



Overview
Quelle: Jonathan F. Blaize1, Elizabeth Suter1, und Christopher P. Corbo1
1 Department of Biological Sciences, Wagner College, 1 Campus Road, Staten Island NY, 10301
Die quantitative Bewertung von Prokaryoten kann angesichts ihres Überflusses, ihrer Neigung zur exponentiellen Proliferation, der Artenvielfalt innerhalb einer Population und der spezifischen physiologischen Bedürfnisse belastend sein. Diese Herausforderung verschlimmert sich in der vierphasigen Natur, in der sich Bakterien vermehren (Lag, Log, stationär und Tod). Die Fähigkeit, die Konzentration von Mikroorganismen genau abzuschätzen, ist für eine erfolgreiche Identifizierung, Isolierung, Kultivierung und Charakterisierung erforderlich (6). Daher haben Mikrobiologen seit über einem Jahrhundert serielle Verdünnungs- und verschiedene Beschichtungstechniken eingesetzt, um die bakterielle und virale Belastung in klinischen, industriellen, pharmazeutischen und akademischen Laborumgebungen zuverlässig zu quantifizieren (2,4,6). Beschreibungen dieser Methode erschienen erstmals 1883, als der deutsche Wissenschaftler und Arzt Robert Koch seine Arbeit über infektionserregende Erreger veröffentlichte (2). Kochs vorgenannte Techniken, die oft als Vater der modernen Bakteriologie bezeichnet werden, sind weltweit zum Goldstandard für die Aufzählung von Mikroorganismen geworden, die kultivierbar oder nicht mehr sind.
Serielle Verdünnung ist eine systematische Reduktion einer bekannten oder unbekannten Einheit (ein Gelöster, Organismus usw.) durch sukzessive Wiedersuspension einer Ausgangslösung (Lösung0) in feste Volumina eines flüssigen Verdünnungsmittels (Rohlinge). Diese Rohlinge bestehen in der Regel aus 0,45% Saline, obwohl die Zusammensetzung variiert werden kann (7). Während ein Experimentator ein beliebiges Volumen für jedes Verdünnungsmittel auswählen kann, ist es meistens ein Vielfaches von 10, was eine logarithmische Reduktion der Probe erleichtert. Beispielsweise enthält Lösung0 insgesamt 100 E. coli-Zellen, die in 10 ml Nährstoffbrühe suspendiert sind. Wenn 1 ml Lösung0 entfernt und zu 9 ml Saline (Verdünnungsstoff1)hinzugefügt wird, würde die neue Lösung (Lösung1) 1/10der Anfangskonzentration von E. colienthalten. In diesem Beispiel würde die neue Lösung (Lösung1) 10 E. coli-Zellen enthalten. Eine Wiederholung dieses Prozesses durch Entfernen von 1 ml Lösung1 und Hinzufügen zu weiteren 9 ml Saline (Verdünnungsmittel2) würde Lösung2ergeben, die nur eine einzelne E. coli-Zelle enthält. Da jede neue Lösung (9 ml Verdünnungsstoff + 1 ml Lösung) insgesamt 10 ml enthält, können wir schlussfolgern, dass der Verdünnungsfaktor für diese Reduktion 10 beträgt oder dass es sich um eine 10-fache serielle Verdünnung handelte (Abbildung 1). Da wir in diesem Beispiel erst mit 100 Zellen begonnen haben und wir um den Faktor 10 verdünnen, sind nur zwei Schritte erforderlich, um die absolute Mindestkonzentration von 1 Zelle zu erreichen.
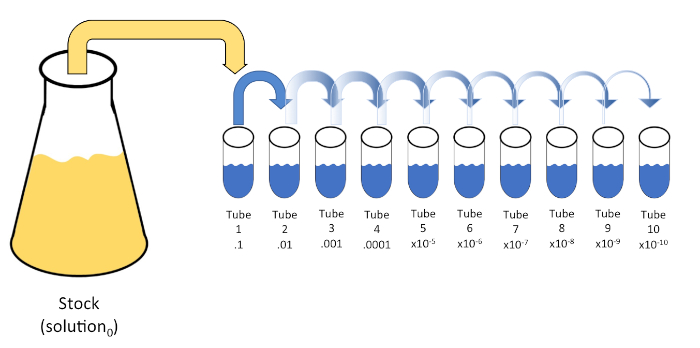
Abbildung 1: Serielle Verdünnung einer Lagerlösung. Ein 1 ml Aliquot der Stammlösung (Lösung0) wird zu Tube 1 hinzugefügt, die 9 ml mit 0,45% Salzsalze (dilent1)enthält; das Produkt dieser Mischung ist Lösung1. Wiederholen Sie dies, indem Sie 1 ml der neu erstellten Lösung1 aliquotieren und zu Tube 2 hinzufügen. Die Aliquotierung und Resuspension setzt sich auf diese Weise fort, bis die endgültige Röhre erreicht ist, wodurch die Lagerkonzentration mit jedem Schritt um den Faktor 10 verwässert wird. