Note: Use data sheets to track all steps of the protocol; example data sheets are given in the supplemental materials Tables S2-S4.
1. Standard Curve Preparation
- Prepare a working stock of the standard curve reagent (e.g., Armored RNA EPA-1615) by diluting it from the concentration supplied by the manufacturer to a concentration of 2.5 x 108 particles/ml (2.5 x 108 GC/ml) using TSM III buffer. Divide the working stock into 250 µl aliquots using 1.5 ml microcentrifuge tubes and store at -20 °C.
NOTE: See supplemental materials protocol Step S1 for instructions on the preparing working stocks of virus and plasmids for use as alternative standard curve reagents. - Thaw one or more of the working stock aliquots. Prepare five 10-fold serial dilutions using 1.5 ml microcentrifuge tubes, giving concentrations of 2.5 x 107, 2.5 x 106, 2.5 x 105, 2.5 x 104, and 2.5 x 103 GC/ml.
- Prepare the first dilution by adding 25 µl of the 2.5 x 108 GC/ml working stock to 225 µl of TSM III buffer. Mix for 5–15 sec using a vortex mixer.
- Prepare the next dilution by adding 25 µl of the dilution prepared in Step 1.2.1 to 225 µl of TSM III buffer. Mix again and continue a similar process to prepare the next three 10-fold dilutions.
- Extract the RNA from the standard curve working stock and the five dilutions immediately using the procedure in Section 3.
2. Tertiary Concentration
- Prepare a centrifugal concentrator (30,000 molecular weight cutoff) for each sample collected by adding at least 10 ml of 1x PBS, 0.2% bovine serum albumin (BSA) to the upper sample chamber. Ensure that solution has filled the thin channel concentration chamber, and then hold O/N at 4 °C.
- Discard the fluid. Rinse the concentrator one time with at least 10 ml of sterile reagent grade water to remove excess BSA and then discard the water.
- Add an amount of secondary water concentrate from each test sample equal to S, the Assay Sample Volume into separate centrifugal concentrators.
- Calculate S using Equation 1,

