To investigate whether the serine protease plasmin can activate ENaC-mediated currents, ΔIami of individual ENaC-expressing oocytes was determined before and after 30 min incubation of the oocytes in protease-free (control) (Figure 2A) or plasmin containing solution (Figure 2B) using the two-electrode voltage-clamp technique (see Figure 1). Exposure to plasmin increased ΔIami in every oocyte measured. In contrast, in control experiments, 30 min incubation of ENaC-expressing oocytes in protease-free solution had a negligible effect (Figure 2 C,D). Thus, by using this method a stimulation of ENaC-mediated current by plasmin can be detected.
To study the effects of mutating putative cleavage sites upon the activation of ENaC-mediated currents, as well as upon channel cleavage, the effect of chymotrypsin on WT-ENaC was compared with that on a mutant ENaC with mutated prostasin and plasmin cleavage sites (γRKRK178AAAA;K189A). The time course of channel activation by chymotrypsin as well as the appearance of ENaC cleavage products at the cell surface was investigated by using different protease incubation times (Figure 4A). It was demonstrated that the mutant channel delays and reduces the activation of ENaC-mediated current by chymotrypsin. This is paralleled by a delayed appearance of a lower molecular weight γENaC cleavage fragment of 67 kDa corresponding to the fully cleaved subunit. Cleavage fragments were detected using a γENaC antibody directed against an epitope in the C-terminus (Figure 3). This methodological approach demonstrates that the time course of proteolytic activation of ENaC-mediated currents correlates with the appearance of a 67 kDa γENaC cleavage product at the cell surface (Figure 4 B,C). This supports the concept of a causal link between proteolytic channel cleavage and channel activation13. Moreover, by combining current measurements and the detection of γENaC fragments at the cell surface it was demonstrated that the mutated cleavage sites are functionally relevant for proteolytic channel activation.

Figure 1. Procedure of determining the stimulatory effect of a protease on ENaC heterologously expressed in Xenopus laevis oocytes. ENaC activity is estimated by measuring the amiloride-sensitive whole-cell current component ΔIami.

Figure 2. Plasmin stimulates ENaC-mediated currents in oocytes expressing ENaC. (A-D) Oocytes expressing human ENaC were incubated for 30 min in protease-free solution (control) or in solution containing plasmin (10 μg/ml). To determine ΔIami before (-) and after (+) incubation, oocytes were clamped at a holding potential of -60 mV. (A,B) Four representative whole-cell current traces from one batch of oocytes. Amiloride (ami) was present in the bath solution to specifically inhibit ENaC as indicated by black bars. (C) Data points obtained from an individual oocyte are connected by a line. (D) Summary of similar experiments as shown in C. Columns represent relative stimulatory effect on ΔIami calculated as the ratio of ΔIami measured after a 30 min incubation (ΔIami 30 min) to the initial ΔIami (ΔIami initial) measured before incubation. Numbers inside the columns indicate the number of individual oocytes measured. N indicates the number of different batches of oocytes. (This figure has been modified from [Haerteis et al. 2012 J Gen Physiol 140, 375-389, doi:10.1085/jgp.201110763])