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Serielle Verdünnung ist die einfachste Technik zur Erzielung überschaubarer Konzentrationen eines gewünschten Organismus und wird durch Petrischalenstreifen und -streuungen ergänzt, nur zwei von vielen Beschichtungstechniken, die von Mikrobiologen verwendet werden. Dieser Vorteil dieses Ansatzes besteht darin, dass der Experimentator reine Stämme einer einzelnen Art oder separate Stämme von einer gemischten Population ernten kann (7). Streaking wird durch die Einführung eines Organismus in ein festes Medium (in der Regel bestehend aus Agarose) erreicht, auf dem er wachsen wird, wenn die entsprechenden Nährstoffe verfügbar sind. Das sanfte Schwenken einer sterilen Impfschleife über das Medium (so dass ein subtiler Streifen bleibt) in einem starren sinusförmigen Muster verteilt den Organismus proportional zur Frequenz der Wellenform des Experimentators. Die Aufteilung der Petrischale in Drittel oder Viertel (Quadrantstreifen) und die Verringerung der Häufigkeit jedes Streifens, wenn eine neue Region der Schale eingegeben wird, wird die Anzahl der Mikroorganismen, die diese Region besetzen können, allmählich reduzieren und einzelne Kolonien anstelle einer nicht quantifizierbaren bakteriellen Rasen. Die Streubeschichtung verdünnt die Proben nicht zusätzlich; ein steriler Glasstreuer wird verwendet, um ein Aliquot von Suspensionsmedien über eine ganze Petrischale zu verteilen (Abbildung 2). Die Kolonien, die auf der Streuplatte wachsen, entstehen aus einer einzigen Zelle und jede Kolonie auf der Schale kann gezählt werden, um die Anzahl der koloniebildenden Einheiten pro Milliliter (KBE) in einer gegebenen Suspension zu schätzen, dargestellt als KBE/ml (6) (Abbildung 3) Weicher Agar und Nachbau Beschichtungen sind Variationen der oben genannten Techniken und ermöglichen die Isolierung von Bakteriophagen bzw. Mutantenscreening (1,7).
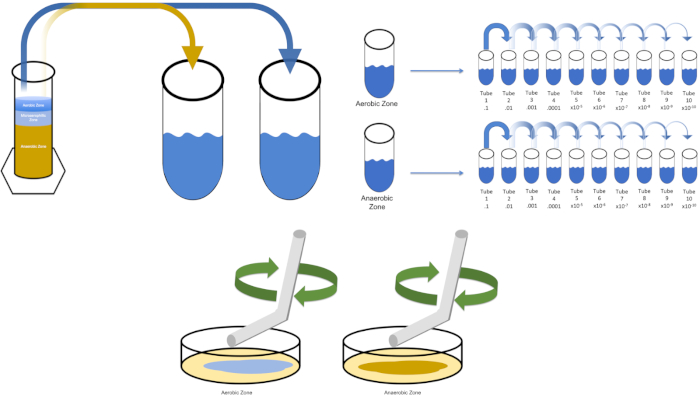
Abbildung 2: Plattenstreifen für bakterielle Aufzählung und Dehnungsisolierung. Beschriften Sie den Boden einer Petrischale mit Identifikationsinformationen (Bakterienname, Datum, Medien) und teilen Sie sie in Drittel auf. Nach der Auswahl einer geeigneten Verdünnung der Stammprobe eine sterile (Einweg- oder Flammen)-Impfschleife nehmen und das Reagenzglas untertauchen (hier, T3). Die Petrischalenabdeckung auf einer Seite leicht anheben, so dass nur die Impfschlaufe auf den Agar zugreifen kann. Gleiten Sie die Impfschleife über die Oberseite der Medien in einer Zick-Zack-Manier, um darauf zu achten, den Agar nicht zu kompromittieren. Drehen Sie die Platte um etwa 1/3rd (ca. 118°) und reduzieren Sie die Frequenz der Zick-Zack-Bewegung. Drehen Sie ein letztes Mal und reduzieren Sie die Zick-Zack-Frequenz noch einmal. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
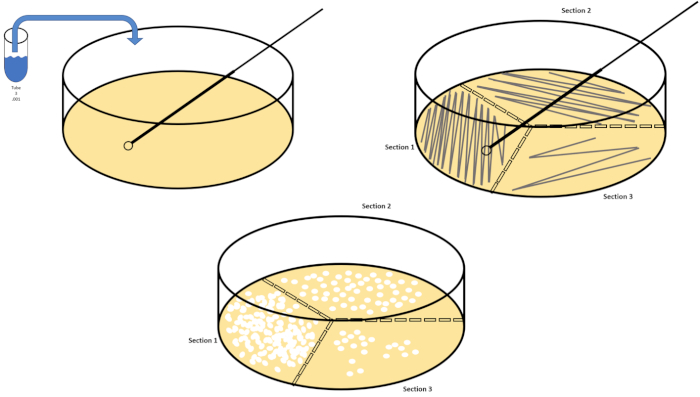
Abbildung 3: Streubeschichtung. 1 g der Aerobic-Zone wurde in T1 resuspendiert und dann seriell verdünnt. Ein steriler Glas- oder Kunststoff-Einweg-Streustab wird verwendet, um Inoculum in jeder Schale zu verteilen. Dies wurde mit 1 g der anaeroben Zone wiederholt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Wie bei seriellen Verdünnungen wird eine logarithmische Skala verwendet, um die Konzentration des Organismus auszudrücken. Die Anzahl der Kolonien, die in Standard-Petrischalen mit einer Größe von 100 mm x 15 mm angebaut werden, kann manuell (oder automatisiert mit Hilfe der Computerverarbeitung) aufgezählt werden, indem isolierte Wachstumscluster identifiziert werden. Zählt, die insgesamt weniger als 30 oder mehr als 300 wert sind, sollten als zu wenig zum Zählen (TFTC) oder zu zahlreich für die Anzahl (TNTC) definiert werden. Im letzteren Fall sollte eine serielle Verdünnung durchgeführt werden, um die Konzentration zu reduzieren, bevor eine neue Petrischale wieder aufgelegt wird. Die Mittelung der Anzahl der in sich geschlossenen Kolonien, die aus drei getrennten Petrischalen identifiziert wurden, und multiplizierte den Mittelwert mit dem Verdünnungsfaktor ergibt CFU/ml; Das Zeichnen des Protokolls10 von KFU/ml gegen die Zeit wird die mittlere Erzeugungszeit des Organismus offenbaren (7).