Where D is the Volume of Original Water Sample Assayed, TSV is the Total Sample Volume and FCSV is the Final Concentrated Sample Volume. See supplemental materials S2 for an example of the calculation of S.
- Calculate S using Equation 1,
- Centrifuge each test sample at 3,000–6,000 x g at 4 °C in a swinging bucket rotor for 20 – 30 min. Check the volume in the thin channel concentration chamber.
- If the volume is greater than 400 µl, centrifuge again for 20 min or longer. Continue centrifuging until the sample in the thin channel concentration chamber has been reduced to less than 400 µl. Do not remove the supernatant.
- Wash the sides of the centrifugal concentrator with 1 ml of sterile 0.15 M sodium phosphate, pH 7–7.5 to increase virus recovery. Centrifuge again at 3,000–6,000 x g and 4 °C until the sample has been reduced to less than 400 µl. Repeat this wash step one additional time.
- Using a 100-200 µl micropipette, carefully measure and transfer each concentrated sample to a 1.5 ml microcentrifuge tube (i.e., transfer 200 µl to the microcentrifuge tube and then measure the remaining concentrate by adjusting the micropipette until the remaining fluid can be completely drawn up into the pipettor tip). Add 0.15 M sodium phosphate, pH 7–7.5, to adjust the final volume to 400 ± 2 µl.
- Extract nucleic acid immediately by proceeding to step 3. Hold any concentrated samples that cannot be processed immediately at 4 °C for no more than 24 hr.
3. Nucleic Acid Isolation
- Add 200 µl of extraction buffer prepared as described in Step 3.3 and 200 µl of the tertiary concentrate from each test sample from Step 2.5 or standard curve dilution from Step 1.3 to separate labeled 1.5 ml microcentrifuge tubes. Freeze remaining tertiary concentrates at or below -70 °C. Extract the nucleic acids from each sample according to the nucleic acid extraction kit manufacturer’s instructions for the spin protocol for blood samples with the following exceptions.
- Do not add protease to the water samples or use the extraction buffer supplied with the nucleic acid extraction kit.
- Prepare extraction buffer with carrier RNA
- Add 310 µl of carrier RNA dilution buffer to a vial containing 310 µg of carrier RNA. Mix to dissolve and then divide into 6 aliquots containing about 50 µl. Store at -20 °C.
- Add 28 µl of thawed carrier RNA per ml of the extraction buffer to obtain a carrier RNA concentration of 0.027 µg/µl. Use the carrier RNA-amended extraction buffer in place of that supplied with the extraction kit.
- Prepare a master solution of elution buffer by adding ribonuclease (RNase) Inhibitor to a final concentration of 400 units/ml to the elution buffer supplied with the extraction kit.
- Elute RNA from the nucleic acid binding spin column by placing 50 µl of elution buffer with RNase inhibitor into the column. Wait for 1 min, and then centrifuge at 6,000 x g for 1 min at RT.
- Repeat Step 3.4.1 and then remove and discard the column.
- Prepare aliquots of the RNA extracts from Step 3.4.2. Prepare 6 aliquots of the standard curve working stock and each standard curve dilution containing at least 15 µl each. Prepare 4 aliquots of all other RNA samples containing at least 22 µl each. Store one aliquot of each sample and standard curve controls at 4 °C if they can be processed by reverse transcription within 4 hr; otherwise, store all aliquots at or below -70 °C.
4. Reverse Transcription (RT)
- Prepare 100 µM stock solutions of each oligonucleotide primer and probe listed in Table 1 by adding a volume of PCR-grade water to each vial using an amount (in microliters) equal to ten times the number of nanomoles (nmol) of oligonucleotide present in the vial (as shown on the vial label or in the manufacturer’s specification sheet (e.g., resuspend a primer containing 36.3 nmol in 363 µl). Mix to dissolve.
- Dilute the 100 µM solutions 1:10 with PCR-grade water to prepare 10 µM working solutions.
- Prepare RT Master Mix 1 and 2 in a clean room using the guide in Table 2. Pipette 16.5 µl of RT Master Mix 1 to each PCR plate well using a multichannel pipette.
- Thaw, if frozen, the nucleic acid extracts from each field sample and Lab Fortified Sample Matrix (LFSM; i.e., seeded water matrix sample).
- Dilute each field and LFSM sample 1:5 and 1:25 in elution buffer containing 400 units/ml of RNase inhibitor.
- Thaw, if frozen, but do not dilute Lab Fortified Blank (LFB; i.e., a positive quality control using seeded reagent grade water), Lab Reagent Blank (LRB; i.e., a negative quality control using reagent grade water), Performance Evaluation (PE; i.e., seeded reagent grade water samples used to evaluate laboratory performance prior to the start of a study), Performance Test (PT; i.e., seeded reagent grade water samples with titers unknown to an analyst that are used to evaluate laboratory performance during a study), NA Batch negative extraction control, or extracted RNA from the standard curve set.
- Place 6.7 µl of the RNA from every test sample, control, and standard curve into separate PCR plate wells, using triplicate wells for test samples and controls and duplicate wells for standard curves (see Figure S1 for an RT plate example).
- Place 6.7 µl of elution buffer into separate PCR plate wells for no template controls (NTC). Include 2-8 NTC per RT plate, using two for the first sample and then two more for every fourth additional sample.
- Distribute the negative extraction and NTC controls throughout the plate.
- Seal the PCR plate with a heat resistant plate sealer. Mix the samples for 5-10 sec and then centrifuge at ≥ 500 x g briefly.
- Incubate the plate for 4 min at 99 °C and then cool rapidly to 4 °C in a thermal cycler. Centrifuge again at ≥ 500 x g briefly.
- Carefully remove the plate seal and then add 16.8 µl of RT Master Mix 2 to each well. Seal the plate again with a heat resistant plate sealer, followed by mixing and a brief centrifugation at ≥ 500 x g.
- Place the plate in a thermal cycler and run for 15 min at 25 °C, followed by 60 min at 42 °C, 5 min at 99 °C, and then by a 4 °C hold cycle.
- Process immediately or within 8 hr by qPCR (Step 5), or store samples at or below -70 °C until they can be processed. Store samples that can be processed within 8 hr at 4 °C.
5. Real-time Quantitative PCR (qPCR)
- Determine the mean hepatitis G Cq value for each lot of hepatitis G reagent prior to running any test samples.
- Run an RT assay using 10 replicates prepared as described for NTC controls (Step 4.1.1). Run the hepatitis G qPCR assay as described below (Steps 5.2 to 5.5.3). Calculate the mean Cq value of the 10 replicates.
- Adjust the hepatitis G reagent quantity in RT Master Mix 1 (Table 2), if necessary, to obtain a mean Cq value between 25 and 32 units. Compensate the amount raised or lowered by changing the volume of water added to keep the RT Master Mix 1 final volume at 16.5 µl per assay.
- Confirm any adjustments by repeating Steps 5.1.1 to 5.1.2 and change Table 2 to reflect the adjusted quantities.
- Prepare qPCR master mixes in a clean room using the guides in Table 3 for enterovirus, Table 4 and Table 5 for norovirus genogroup I, Table 6 for norovirus genogroup II, Table 7 for murine norovirus (norovirus genogroup V), and Table 8 for hepatitis G. Mix each master mix and then centrifuge at ≥ 500 x g briefly.
- Add the PCR master mixes to the appropriate wells of a labeled optical reaction plate, using 14 µl per well and separate plates for each qPCR assay (see Figure S2 for a possible layout for a qPCR assay based upon the RT layout in Figure S1).
- Thaw the RT plate from Step 4.8 at RT, if frozen. Mix using a plate mixer and then centrifuge at ≥ 500 x g briefly.
- Dispense 6 µl of the appropriate cDNA to the appropriate wells of the optical reaction plate. Mix the samples in the optical reaction plate and centrifuge at ≥ 500 x g briefly.
- Run the hepatitis G qPCR assay on the undiluted and diluted field and LFSM samples before running all other qPCR assays. Use the lowest dilution of field or LFSM sample that is <1 Cq value greater than the mean hepatitis G Cq value for the enterovirus and norovirus qPCR assays.
- Set up the quantitative PCR thermal cycler software according to the manufacturer’s instructions. Identify the standard curve samples as standards and for each standard curve dilution, enter the genomic copy values shown in Table 9.
- Run the plate in the quantitative PCR thermal cycler for 10 min at 95 °C, followed by 45 cycles of 15 sec at 95 °C and 1 min at 60 °C.
- Determine whether each standard curve meets the acceptable values given in Table 10. See supplemental materials section S3 for examples.
- Calculate the overall standard deviation (StdDev) for the standard curve using Equation 2,

