

Summary
This work shows how to form an RNA/DNA hybrid at the chromosomal level and reveal transfer of genetic information from RNA to genomic DNA in yeast cells.
Abstract
Synthetic short nucleic acid polymers, oligonucleotides (oligos), are the most functional and widespread tools of molecular biology. Oligos can be produced to contain any desired DNA or RNA sequence and can be prepared to include a wide variety of base and sugar modifications. Moreover, oligos can be designed to mimic specific nucleic acid alterations and thus, can serve as important tools to investigate effects of DNA damage and mechanisms of repair. We found that Thermo Scientific Dharmacon RNA-containing oligos with a length between 50 and 80 nucleotides can be particularly suitable to study, in vivo, functions and consequences of chromosomal RNA/DNA hybrids and of ribonucleotides embedded into DNA. RNA/DNA hybrids can readily form during DNA replication, repair and transcription, however, very little is known about the stability of RNA/DNA hybrids in cells and to which extent these hybrids can affect the genetic integrity of cells. RNA-containing oligos, therefore, represent a perfect vector to introduce ribonucleotides into chromosomal DNA and generate RNA/DNA hybrids of chosen length and base composition. Here we present the protocol for the incorporation of ribonucleotides into the genome of the eukaryotic model system yeast /Saccharomyces cerevisiae/. Yet, our lab has utilized Thermo Scientific Dharmacon RNA-containing oligos to generate RNA/DNA hybrids at the chromosomal level in different cell systems, from bacteria to human cells.
Protocol
Rationale
Exploiting the use of Thermo Scientific Dharmacon oligos, 50 to 80-mers, here we present a procedure, based on a gene correction assay, by which oligos can transfer genetic information to genomic DNA of yeast cells, following annealing to the target chromosomal DNA. Successful targeting by oligos is scored by the appearance of yeast colonies displaying the expected phenotype. If the desired genetic modification is carried within one or more ribonucleotides incorporated in the oligo sequence, the genetic information flows directly from the RNA tract to the chromosomal target DNA, thus, an RNA/DNA hybrid forms at the chromosomal level during the targeting process. The ribonucleotide/s of a Thermo Scientific Dharmacon RNA-containing oligo can serve as template for gene targeting via DNA repair synthesis or can be embedded into the DNA and serve as template during DNA replication. In these experiments we use the yeast /Saccharomyces cerevisiae/ eukaryotic model system; however, a similar approach can also be applied in other organisms or cell types. In this video, we use a Thermo Scientific Dharmacon oligo designed with homology to a target yeast genomic locus and containing one ribonucleotide in the middle to correct a mutation in the target gene. After transformation with the RNA-containing oligo, we observe the correction of the targeted site at a certain frequency, which indicates that the RNA tract of the oligo could be incorporated into the chromosomal DNA and serve as template for DNA synthesis during DNA replication. The genetic information carried within the RNA tract is stably transmitted to the following cell generations. Here we describe the steps of yeast cell transformation by the Thermo Scientific Dharmacon RNA-containing oligo and the approach to detect the RNA information transferring into chromosomal DNA.
A. Design of the RNA-containing Oligo used in this Experiment
We have designed an RNA-containing oligo to correct a genetic defect in the yeast S. cerevisiae chromosomal DNA at the trp5 locus. The yeast strain used in the protocol contains a mutant trp5 gene with a two-base deletion and one nonsense mutation (Figure 1A). Such yeast strain is a tryptophan auxotrophic mutant and does not form colonies on media without tryptophan. The DNA sequence of the TRP5 gene is available at the Saccharomyces Genome Database (http://www.yeastgenome.org/). The Thermo Scientific Dharmacon RNA-containing oligo used in this protocol is a 65-mer with a 2-base DNA insertion, designed to correct the deletion mutation and one ribonucleotide designed to correct the nonsense mutation of the trp5 allele (Figure 1A). The sequence of the oligo is designed as follows:
5'-AAAAGGGTTTTGATGAAGCTGTCG-CG-GATCCCACATTCTG-rG-GAAGACTTCAAATCCTTGTATTCT. This oligo is synthesized by Thermo Scientific Dharmacon (Lafayette, CO) at 200 nM scale, and is desalted, deprotected and used without PAGE purification.