Procedure
1. Einrichtung
- Ein Flussdiagramm mit allen Materialien, einem schrittweisen Versuchsprotokoll und einer Methode zum Entsorgen von Verbrauchsmaterialien sollte in ein Labornotizbuch geschrieben und in der Nähe des experimentellen Arbeitsbereichs aufbewahrt werden.
- Arbeitsbereiche sollten mit einem geeigneten Antiseptikum (70% Ethanol) sterilisiert werden, und der Experimentator sollte das Kontaminationsrisiko durch das Tragen sauberer Laborkleidung verringern, die sie auch vor Expositionsanomalien schützt. Geeignete Kleidungsstücke umfassen, sind aber nicht beschränkt auf laborkleidung, Latex- oder Nitrilhandschuhe, Googles, Atemschutzgeräte und geschlossene Schuhe. Es ist wichtig, die aseptische Technik jederzeit aufrechtzuerhalten.
- Bereiten Sie 90 ml mit 0,45% Saline vor. Mit einem sauberen abgestuften Zylinder 90 ml steriles Wasser messen und in einen sauberen Erlenmeyerkolben mit der Bezeichnung 0,45% Salzwassergeben. Wiegen Sie .405 g Natriumchlorid (Sigma-Aldrich NaCl S9888) und fügen Sie es dem Kolben mit der Bezeichnung 0,45% Saline hinzu. Immer wieder wirbeln, bis keine Lösung sichtbar bleibt.
- Nach Abschluss sollte der Experimentator alle Oberflächen neu sterilisieren und unerwünschte Organismen, Verdünnungsvorräte, Petrischalen oder Einweg-Impfschleifen gemäß den OSHA-Richtlinien entsorgen. Laborkleidung kann vor dem Händewaschen entfernt werden.
2. Medienvorbereitung
- Wählen Sie Medien aus, die für die Kultivierung eines gewünschten Organismus geeignet sind. In den meisten Szenarien würde eine Brühe ein ausreichendes Bakterienwachstum ermöglichen. Da hier Organismen aus einem Winogradsky-Protokoll gewünscht werden, wurde eine Kolonne aus Calciumcarbonat, Schwefel, Zellulose und Schlamm zusammengebaut und 7 Tage lang ungestört gelassen. Die vorgenannte Säule ist in aerobe, mikroaerophile und anaerobe Abschnitte unterteilt.
- Wählen Sie ein Medium, das für die Beschichtung des Organismus von Interesse geeignet ist. Die Supplementierung von flüssigen Medien mit mikrobiologischen Agar wird in der Regel als Erstarrungsmittel verwendet. LB Medium/Agar ist ausreichend, wenn Proben aus aeroben, mikroaerophilen und anaeroben Regionen der vorgenannten Säule geerntet werden. Hinweis: Proben aus der mikroaerophilen Region wurden für dieses Verfahren nicht geerntet. Diese Organismen sollten jedoch in Kerzengläsern angebaut werden. Die Einführung einer Kerze in diese Kultivierungskammer vor dem Versiegeln schafft eine sauerstoffarme Umgebung, die für die mikroaerophile Proliferation geeignet ist.
- Da wir 250 ml vorbereiten möchten, verwenden Sie 500 ml (oder größere) Erlenmeyerkolben, um ein Überkochen beim Autoklavieren zu verhindern. Beschriften Sie eine "Broth" und die andere "Agar".