Figure 3. Model of the γENaC subunit showing cleavage sites for proteolytic activation and the binding site of the antibody used. Proteolytic cleavage by the Golgi-associated convertase furin is important for ENaC maturation in the biosynthetic pathway before the channel reaches the plasma membrane. After cleavage by furin a 76 kDa fragment can be detected at the cell surface using a biotinylation approach and an antibody against an epitope in the C-terminus of the γ-subunit. The pivotal final step in proteolytic ENaC activation probably takes place at the plasma membrane where γENaC is cleaved by extracellular proteases (e.g. plasmin or chymotrypsin) in a region distal to the furin site resulting in a 67 kDa cleavage fragment. (This figure has been modified from [Haerteis et al. 2012 J Gen Physiol 140, 375-389, doi:10.1085/jgp.201110763])
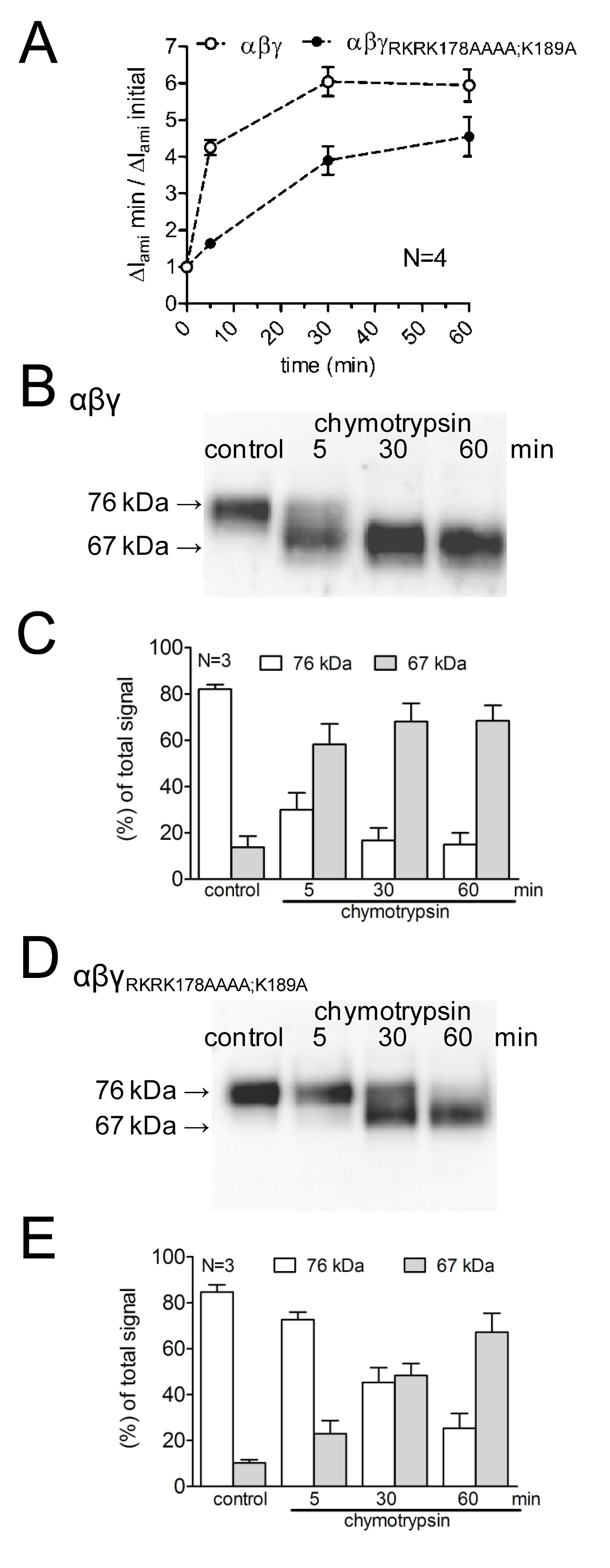
Figure 4: Mutating both the plasmin (K189) and the prostasin cleavage site (RKRK178) delays the activation of ENaC-mediated currents and the appearance of a 67 kDa cleavage product of the channel’s γ-subunit. Oocytes expressing WT (open symbols) and γRKRK178AAAA;K189AENaC mutant channel (closed symbols) were incubated for 30 min in protease-free solution (control) or for 5, 30, or 60 min in a solution containing chymotrypsin (2 μg/ml). (A) To determine ΔIami before and after incubation, oocytes were clamped at a holding potential of -60 mV. Circles represent the ratio of ΔIami measured after 5, 30, or 60 min incubation (ΔIami min) to the initial ΔIami (ΔIami initial) measured before incubation. Each data point represents the mean ΔIami measured in 22-24 individual oocytes of four different batches. (B-D) In parallel to the detection of ΔIami, expression of biotinylated γENaC at the cell surface was analyzed by SDS-PAGE. γENaC was detected with an antibody against an epitope in the C-terminus of human γENaC. Representative western blots from one batch of oocytes are shown. (C-E) Densitometric analysis of three western blots similar to those shown in B or D. For each lane, the signals detected in the regions of 76 kD (open columns) and 67 kD (gray columns) were determined and normalized to the sum of the total signal detected. N indicates the number of different batches of oocytes. Click here to view larger image.
