 30 or greater than 300 should be defined as too few to count (TFTC) or too numerous to count (TNTC), respectively. In the case of the latter, a serial dilution should be performed to reduce concentration before restreaking a new petri dish. Averaging the number of self-contained colonies identified from three separate petri dishes and multiplying the mean by the dilution factor will yield CFU/mL; plotting the log10 of CFU/mL against time will reveal the mean generation time of the organism (7).
Procedure
1. Set-up
- A flow chart listing all materials, stepwise experimental protocol and method for discarding supplies should be written in a laboratory notebook and kept near the experimental workspace.
- Workspaces should be sterilized with an appropriate antiseptic (70% ethanol) and the experimenter should mitigate contamination risk by wearing clean laboratory garments that also protect them from exposure anomalies. Suitable garments include but are not limited to a lab coat, latex or nitrile gloves, googles, respirators, and closed shoes. It is critical to maintain aseptic technique at all times.
- Prepare 90 mL of 0.45% saline. Using a clean graduated cylinder, measure 90 mL of sterile water and transfer it to a clean Erlenmeyer flask labeled 0.45% saline. Weigh .405 g of sodium chloride (Sigma-Aldrich NaCl S9888) and add it to the flask labeled 0.45% saline. Swirl repeatedly until no solute remains visible.
- Upon completion, the experimenter should re-sterilize all surfaces and discard any unwanted organisms, diluent stocks, petri dishes, or disposable inoculating loops according to OSHA guidelines. Laboratory garments can be removed before washing hands.
2. Media Preparation
- Select media that is appropriate for cultivation of a desired organism. In most scenarios, a broth would enable sufficient bacterial growth. Since organisms from a Winogradsky protocol are desired here, a column consisting of calcium carbonate, sulfur, cellulose and mud was assembled and left undisturbed for 7 days. The forenamed column is separated into aerobic, microaerophilic, and anaerobic sections.
- Choose a medium appropriate for plating the organism of interest. Supplementation of liquid media with microbiology grade agar is typically employed as a solidifying agent. LB medium/agar is sufficient when harvesting samples from aerobic, microaerophillic and anaerobic regions of the forenamed column. Note: Samples from the microaerophillic region were not harvested for this procedure. However, these organisms should be cultivated in candle jars. Introducing a candle to this cultivation chamber before sealing creates a low oxygen environment that is suitable for microaerophilic proliferation.
- Since we wish to prepare 250 mL, use 500 mL (or larger) Erlenmeyer flasks to prevent boil-over when autoclaving. Label one "Broth" and the other "Agar."
- Determine the amount of media required to create each solution by following the manufacturer concentration recommendations. LB Agar, used here, is prepared by combining 25g/L with ultrapure water. Our volume of 250 mL requires a solution of 6.25 LB Agar/250 mL water. Similarly, LB Broth is prepared by combining the same ratio of LB Broth and water. Since it is not supplemented with a solidifying agent, it will not harden when cooled.
- Weigh the media and mix it with water in proportions consistent with manufacturer recommendations. Add 6.25g of LB Agar to a flask labeled "Agar", and 6.25g of LB Broth to a flask labeled "Broth." Add 250 mL ultrapure water to each flask.
- Wrap aluminum foil over each flask and use an autoclave to sterilize media for a minimum of 15 minutes at 121°C, 15 psi.
- Using a heat resistant glove or pad, remove the flasks from the autoclave when the cycle is complete and place them in a 40-50°C water bath.
- Once the appropriate temperature has been reached, pour the contents of the flask labeled "Broth" into a 250 mL Erlenmeyer, or round-bottom flask. Label the 250 mL flask "solution0".
- Obtain 10, 100mm x 15 mm sterile petri dishes and label them with the date, name, the type of media used and the Winogradsky column zone that organisms will be harvested from.
- Remove the flask labeled "Agar" from the water bath and begin pouring into each of the 10 petri dishes. No more than 15 mL should be added to each dish. This may also be performed with a pipettor and 25 mL serological pipette to improve accuracy. Use a sterile pipette tip to remove any bubbles present, and then cover with the plate lids and leave to solidify overnight.
3. Diluent Preparation
- Prepare ten test tubes capable of storing 20 mL or more in a rack and label them T1-T10. Each tube number is consistent with the dilution factor it corresponds to (i.e., T4 = 1x10-4 or 0.0001 or 1/10,000th of stock concentration).
- Pipet 9 mL of 0.45% saline into each of the 10 test tubes.
- Saline blanks are now ready to be sterilized by autoclave. Use aluminum foil to cover each of the 10 test tubes and then transfer them to an autoclave compatible test tube rack. Sterilize for a minimum of 15 minutes at 121°C, 15 psi.