- Bestimmen Sie die Menge an Medien, die zum Erstellen jeder Lösung erforderlich sind, indem Sie den Konzentrationsempfehlungen des Herstellers folgen. LB Agar, hier verwendet, wird durch die Kombination von 25g/L mit Reinstwasser hergestellt. Unser Volumen von 250 ml erfordert eine Lösung von 6,25 LB Agar/250 ml Wasser. In ähnlicher Weise wird LB Broth durch die Kombination des gleichen Verhältnisses von LB Broth und Wasser hergestellt. Da es nicht mit einem Erstarrungsmittel ergänzt wird, wird es nicht verhärten, wenn es gekühlt wird.
- Wiegen Sie die Medien und mischen Sie es mit Wasser in Proportionen, die den Herstellerempfehlungen entsprechen. Fügen Sie 6,25 g LB Agar zu einem Kolben mit der Bezeichnung "Agar" und 6,25 g LB Broth zu einem Kolben mit der Bezeichnung "Broth" hinzu. Fügen Sie 250 ml Reinstwasser in jeden Kolben.
- Aluminiumfolie über jeden Kolben wickeln und mit einem Autoklaven Medien mindestens 15 Minuten bei 121°C, 15 psi sterilisieren.
- Mit einem hitzebeständigen Handschuh oder Pad entfernen Sie die Kolben aus dem Autoklaven, wenn der Zyklus abgeschlossen ist, und legen Sie sie in ein 40-50°C Wasserbad.
- Sobald die entsprechende Temperatur erreicht ist, gießen Sie den Inhalt des Kolbens mit der Aufschrift "Broth" in einen 250 ml Erlenmeyer oder Rundbodenkolben. Beschriften Sie den 250 ml Kolben als "Lösung0".
- Erhalten Sie 10, 100mm x 15 mm sterile Petrischalen und kennzeichnen Sie sie mit dem Datum, dem Namen, der Art der verwendeten Medien und der Winogradsky-Säulenzone, aus der Organismen geerntet werden.
- Entfernen Sie den Kolben mit der Aufschrift "Agar" aus dem Wasserbad und beginnen Sie, in jede der 10 Petrischalen zu gießen. Jedes Gericht sollte nicht mehr als 15 ml zugesetzt werden. Dies kann auch mit einem Pipettentor und einer 25 ml serologischen Pipette durchgeführt werden, um die Genauigkeit zu verbessern. Verwenden Sie eine sterile Pipettenspitze, um vorhandene Blasen zu entfernen, und decken Sie dann mit den Plattendeckeln ab und lassen Sie sie über Nacht erstarren.
3. Verdünnende Zubereitung
- Bereiten Sie zehn Reagenzgläser vor, die 20 ml oder mehr in einem Rack lagern können, und beschriften Sie sie T1-T10. Jede Rohrnummer entspricht dem Verdünnungsfaktor, dem sie entspricht (d. h. T4 = 1x10-4 oder 0,0001 oder 1/10.000der der Lagerkonzentration).
- Pipetten 9 ml von 0,45% Saline in jedes der 10 Reagenzgläser.
- Saline-Rohlinge sind nun bereit, durch Autoklaven sterilisiert zu werden. Verwenden Sie Aluminiumfolie, um jedes der 10 Reagenzgläser zu bedecken und dann in ein autoklavkompatibles Reagenzglasregal zu übertragen. Mindestens 15 Minuten bei 121°C, 15 psi sterilisieren.
- Entfernen Sie Die Rohlinge mit hitzebeständigen Handschuhen und lassen Sie sie abkühlen. Bedecken und lagern Sie bei 4°C, bis sie benötigt werden, wenn die Rohre Raumtemperatur erreicht haben, oder wenn sie zum Anfassen abkühlen.
4. Kultivierung des Zielorganismus
- "Lösung0" mit einer einzigen Kolonie aus einer zuvor gestreiften Platte oder 50 l eines gefrorenen Bestands impfen. Erlauben Sie dem Zielorganismus Zeit, sich zu replizieren, indem Sie geimpfte "Lösung0"über Nacht mit Schütteln (falls erforderlich) in einen 37°C-Inkubator legen. (Hinweis: Der Kolben sollte abgedeckt werden, um eine Kontamination zu verhindern. Wenn der Zielorganismus aerob ist, verwenden Sie sterile Gaze und Baumwollstecker, um eine Kontamination zu verhindern. Wenn Sie Regionen der Winogradsky-Säule bewerten, entfernen Sie einfach 1 Gramm aus jeder gewünschten Zone (aerobe und anaerobe für die Zwecke dieser Studie) und setzen Sie in T1 erneut ab, bevor Sie mit Schritt 5.3 fortfahren.
5. Serielle Verdünnung
- Den Kolben mit der Aufschrift "Nährstoffbrühe" aus dem Inkubator beziehen und kräftig schütteln.
- Pipetten 1 ml "Lösung0"in das Reagenzglas mit der Bezeichnung T1. Vortex T1. Bei der Bewertung von Winogradsky-Explants 1 Gramm der gewünschten Zone wiegen und vor dem Wirbeln zu T1 hinzufügen. (Hinweis: Hier wird aus Gründen der Einfachheit 1 ml verwendet - es können auch kleinere oder größere Verdünnungsvolumina verwendet werden).
- Entfernen Sie 1 ml aus dem Reagenzglas T1 und fügen Sie es dem Reagenzglas T2 hinzu. Vortex T2.