where Cq is the value reported for each standard curve replicate, Cq is the mean value for each set of replicates, #Cq is the total number of Cq values for all standard control replicates that have positive values (i.e., not undetermined), and #Stds is the number of standard controls that have positive values. - If the quantitative PCR thermal cycler software does not calculate the slope for each standard curve, calculate the slope using Equation 3,

where Cq is the mean value of the highest and lowest dilutions used and log GC is the log of the genomic copy value for the highest and lowest dilutions used from Table 9. - Calculate the R2 value using Equation 4.

where Cq is the mean of all Cq values and Log GC is the mean Log GC value for each replicate. - Calculate the % Efficiency using Equation 5:

- Calculate the overall standard deviation (StdDev) for the standard curve using Equation 2,
- Record the GC values calculated by the thermal cycler software for all test samples based upon standard curves that meet the criteria specified in Table 10 and the mean GC values for each sample. Rerun any samples with standard curves that do not meet the criteria in Table 10 or where any negative controls (LRB, NA Batch negative extraction control, or NTC) are positive. Reprocess any samples that fail to meet the criteria or have false positive controls during the rerun.
- Determine the GC per liter (GCL) for each test sample using Equation 6:

where GC is the mean genomic copy number from step 5.7, the factor '199' is the total dilution factor for the volume reductions that occur during the tertiary concentration, RNA extraction and RT-qPCR steps, DF is the dilution factor that compensates for inhibition, and D is the Volume of Original Water Sample Assayed in liters. See supplemental materials section S4 for an example of the calculation of GCL. - Compute the total GC of LFB and LRB samples by multiplying the mean GC value from Step 5.5 by 199 and dividing by 0.3.
Overall virus recovery was determined using paired field and LFSM ground water samples. A total of seven sample sets were analyzed using two sets collected on separate occasions from three public treatment plants, and one sample set collected from the private well. Seed levels for the LFSM samples were 3 x 106 MPN of Sabin poliovirus serotype 3 and 5 x 106 PFU of murine norovirus. Murine norovirus was used as a surrogate in the method evaluation due to a lack of human norovirus stocks with a virus concentration sufficient for LFSM samples. For groundwater samples the mean poliovirus recovery was 20%, with a standard error of 2%, 14 while mean murine norovirus recovery was 30%, with a standard error of 3% (Figure 3). The regular field groundwater sample for each LFSM had no detectable enterovirus or norovirus.
LFB and LRB samples were measured using seeded and unseeded reagent-grade water. All LRB samples were negative (data not shown). Poliovirus recovery averaged 44% with a standard error of 1% (Figure 3), while murine norovirus recovery averaged 4% with a standard error of 0.5%.
RT-qPCR requires the use of adequate standard curve reagents. Figure 4 shows a typical standard curve for enterovirus and norovirus GII. The norovirus GII curve meets the standard curve performance criteria (Table 10) with a R2 value of 0.9987, an overall standard deviation of 0.14, and 101% efficiency. Norovirus GIA and GIB curves (not shown) are nearly identical to that of norovirus GII. The enterovirus curve meets the method performance criteria with a R2 value of 0.9874, an overall standard deviation of 0.58, and 103% efficiency, but has about a hundred fold less sensitivity and thus a higher detection limit than the norovirus curves.