- Remove blanks using heat resistant gloves and allow to cool. Cover and store at 4°C until needed when tubes have reached room temperature, or when cool to the touch.
4. Cultivation of Target Organism
- Inoculate "solution0" with a single colony from a previously streaked plate or 50 µL of a frozen stock. Allow the target organism time to replicate by placing inoculated "solution0" into a 37°C incubator overnight with shaking (if necessary). (Note: The flask should be covered to prevent contamination. If the target organism is aerobic, use sterile gauze and cotton plugs to prevent contamination. If evaluating regions of Winogradsky column, simply remove 1 gram from each desired zone (aerobic and anaerobic for the purposes of this study) and resuspend in T1 before proceeding to step 5.3.
5. Serial Dilution
- Obtain the flask labeled "Nutrient broth" from the incubator and shake vigorously.
- Pipet 1 mL of "solution0" into the test tube labeled T1. Vortex T1. If evaluating Winogradsky explants, weigh 1 gram of the desired zone and add it to T1 prior to vortexing. (Note: 1 mL is used here for simplicity- smaller or larger volumes of diluent may also be used).
- Remove 1 mL from test tube T1 and add it to test tube T2. Vortex T2.
- Remove 1 mL from test tube T2 and add it to test tube T3. Vortex T3.
- Remove 1 mL from test tube T3 and add it to test tube T4. Vortex T4.
- Remove 1 mL from test tube T4 and add it to test tube T5. Vortex T5.
- Remove 1 mL from test tube T5 and add it to test tube T6. Vortex T6.
- Remove 1 mL from test tube T6 and add it to test tube T7. Vortex T7.
- Remove 1 mL from test tube T7 and add it to test tube T8. Vortex T8.
- Remove 1 mL from test tube T8 and add it to test tube T9. Vortex T9.
- Remove 1 mL from test tube T9 and add it to test tube T10.
6. Spread Plating
- Pipet 100 µL of a diluted sample from T1 directly onto a petri dish. This step can be, but does not need to be, repeated for each tube.
- Obtain a sterile, disposable spreading rod or flame sterilize a glass spreading rod. In a clockwise/counterclockwise motion, glide the horizontal portion of the spreading rod to equally distribute the sample within the petri dish.
- Repeat for each zone of the Winogradsky column that is to be evaluated.
- Incubate plates in a 37°C incubator for 24 hours. For anaerobic organisms, use an anaerobic chamber.
7. Streaking
- Select an appropriate dilution of your target organism. For example, solution4 will yield a 1/10,000th dilution of your initial concentration. Typically, dilutions of 1/1,000th (T3/Solution), 1/1,000,000th (T6/Solution6) and 1/1,000,000,000th (T9/Solution9) are evaluated to enumerate microbes.
- Using a plastic sterile disposable inoculating loop or a reusable metal inoculating loop that has been under fire for no less than 10 seconds, immerse in the desired solution from step 5. Calibrated inoculating loops should transfer 0.01 mL. (Caution: Do not allow flamed loop to contact bacteria immediately after removing from heat)
- Slightly raise the petri dish cover on one side so that only the inoculating loop can access the agar. Glide the inoculating loop across the top of the media in a zig-zag fashion being careful not to compromise the agar. Lower the petri dish lid.
- Use a new disposable inoculating loop or re-sterilize your reusable loop.
- Rotate the plate by roughly 1/3rd (~118°) and reduce the frequency of zig-zag motion.
- Again, use a new disposable loop or re-sterilize a metal loop before rotating a final time and reduce zig-zag frequency once more. Lower the petri dish lid.
- Repeat steps 7.2 - 7.6 until at least three petri dishes have been streaked for three different dilutions, using a new disposable loop or by re-flaming a reusable loop (Figure 2).
- Place streaked petri dishes in a 37°C incubator overnight. For anaerobic organisms, use an anerobic chamber.
8. Data Analysis and Results
- Cultures were harvested from the oxic and anoxic zones of a 7-day Winogradsky column. These zones are suitable for heterotrophic and iron-oxidizing anaerobes, respectively.
- Column explants were serially diluted prior to streaking or spreading on LB Agar plates.
- Streaking revealed a mixed population from each of the Winogradsky zones evaluated. Spread plates produced similar results.
- To calculate CFU/mL or CFU/g, average the number of counted colonies from three plates. Multiply the average number of colonies by the dilution factor and divide by the amount aliquoted. For example, if an average of 65 colonies were counted on plates inoculated with 0.1 mL of solution6 (T6) the formula described previously would equal 650,000,000 CFU/mL.