- Entfernen Sie 1 ml aus dem Reagenzglas T2 und fügen Sie es dem Reagenzglas T3 hinzu. Vortex T3.
- Entfernen Sie 1 ml aus dem Reagenzglas T3 und fügen Sie es dem Reagenzglas T4 hinzu. Vortex T4.
- Entfernen Sie 1 ml aus dem Reagenzglas T4 und fügen Sie es dem Reagenzglas T5 hinzu. Vortex T5.
- Entfernen Sie 1 ml aus dem Reagenzglas T5 und fügen Sie es dem Reagenzglas T6 hinzu. Vortex T6.
- Entfernen Sie 1 ml aus dem Reagenzglas T6 und fügen Sie es dem Reagenzglas T7 hinzu. Vortex T7.
- Entfernen Sie 1 ml aus dem Reagenzglas T7 und fügen Sie es dem Reagenzglas T8 hinzu. Vortex T8.
- Entfernen Sie 1 ml aus dem Reagenzglas T8 und fügen Sie es dem Reagenzglas T9 hinzu. Vortex T9.
- Entfernen Sie 1 ml aus dem Reagenzglas T9 und fügen Sie es dem Reagenzglas T10 hinzu.
6. Verbreitung Plating
- 100 l einer verdünnten Probe aus T1 direkt auf eine Petrischale. Dieser Schritt kann, muss aber nicht für jedes Rohr wiederholt werden.
- Erhalten Sie einen sterilen Einweg-Streustab oder Flamme sterilisieren eine Glasstreustange. Gleiten Sie in einer Bewegung im Uhrzeigersinn/gegen den Uhrzeigersinn den horizontalen Teil der Streustange, um die Probe gleichmäßig innerhalb der Petrischale zu verteilen.
- Wiederholen Sie diesen Vorgang für jede Zone der Winogradsky-Spalte, die ausgewertet werden soll.
- Inkubieren Sie Platten in einem 37°C Inkubator für 24 Stunden. Verwenden Sie für anaerobe Organismen eine anaerobe Kammer.
7. Streaking
- Wählen Sie eine geeignete Verdünnung Ihres Zielorganismus aus. Beispielsweise ergibt Lösung4 eine 1/10.000. Verdünnung Ihrer Anfangskonzentration. Typischerweise werden Verdünnungen von 1/1.000th (T3/Lösung), 1/1.000.000th (T6/Lösung6) und 1/1,000,000,000th (T9/Lösung9) ausgewertet, um Mikroben aufzuzählen.
- Mit einer sterilen Einweg-Impfschleife aus Kunststoff oder einer wiederverwendbaren Metall-Impfschleife, die seit nicht weniger als 10 Sekunden unter Beschuss steht, tauchen Sie ab Schritt 5 in die gewünschte Lösung ein. Kalibrierte Impfschleifen sollten 0,01 ml übertragen. (Achtung: Lassen Sie nicht zulassen, dass geflammte Schleife Bakterien sofort nach dem Entfernen von der Hitze kontaktieren)
- Die Petrischalenabdeckung auf einer Seite leicht anheben, so dass nur die Impfschlaufe auf den Agar zugreifen kann. Gleiten Sie die Impfschleife über die Oberseite der Medien in einer Zick-Zack-Manier, um darauf zu achten, den Agar nicht zu kompromittieren. Senken Sie den Petrischalendeckel.
- Verwenden Sie eine neue Einweg-Impfschleife oder sterilisieren Sie Ihre wiederverwendbare Schleife erneut.
- Drehen Sie die Platte um etwa 1/3rd (ca. 118°) und reduzieren Sie die Frequenz der Zick-Zack-Bewegung.
- Verwenden Sie erneut eine neue Einwegschleife oder sterilisieren Sie eine Metallschleife, bevor Sie sich ein letztes Mal drehen, und reduzieren Sie die Zick-Zack-Frequenz erneut. Senken Sie den Petrischalendeckel.
- Wiederholen Sie die Schritte 7.2 - 7.6, bis mindestens drei Petrischalen für drei verschiedene Verdünnungen gestreift wurden, indem sie eine neue Einwegschleife verwenden oder eine wiederverwendbare Schleife erneut entzünden (Abbildung 2).
- Die gestreiften Petrischalen über Nacht in einen 37°C-Inkubator geben. Für anaerobe Organismen verwenden Sie eine anerobische Kammer.
8. Datenanalyse und Ergebnisse
- Kulturen wurden aus den oxischen und anoxischen Zonen einer 7-tägigen Winogradsky-Säule geerntet. Diese Zonen eignen sich für heterotrophe bzw. eisenoxidierende Anaeraare.
- Säulenexplants wurden vor dem Streichen oder Ausbreiten auf LB Agar-Platten seriell verdünnt.
- Streaking ergab eine gemischte Population aus jeder der bewerteten Winogradsky-Zonen. Streuplatten führten zu ähnlichen Ergebnissen.