B. Preparation of the RNA-containing Oligo for Yeast Transformation (modified from Storici et al., 2007 1)
- Wipe materials that will be used for the experiment including oligo tubes, pipettes, vortex, racks, experimental area and gloves worn by the investigator with RNase decontamination solution to remove potential RNase contamination before everything starts. Use RNase-free water, chemical reagents, tubes and pipette tips in all steps. Every step in this experiment should be RNase-free.
- Resuspend the Thermo Scientific Dharmacon RNA-containing oligo received from the company to 250 pmoles/μl stock solution with RNase-free water and vortex vigorously to dissolve the pellet. Store at -80°C.
- Immediately before the transformation, thaw the RNA-containing oligo on ice and dilute to 50 pmoles/μl with RNase-free water in RNase-free tubes. Each transformation requires 1 nmole of the RNA-containing oligos.
- Denature a chosen amount of the RNA-containing oligos on the 100 °C heat block for 2 min to eliminate secondary structures of the oligos.
- Immediately after denaturation place the tube on ice. Keep on ice till transformation.
- As control in the experiment a corresponding DNA-only oligo is used. This oligo is thawed from -20 °C and prepared for the transformation as the RNA-containing oligo, as described above.
C. Transformation of Yeast Cells using the RNA-containing Oligo
- Inoculate 5 ml of rich YPD liquid medium with the trp5 mutant yeast cells and grow at 30 °C overnight (see Materials).
- Transfer 1.5 ml of the overnight culture into 50 ml of YPD liquid medium.
- Incubate cells in a 30 °C shaker (225 rpm) for 4h.
- Prepare Solution 1 and Solution 2 immediately before transformation in RNase-free tubes (see Materials).
- Transfer the cell culture to a 50 ml RNase-free tube and spin at 3000 rpm, corresponding to 1562 g, for 2 min. The pellet of the cell precipitation is approx. 0.3 cm3.
- Remove the supernatant and wash cells with 50 ml of RNase-free water and spin at 3000 rpm for 2 min.
- Repeat step 6 for 5 times to get rid of the culture medium and RNases that could be present in the medium as much as possible.
- Remove the supernatant and resuspend cells in 5 ml of Solution 1 and spin at 3000 rpm for 2 min.
- Remove supernatant and resuspend cells in 250 μl of Solution 1. This amount of cells is sufficient for 7-8 transformations.
- Aliquot 50 μl of the cell suspension in RNase-free microcentrifuge tubes, add 20 μl of RNA-containing oligo working solution (1 nmole), or 20 μl of DNA-only oligo working solution (1 nmole), or 20 μl of sterile water with no oligo for the negative control. Then add 300 μl of Solution 2 for each transformation reaction. There is no need to add salmon sperm DNA in the process of transformation, as the oligos act as carrier themselves.
- Vortex vigorously to mix components homogenously.
- Incubate transformation reactions at 30 °C for 30 min in a shaker.
- Heat shock at 42 °C for 15 min.
- Spin down cells at 5000 rpm, corresponding to 2236 g, for 4 min.
- Remove supernatant and resuspend cells in 100 μl of sterile water.
- Take an aliquot of this cell suspension and dilute it with sterile water by 100,000 fold and plate cells on one YPD plate using approx. 15 sterile glass beads and incubate at 30 °C for 2 days.
- Plate all resuspended cells from each transformation reaction on one Petri dish of synthetic complete solid medium without tryptophan (SC-Trp) using approx. 15 sterile glass beads and incubate at 30 °C for 4-5 days (Figure 2).
D. Analysis of Gene Correction by the RNA-containing Oligo
- Count the number of colonies grown on the selective medium (Figure 2) as well as on YPD medium to calculate the gene correction frequency for the RNA-containing oligo, the DNA-only oligo and for the no-oligo control. Compare the number obtained. Spontaneous reversion rate of the trp5 alleles with the two-base deletions and the nonsense mutation is less than 10-9 in the used yeast strain, thus we expect no colonies formation on the selective medium when no oligos are added to the cells.
- Streak out several randomly picked transformant colonies onto YPD medium to obtain single colony isolates. Wait two days for colony growth, then take several (at least 5) single colonies and make patches on YPD and on selective medium.