- Isolated colonies can now be chosen from each plate for use in enrichment assays to determine species identity.
Sometimes, in order to identify and study bacteria we first need to isolate and enrich them from a sample. For example, samples obtained from a Winogradsky Column are mixed, meaning they contain multiple species or strains of bacteria, so studying an individual bacterium or enumerating the different kinds present can be challenging. To this end, serial dilution and plating techniques are typically employed to reliably quantify bacterial load and isolate individual colonies.
Serial dilution is a process through which the concentration of an organism, bacteria in this example, is systematically reduced through successive resuspension in fixed volumes of liquid diluent. Usually the volume of the diluent is a multiple of 10 to facilitate logarithmic reduction of the sample organism. For example, one gram of sediment is first removed from the Winogradsky zone of interest and added to 10 milliliters of an appropriate liquid medium. Then, one milliliter of this first dilution is added to another tube containing nine milliliters of medium. The process can be repeated until several different concentrations of bacteria have been prepared. Serial dilution is the key to enumeration of bacteria in this example, since mixed samples from a Winogradsky Column contain an unknown, often large, number of bacteria.
Next, streak plating and spread plating enable the isolation and enumeration of bacteria within a sample, respectively. Streaking is accomplished by introducing a diluted sample to one section of the solid medium supplemented with nutrient, which is divided into thirds. This inoculum is then spread over each third of the plate in a zig-zag pattern. As different sections of the plate are streaked, crossing from the previous sample only once, the sample is spread more thinly. This means that you may only need to streak from one dilution to achieve individual colonies in the later sections. After incubation, the streaked plates allow for observations of colony morphology, information that can help differentiate between different bacterial species.
Alternatively, if the main goal is the enumeration of the bacteria in the sample spread plating may be used. In spread plating, an aliquot of a single sample is spread evenly over the entire surface of solid medium. Typically, because we don't know the bacterial numbers in the mixed sample, a spread plate is made for each of the dilutions or a representative sample of them. After incubation, enumeration can be performed using these spread plates. Any plates with colony counts fewer than 30 should be discarded, since small counts are subject to greater error. Similarly, any counts over 300 should be discarded because colony crowding and overlapping can lead to underestimation of colony count. If the colony counts of each of these remaining dishes is recorded and multiplied by the dilution factor, and then divided by the volume plated, this yields the colony forming units, or CFUs, per milliliter of suspension. In this video, you will learn how to qualitatively and quantitatively evaluate a sample containing a known bacterium, and the microbial communities contained in various regions of a Winogradsky Column via serial dilution, spread plating, and streak plating.
First, put on any appropriate personal protective equipment including a lab coat, gloves, and goggles. Next, sterilize the workspace with 70% ethanol and wipe down the surface. Next, gather two 500 milliliter Erlenmeyer flasks and label one broth and the other agar. To prepare LB agar solution, mix approximately 6.25 grams of LB agar, three grams of technical agar, and 250 milliliters of distilled water in the flask labeled agar.
Then, prepare LB broth by combining 2. 5 grams of LB media and 100 milliliters of distilled water in the flask labeled broth. After autoclaving the flasks, use a heat resistant glove to remove the flasks from the autoclave and place them in a 40 to 50 degree Celsius water bath. Once the flasks are 50 degrees Celsius, carefully prepare three 100 milliliter aliquots of the broth solution and label each aliquot solution zero. Next, gather 10 sterile petri dishes and label them with the date, name, type of media used, and the Winogradsky Column zone that the organisms will be harvested from. Pipette 15 milliliters of agar from the agar flask into each petri dish. Then, use the pipette tip to remove any bubbles, replace the plate lids, and allow them to solidify on the bench top overnight.
The next day, wipe down the bench top with 70% ethanol. Next, label 10 20 milliliter test tubes T1 through T10 and place them in a rack. Pipette nine milliliters of .45% saline into each tube. Now, cover each of the 10 test tubes loosely with their caps and transfer them to an autoclave-compatible test tube rack. After the cycle is complete, remove the saline blanks using heat resistant gloves and allow them to cool. Store the tubes at room temperature until they have reached approximately 22 degrees Celsius.
To cultivate a known target organism, E. coli in this example, inoculate 100 milliliters of solution zero with a single colony from a previously streaked plate. Then, cover the tube and incubate it over night at 37 degrees Celsius. To evaluate the regions of a Winogradsky Column, add approximately one gram of material from the aerobic zone to T1 and resuspend by vortexing. Then, repeat this process with one gram of material from the anaerobic zone.