- Um KBE/ml oder KBE/g zu berechnen, durchschnittlich die Anzahl der gezählten Kolonien aus drei Platten. Multiplizieren Sie die durchschnittliche Anzahl der Kolonien mit dem Verdünnungsfaktor und dividieren Sie durch den Betrag aliquoted. Wenn beispielsweise durchschnittlich 65 Kolonien auf Platten gezählt würden, die mit 0,1 ml Lösung6 (T6) geimpft wurden, würde die zuvor beschriebene Formel 650.000.000 KBE/ml entsprechen.
- Isolierte Kolonien können nun aus jeder Platte ausgewählt werden, um sie in Anreicherungstests zu verwenden, um die Artenidentität zu bestimmen.
Manchmal müssen wir, um Bakterien zu identifizieren und zu untersuchen, sie zunächst isolieren und aus einer Probe anreichern. Zum Beispiel werden Proben aus einer Winogradsky-Säule gemischt, was bedeutet, dass sie mehrere Arten oder Bakterienstämme enthalten, so dass die Untersuchung eines einzelnen Bakteriums oder die Aufzählung der verschiedenen vorhandenen Arten eine Herausforderung darstellen kann. Zu diesem Zweck werden in der Regel serielle Verdünnungs- und Beschichtungstechniken eingesetzt, um die bakterielle Belastung zuverlässig zu quantifizieren und einzelne Kolonien zu isolieren.
Die serielle Verdünnung ist ein Prozess, durch den die Konzentration eines Organismus, Bakterien in diesem Beispiel, systematisch durch sukzessive Resuspension in festen Mengen flüssiger Verdünnungsmittel reduziert wird. Normalerweise ist das Volumen des Verdünnungsmittels ein Vielfaches von 10, um die logarithmische Reduktion des Probenorganismus zu erleichtern. Zum Beispiel wird zunächst ein Gramm Sediment aus der Winogradsky-Zone entfernt und 10 Milliliter eines geeigneten flüssigen Mediums zugegeben. Dann wird einem anderen Rohr, das neun Milliliter Medium enthält, ein Milliliter dieser ersten Verdünnung zugesetzt. Der Prozess kann wiederholt werden, bis mehrere verschiedene Konzentrationen von Bakterien vorbereitet wurden. Serielle Verdünnung ist der Schlüssel zur Aufzählung von Bakterien in diesem Beispiel, da gemischte Proben aus einer Winogradsky-Säule eine unbekannte, oft große Anzahl von Bakterien enthalten.
Als nächstes ermöglichen die Streifenbeschichtung und die Streubeschichtung die Isolierung und Aufzählung von Bakterien innerhalb einer Probe. Streaking wird durch die Einführung einer verdünnten Probe in einen Abschnitt des festen Mediums mit Nährstoff entemiert erreicht, der in Drittel unterteilt ist. Dieses Inokulum wird dann in einem Zick-Zack-Muster über jedes Drittel der Platte verteilt. Da verschiedene Teile der Platte gestreift sind und sich nur einmal von der vorherigen Probe kreuzen, wird die Probe dünner verteilt. Dies bedeutet, dass Sie möglicherweise nur von einer Verdünnung streifen müssen, um einzelne Kolonien in den späteren Abschnitten zu erreichen. Nach der Inkubation ermöglichen die gestreiften Platten Beobachtungen der Koloniemorphologie, Informationen, die helfen können, zwischen verschiedenen Bakterienarten zu unterscheiden.
Alternativ kann, wenn das Hauptziel ist, die Aufzählung der Bakterien in der Probenausbreitung Beschichtung verwendet werden. Bei der Streubeschichtung wird ein Aliquot einer einzelnen Probe gleichmäßig über die gesamte Oberfläche des festen Mediums verteilt. In der Regel, weil wir die Bakterienzahlen in der gemischten Probe nicht kennen, wird für jede der Verdünnungen oder eine repräsentative Probe von ihnen eine Streuplatte hergestellt. Nach der Inkubation kann die Aufzählung mit diesen Streuplatten durchgeführt werden. Alle Platten mit Koloniezahlen von weniger als 30 sollten verworfen werden, da kleine Zahlen größeren Fehlern unterliegen. Ebenso sollten alle Überzahlwerte über 300 verworfen werden, da das Übervervölkern und Überlappen von Kolonien zu einer Unterschätzung der Kolonieanzahl führen kann. Wenn die Koloniezahlen jedes dieser verbleibenden Schalen aufgezeichnet und mit dem Verdünnungsfaktor multipliziert und dann durch das plattierte Volumen dividiert werden, ergibt dies die Kolonie bildenden Einheiten, oder KBE, pro Milliliter Suspension. In diesem Video erfahren Sie, wie Sie eine Probe, die ein bekanntes Bakterium enthält, und die mikrobiellen Gemeinschaften, die in verschiedenen Regionen einer Winogradsky-Säule enthalten sind, durch serielle Verdünnung, Streuung und Streifenbeschichtung qualitativ und quantitativ bewerten.