- Design a pair of primers to amplify the region (250-1,000 bp) targeted by the RNA-containing oligo by colony PCR (Figure 1B). The procedure for colony PCR is as follows (modified from Storici and Resnick, 2006 2).
- Resuspend cells (approx. 1 mm3) taken from the individual patches in 50 μl of water and add 1 unit of lyticase. Incubate at room temperature for 10 min, followed by incubation in a heat block at 100 °C for 5 min to break the cells and release genomic DNA in solution.
- PCR conditions: The PCR reaction includes 10 μl of the cell resuspension solution, 50 pmoles each of forward and reverse primers, 1 μl of 10 mM dNTPs, 1 unit of Taq polymerase, 5 μl of 10x buffer and is adjusted with sterile water to a final volume of 50 μl. The PCR program is 3 min at 95 °C; 30 cycles of 30 s at 95°C, 30 s at 55 °C, and 1 min at 72 °C; a final extension time of 7 min at 72 °C; then samples are kept at 4 °C. An extension time of 1 min/kb is assumed for this reaction.
- Following PCR, samples are run on a 1% or 2% (depending on the expected size of the PCR product) agarose gel for observation of the PCR product (Figure 3).
- If the genetic information transferred by the RNA-containing oligo generates a new restriction site in the yeast genomic target region (Figure 1B), it is possible to verify the correct transfer of information by digesting the PCR product with the appropriate restriction enzyme. If no restriction site is generated by the RNA-containing oligo, go to step 6. Digest PCR products using a specific restriction enzyme. The digestion reaction includes 6 μl of PCR product, buffer, BSA (may not be needed for some enzymes, see instruction for the enzyme used), 0.5 μl of restriction enzyme, and sterile water to 15 μl. Samples are incubated for 1 h at the temperature specific for the enzyme used.
- Run an undigested sample together with the digested samples on the same row of a 2% agarose gel to observe the genetic modification transferred by the RNA tract of the RNA-containing oligo (Figure 3).
- Purify PCR products by using a PCR purification kit and prepare them for DNA sequencing. Submit samples for sequencing with the same primers used to amplify the product.
- Analyze DNA sequencing results using software that allows alignment of multiple sequences with a chosen reference sequence (Figure 4).
E. Alkali Treatment of the RNA-containing oligo (Figure 5)
- For each reaction, add 1 nmole (4 μl of 250 pmoles/μl stock solution) of the RNA-containing oligo, or DNA-oligo into a 1.5 ml tube.
- Add 4 μl of 1 M NaOH for hydrolysis, or alternatively add 4 μl H2O as negative control, and incubate at 65° C in a water bath for 1 h. Then move from the water bath to ice.
- Neutralize with 2 μl of 1.2 M HCl, 4 μl of 1 M Tris-HCl, and 4 μl of H2O, or alternatively 6 μl of H2O and 4 μl of 1 M Tris-HCl for negative control. Keep on ice till transformation.

Figure 1. Schematic diagram of the defective trp5 gene and of the TRP5 allele corrected by the RNA-containing oligo. A) The trp5 mutant gene contains a 2-base deletion (black triangles) and 1-base nonsense mutation (asterisk). The single-strand RNA-containing oligo with 2-base insertion (blue loop) and 1-RNA base substitution (red rectangle) generating a Van91I restriction enzyme site (shown by the bracket) is transformed into yeast cells to correct the genetic defects of the trp5 gene. B) After the TRP5 gene is repaired by the RNA-containing oligo (corrected bases are indicated as blue rectangles), the Van91I site is generated into the TRP5 gene. A 278 bp fragment including only one Van91I restriction site in TRP5 gene is PCR amplified by a pair of primers (P1 and P2). The 177 bp and 101 bp fragments generated after digestion of the P1 and P2 PCR product by Van91I are also shown.

Figure 2. Transformation result with the RNA-containing oligo. A) Yeast cells transformed with no oligo do not form any colony on the SC-Trp medium, see plates a-b. B) Plates c-g show yeast colonies growing on SC-Trp after cells are transformed with 1 nmole of the RNA-containing oligo. C) Plates h-l show yeast colonies growing on SC-Trp after cells are transformed with 1 nmole of the corresponding DNA-only control oligo.