Remove the tube containing solution zero inoculated with E. coli from the incubator and shake it. Then, pipette one milliliter of the solution into a T1 test tube and vortex to mix. Remove one milliliter of solution from T1 and transfer it to T2, vortexing to mix. Repeat this process through tube T10. To evaluate the aerobic and anaerobic zones of the Winogradsky Column, remove one milliliter of solution from each of the previously prepared T1 tubes and transfer it to the appropriate T2 tubes. Then, continue the serial dilutions through the T10 tubes as previously demonstrated.
To spread plate, pipette 100 microliters of the diluted sample from each T3 tube on to the corresponding petri dish. Then, use a sterile spreading rod to gently distribute the sample on to the petri dish and replace the plate lid. Repeat this process for the T6 and T9 dilutions, as previously demonstrated. Incubate the plates containing aerobic organisms in a 37 degree Celsius incubator for 24 hours. Incubate the plates containing anaerobic organisms in an anaerobic chamber set to 37 degrees Celsius for 24 hours. The next day remove the T3, T6, and T9 dilution plates from the incubator and the anaerobic chamber and transfer them to the bench top. Working with one plate at a time, glide a sterile inoculating loop across the top of the media in a zig-zag pattern. Then, replace the petri dish lid. Next, rotate the plate by 1/3 and sterilize the loop to reduce the frequency of the previously made zig-zag pattern. Again, after sterilizing the loop, rotate the plate by 1/3, reduce the frequency of the zig-zag pattern one last time, and replace the lid. Repeat this streaking method for the remaining plates, as previously shown. Then, place the streaked plates containing aerobic organisms in a 37 degree Celsius incubator overnight and the streaked plates containing anaerobic organisms in an anaerobic chamber set to 37 degrees Celsius overnight.
Cultures were harvested from the aerobic and anaerobic zones of a seven day Winogradsky Column. Then, the cultures were serially diluted prior to streaking and spreading on LB agar plates. Streaking revealed a mixed population from each of the evaluated Winogradsky zones, and the spread plates produced similar results. A plate streaked from a mixed population will result in bacterial colonies of different shapes, sizes, textures, and colors. In contrast, the streaked and spread plates containing the known organism, E. coli, demonstrated a homologous population. Generally, it is best to calculate CFUs per milliliter using the average colony count of three plates spread with the same sample and dilution factor. Multiply the average number of colonies by the dilution factor and divide by the amount aliquoted. Finally, isolated colonies chosen from each plate can be used in further enrichment assays to determine species identity.
Applications and Summary
Bacterial enumeration and strain isolation by plating requires manageable concentrations of target organisms. Successful plating is therefore contingent upon serial dilution. As such, the aforementioned techniques remain the cornerstone of microbiological examination and experimentation. Though simple by design, dilution factors and plating technique can be modified to by the experimenter to bolster outcomes without compromising the integrity of each method. Plotting the four phases of bacterial growth can be helpful when characterizing desired microbes. These phases, lag, log, stationary, and death, are marked by changes in bacterial replication. The lag phase features slow growth due to physiological adaptation, the log phase is the period of maximum proliferation featuring an exponential rise in viable cells, stationary phase is then reached due to environmental limitations and accumulations of toxins, before the death phase where cell counts begin to fall. This can be accomplished by serially diluting (or 1-step diluting to avoid confusion) Solution0 every hour for a total of 8 hours, beginning at Time0 (Solution0 should be returned to a shaking incubator after each dilution). Calculate the log10 of CFU/ml for a single diluent of Time0 and plot on the Y-axis. Repeat this calculation for the sample Time1 (make sure calculate CFU/mL using the same dilution factor as Time0). Repeat until each time (Time1-Time8) are plotted on the X-axis.
References
- Allen, M.E., Gyure, R.A. (2013) An Undergraduate Laboratory Activity Demonstrating Bacteriophage Specificity. Journal of Microbial Biological Education 14: 84-92.
- Ben-David, A., Davidson, C.E. (2014) Estimation Method for Serial Dilution Experiments. Journal of Microbiological Methods 107:214-221.
- Goldman, E., Green, L.H. (2008) Practical Handbook of Microbiology.
- Koch, R. (1883) New Research Methods for Detection of Microcosms in Soil, Air and Water.
- Lederberg, J., Lederberg, E.M. (1952) Replica Plating and Indirect Selection of Bacterial Mutants. Journal of Bacteriology 63:399-406
- Pepper, I., Gerba, C., Ikner, L. (2019) Bacterial Growth Curve Analysis and its Environmental Changes. JoVE Science Education Database. Environmental Microbiology.
- Sanders., E.R. (2012) Aseptic Laboratory Technique: Plating Methods. JoVE 63:e3063.