Ziehen Sie zunächst alle geeigneten persönlichen Schutzausrüstungen an, einschließlich Labormantel, Handschuhe und Schutzbrille. Als nächstes sterilisieren Sie den Arbeitsbereich mit 70% Ethanol und wischen Sie die Oberfläche ab. Als nächstes zwei 500-Milliliter-Erlenmeyer-Flaschen sammeln und eine Brühe und den anderen Agar beschriften. Zur Zubereitung der LB-Agar-Lösung etwa 6,25 Gramm LB-Agar, drei Gramm technischea gar und 250 Milliliter destilliertes Wasser in den Kolben mit der Bezeichnung Agar mischen.
Dann bereiten LB Brühe durch Kombination 2. 5 Gramm LB-Medien und 100 Milliliter destilliertes Wasser in der Mitbrühe. Nach dem Autoklavieren der Kolben, verwenden Sie einen hitzebeständigen Handschuh, um die Kolben aus dem Autoklaven zu entfernen und legen Sie sie in einem 40 bis 50 Grad Celsius Wasserbad. Sobald die Kolben 50 Grad Celsius sind, sorgfältig vorbereiten drei 100 Milliliter Aliquots der Brühe Lösung und beschriften jede Aliquot-Lösung Null. Als nächstes sammeln Sie 10 sterile Petrischalen und kennzeichnen sie mit dem Datum, dem Namen, der Art der verwendeten Medien und der Winogradsky-Säulenzone, aus der die Organismen geerntet werden. 15 Milliliter Agar aus dem Agarkolben in jede Petrischale pfeifen. Dann verwenden Sie die Pipettenspitze, um Blasen zu entfernen, ersetzen Sie die Plattendeckel, und lassen Sie sie auf der Bank oben über Nacht zu erstarren.
Am nächsten Tag die Bank mit 70% Ethanol abwischen. Als nächstes 10 20 Milliliter Reagenzgläser T1 bis T10 beschriften und in ein Rack legen. Pipette neun Milliliter von .45% Saline in jedes Rohr. Bedecken Sie nun jedes der 10 Reagenzgläser locker mit ihren Kappen und übertragen Sie sie in ein autoklavkompatibles Reagenzglasregal. Nachdem der Zyklus abgeschlossen ist, entfernen Sie die Saline-Rohlinge mit hitzebeständigen Handschuhen und lassen Sie sie abkühlen. Bewahren Sie die Rohre bei Raumtemperatur auf, bis sie etwa 22 Grad Celsius erreicht haben.
Um einen bekannten Zielorganismus, E. coli, in diesem Beispiel zu kultivieren, impfen 100 Milliliter Lösung Null mit einer einzigen Kolonie aus einer zuvor gestreiften Platte. Dann bedecken Sie die Röhre und bebrüten sie über Nacht bei 37 Grad Celsius. Um die Bereiche einer Winogradsky-Säule auszuwerten, fügen Sie etwa ein Gramm Material aus der Aerobic-Zone zu T1 hinzu und setzen Sie sich durch Wirbel wieder aus. Wiederholen Sie diesen Vorgang dann mit einem Gramm Material aus der anaeroben Zone.
Entfernen Sie die mit E. coli geimpfte Lösung Null aus dem Inkubator und schütteln Sie sie. Dann pipette einen Milliliter der Lösung in ein T1-Reagenzglas und Wirbel zu mischen. Entfernen Sie einen Milliliter Lösung aus T1 und übertragen Sie es auf T2, Wirbel zu mischen. Wiederholen Sie diesen Vorgang durch Rohr T10. Um die aeroben und anaeroben Zonen der Winogradsky-Säule zu bewerten, entfernen Sie einen Milliliter Lösung aus jedem der zuvor vorbereiteten T1-Rohre und übertragen Sie ihn in die entsprechenden T2-Rohre. Fahren Sie dann die seriellen Verdünnungen durch die T10-Rohre fort, wie zuvor gezeigt.
Um Platte zu verteilen, Pipette 100 Mikroliter der verdünnten Probe von jedem T3-Rohr auf die entsprechende Petrischale. Verwenden Sie dann eine sterile Streustange, um die Probe vorsichtig auf die Petrischale zu verteilen und den Plattendeckel zu ersetzen. Wiederholen Sie diesen Vorgang für die Verdünnungen T6 und T9, wie zuvor gezeigt. Inkubieren Sie die Platten, die aerobe Organismen in einem 37 Grad Celsius Inkubator für 24 Stunden enthalten. Inkubieren Sie die Platten mit anaeroben Organismen in einer anaeroben Kammer, die 24 Stunden lang auf 37 Grad Celsius eingestellt ist. Am nächsten Tag entfernen Sie die Verdünnungsplatten T3, T6 und T9 aus dem Inkubator und der anaeroben Kammer und übertragen Sie sie auf die Bankspitze. Wenn Sie mit einer Platte nach der anderen arbeiten, gleiten Sie eine sterile Impfschleife in einem Zick-Zack-Muster über die Oberseite der Medien. Dann ersetzen Sie den Petrischalendeckel. Als nächstes drehen Sie die Platte um 1/3 und sterilisieren die Schleife, um die Frequenz des zuvor hergestellten Zick-Zack-Musters zu reduzieren. Wiederum, nach der Sterilisation der Schleife, drehen Sie die Platte um 1/3, reduzieren Sie die Frequenz des Zick-Zack-Musters ein letztes Mal, und ersetzen Sie den Deckel. Wiederholen Sie diese Streaking-Methode für die verbleibenden Platten, wie zuvor gezeigt. Dann legen Sie die gestreiften Platten mit aeroben Organismen über Nacht in einen 37 Grad Celsius Inkubator und die gestreiften Platten mit anaeroben Organismen in einer anaeroben Kammer, die über Nacht auf 37 Grad Celsius eingestellt ist.