Figure 3. Detection of genetic information transfer from the RNA-containing oligo to yeast chromosomal DNA by restriction digestion of the PCR product amplifying the targeted genomic region. 2% agarose gel electrophoresis of PCR samples amplified by P1 and P2 and digested with Van91I restriction enzyme. Lanes 1, 8 and 15, DNA ladder with sizes of 100, 200, 300, 400 and 500 bp indicated on the left; lane 2, PCR product of trp5 locus amplified from the genomic DNA of the trp5 mutant strain; lane 3, PCR product amplified from genomic DNA derived from a Trp+ colony targeted by the DNA-only oligo; lane 4-7, PCR products amplified from genomic DNA derived from Trp+ colonies targeted by the RNA-containing oligo; lane 9 to 14, Van91I restriction digestion of the PCR products from lanes 2 to 7. The presence of the uncut PCR product bands in lanes 10 to 14 can be explained by partial digestion by Van91I at the TRP5 locus (CCACATTCTGG). Considering that the cutting site for Van91I (CCANNNN NTGG) can have multiple sequences, the site generated in TRP5 may not be the most optimal target for the enzyme. In fact, following DNA sequencing of all the above PCR products we detect no additional changes beyond those carried by the oligos (see Figure 4). The 278 bp PCR band amplified by P1 and P2 primers and the digestion product bands by Van91I of 177 bp and 101 bp are shown by the arrows on the right.

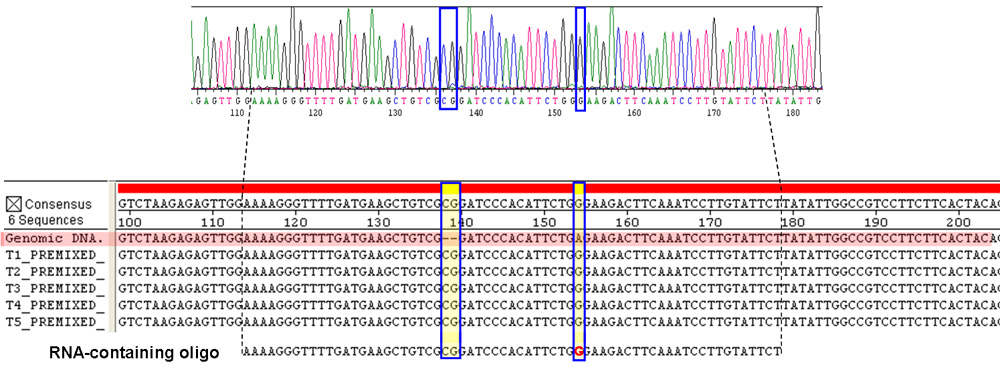
Figure 4. DNA sequencing results showing gene correction by the RNA-containing oligo. A) DNA electropherogram of the genomic region targeted by the RNA-containing oligo. The G in the DNA sequence (blue boxed) derives from the rG on the RNA-containing oligo. Also boxed is the insertion of the CG bases. Sequencing results from all other PCR products are also as clear as this one, with fluorescent signals well above the background. B) Sequences of the TRP5 region of the Trp+ transformants targeted by the DNA-only oligo (T1) and the RNA-containing oligo (T2-T5) match in the consensus sequence on the top and are compared with that of the trp5 mutant cells before targeting by the oligos (Genomic DNA, red shaded). The repair RNA-containing oligo on the bottom has two-base DNA insertion and one-base (G) RNA substitution marked in red. The regions boxed in blue with yellow shade show that the RNA-containing oligo as well as the DNA-only oligo precisely corrected the deletion mutation and nonsense mutation in all tested samples. The dashed lines mark the position of the RNA-containing oligo sequence. Please click here to see a larger version of figure 4.

Figure 5. Alkali treatment prevents gene correction by the RNA-containing oligo. The transformation frequency by the RNA-containing oligo (R) is shown in the red bar and that by the DNA-only oligo (D) is shown in the blue bars. The error bars represent the standard error of the mean for 3 independent transformations for each oligo. The DNA-only oligo displays similar transformation frequency without and with NaOH treatment. Differently, transformation frequency by the RNA-containing oligo drops to 0 following treatment with NaOH. Therefore, the preparation with the RNA-containing oligo is not contaminated with DNA-only oligo, thus, the observed transformation frequency is specific to the RNA-containing oligo.
Discussion
The fact that RNA can transfer genetic information directly to genomic DNA in cells was discovered exploiting the use of synthetic RNA-containing oligos (Dharmacon synthesized oligos) 1. It was the proof of principle that cells can use RNA-containing molecules or RNA-only sequences as templates for DNA synthesis. The use of RNA-containing oligos not only led to the demonstration that genetic information can be transferred directly from RNA to genomic DNA without the need of a reverse transcribed DNA copy intermediate, but also to the proof that RNA can be used as homologous template in the repair of a DNA damage 1,3. The oligos can be made of RNA-only or contain just a single ribonucleotide embedded in a DNA sequence, as in the example we presented here. The RNA-containing oligo has one embedded ribonucleotide designed to correct the nonsense mutation in the genomic defective trp5 allele. The transfer of genetic information from the ribonucleotide to the genomic DNA is revealed by a change in phenotype (cell growing on the medium lacking tryptophan) of the targeted cells. The frequency of gene correction obtained with the RNA-containing oligo is measured and compared with that of a control DNA-only oligo. By performing this gene correction assay in different cell backgrounds, where we mutate various yeast genes, we can detect which factor/s specifically affect targeting by the RNA-containing oligos. Thus, we can reveal mechanisms how cells regulate the stability of RNA/DNA hybrids. By simply designing different variants of the RNA-containing oligos we can determine the likelihood for a particular RNA tract to serve as template in DNA modification and we can determine the stability of specific RNA sequences embedded into DNA. Moreover, we can identify what are the preferred in vivo substrates for factors interacting with RNA/DNA hybrids.
While several in vitro studies, mainly utilizing short RNA-containing oligos, have been conducted to characterize the function of factors that can recognize RNA in a hybrid with DNA, such as the RNase H enzymes 4, the in vivo functions of RNases H as well as the identity of other proteins that could affect RNA/DNA hybrid stability remain mostly unknown. The possibility to utilize RNA-containing oligos of a significant length (50 to 80-mers) and optimal quality (such as Thermo Scientific Dharmacon RNA-containing oligos) has open the way to examining a wide range of molecular processes directly in vivo in the cells of interest. As shown in this video, transformation using RNA-containing oligos requires essentially only a few additional steps compared to transformation using DNA molecules, to prevent degradation by RNases. Thus, transformation using RNA-containing oligos is not limited to the yeast system, but can be applied to any organism or cell type where transformation by DNA oligos is proficient.
In conclusion, targeting cells by RNA-containing oligos provides the opportunity to generate RNA/DNA hybrids and ribonucleotides embedded in DNA in vivo in cells. The stability, function and consequences of these in vivo generated RNA/DNA hybrids can be analyzed and characterized, potentially uncovering unknown mechanisms of DNA repair and revealing novel strategies for gene targeting.
Disclosures
The video production for this article was sponsored by Thermo Fisher Scientific, which produces reagents and instrument used in this publication.
Acknowledgments
This work was supported by the Georgia Cancer Coalition grant-R9028.
Materials
A. Transformation reagents and media (modified from Storici and Resnick, 2006 |
|||
|
|||
B. Colony PCR materials. |
|||
|
|||
C. PCR purification. |
|||
|
|||
D. Gel Electrophoresis. |
|||
|
|||
E. Restriction digestion. |
|||
|
|||
F. Alkali treatment for the RNA-containing oligo. |
|||
|
References
- Storici, F., Bebenek, K., Kunkel, T. A., Gordenin, D. A., Resnick, M. A. RNA-templated DNA repair. Nature. 447, 338-3341 (2007).
- Storici, F., Resnick, M. The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 409, 329-345 (2006).
- Storici, F. RNA-mediated DNA modifications and RNA-templated DNA repair. Curr Opin Mol Ther. 10, 224-230 (2008).
- Cerritelli, S. M., Crouch, R. J. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 276, 1494-1505 (2009).

{kind=link}