Kulturen wurden aus den aeroben und anaeroben Zonen einer siebentägigen Winogradsky-Säule geerntet. Dann wurden die Kulturen seriell verdünnt, bevor sie auf LB-Agarplatten streiften und sich ausbreiteten. Streaking ergab eine gemischte Population aus jeder der bewerteten Winogradsky-Zonen, und die Streuplatten lieferten ähnliche Ergebnisse. Eine Platte, die aus einer gemischten Population gestreift ist, führt zu Bakterienkolonien in verschiedenen Formen, Größen, Texturen und Farben. Im Gegensatz dazu zeigten die gestreiften und ausgebreiteten Platten, die den bekannten Organismus E. colienthielten, eine homologe Population. Im Allgemeinen ist es am besten, KBE pro Milliliter unter Verwendung der durchschnittlichen Koloniezahl von drei Platten zu berechnen, die mit der gleichen Probe und dem gleichen Verdünnungsfaktor verteilt sind. Multiplizieren Sie die durchschnittliche Anzahl der Kolonien mit dem Verdünnungsfaktor und dividieren Sie durch den Betrag aliquoted. Schließlich können isolierte Kolonien, die aus jeder Platte ausgewählt wurden, in weiteren Anreicherungstests verwendet werden, um die Artenidentität zu bestimmen.
Applications and Summary
Die bakterielle Aufzählung und Dehnungsisolierung durch Beschichtung erfordert überschaubare Konzentrationen von Zielorganismen. Eine erfolgreiche Beschichtung ist daher von einer seriellen Verdünnung abhängig. Daher bleiben die genannten Techniken der Eckpfeiler der mikrobiologischen Untersuchung und des Experimentierens. Obwohl einfach durch design, Verdünnungsfaktoren und Beschichtungstechnik kann durch den Experimentator geändert werden, um die Ergebnisse zu stärken, ohne die Integrität jeder Methode zu beeinträchtigen. Das Plotten der vier Phasen des bakteriellen Wachstums kann hilfreich sein, wenn sie gewünschte Mikroben charakterisieren. Diese Phasen, Verzögerung, Protokoll, stationär und Tod, sind durch Veränderungen in der bakteriellen Replikation gekennzeichnet. Die Verzögerungsphase zeichnet sich durch ein langsames Wachstum aufgrund physiologischer Anpassung aus, die Logphase ist die Periode der maximalen Proliferation mit einem exponentiellen Anstieg lebensfähiger Zellen, stationäre Phase wird dann aufgrund von Umwelteinschränkungen und Ansammlungen von Toxinen erreicht. vor der Todesphase, in der die Zellzahl zu sinken beginnt. Dies kann durch serielle Verdünnung (oder 1-stufige Verdünnung, um Verwechslungen zu vermeiden) Lösung0 jede Stunde für insgesamt 8 Stunden erreicht werden, beginnend mit Zeit0 (Lösung0 sollte nach jeder Verdünnung an einen Schüttel-Inkubator zurückgegeben werden). Berechnen Sie das Protokoll10 der KBE/ml für ein einzelnes Verdünnungszeichen von Zeit0 und Plot auf der Y-Achse. Wiederholen Sie diese Berechnung für die Stichprobe Zeit1 (stellen Sie sicher, dass CFU/ml mit dem gleichen Verdünnungsfaktor wie Zeit0berechnet werden). Wiederholen Sie dies, bis jedes Mal (Zeit1-Zeit8) auf der X-Achse geplottet wird.
References
- Allen, M.E., Gyure, R.A. (2013) An Undergraduate Laboratory Activity Demonstrating Bacteriophage Specificity. Journal of Microbial Biological Education 14: 84-92.
- Ben-David, A., Davidson, C.E. (2014) Estimation Method for Serial Dilution Experiments. Journal of Microbiological Methods 107:214-221.
- Goldman, E., Green, L.H. (2008) Practical Handbook of Microbiology.
- Koch, R. (1883) New Research Methods for Detection of Microcosms in Soil, Air and Water.
- Lederberg, J., Lederberg, E.M. (1952) Replica Plating and Indirect Selection of Bacterial Mutants. Journal of Bacteriology 63:399-406
- Pepper, I., Gerba, C., Ikner, L. (2019) Bacterial Growth Curve Analysis and its Environmental Changes. JoVE Science Education Database. Environmental Microbiology.
- Sanders., E.R. (2012) Aseptic Laboratory Technique: Plating Methods. JoVE 63:e3063.