Figure 1. Overview of the Molecular Procedure. The molecular procedure includes additional sample concentration beyond that performed for measuring infectious virus, extraction of nucleic acids, a two-step reverse transcription (RT) protocol, and quantitative PCR (qPCR). The starting volume (S) represents a method-defined proportion of the original water sample.
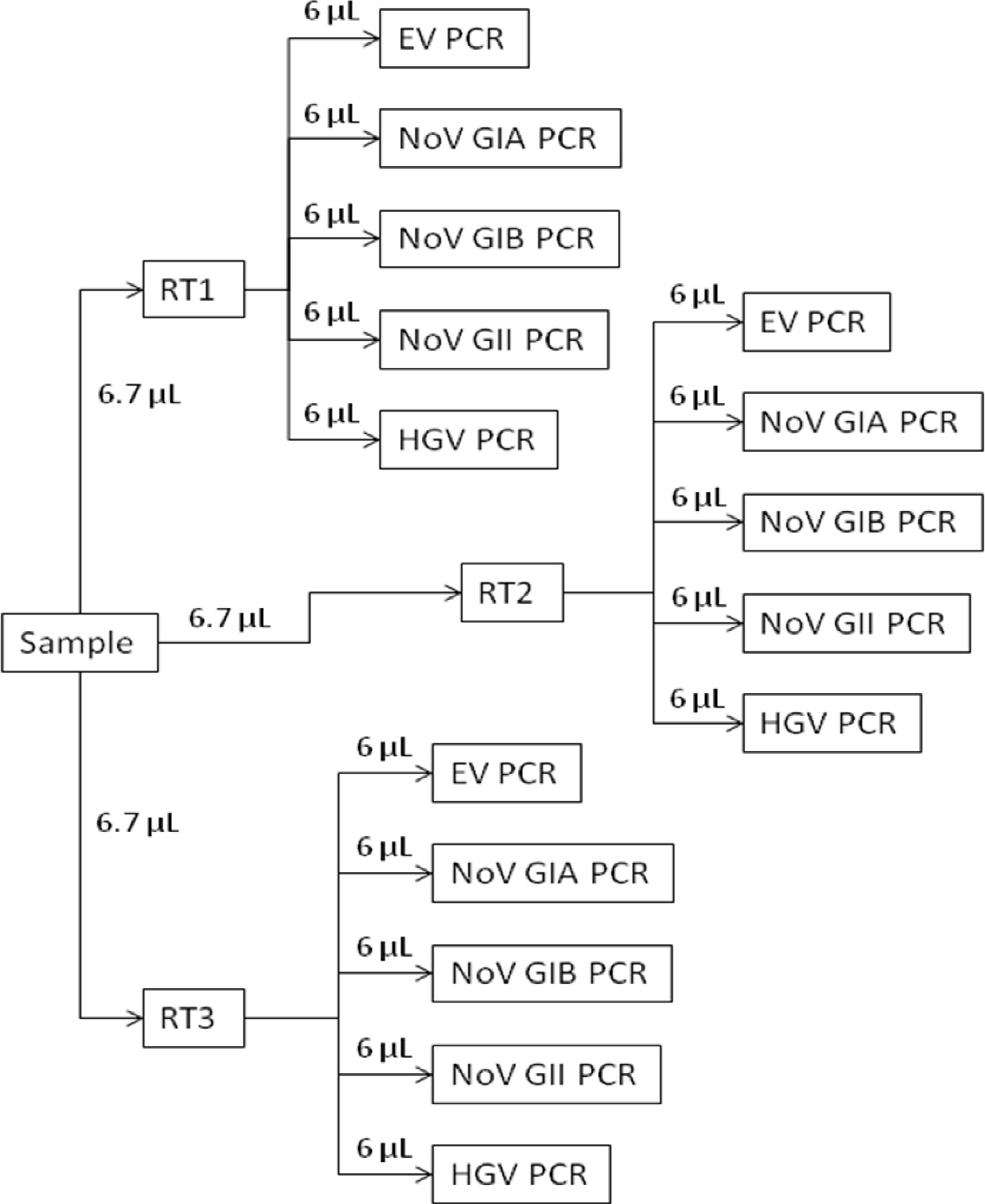
Figure 2. RT-qPCR overview schematic. Each extracted test sample RNA is reverse transcribed using triplicate assays (RT1, RT2, and RT3). The cDNA from each of the triplicate RT assays then is analyzed for specific viruses using separate enterovirus (EV PCR), norovirus genogroup I (NoV GIA PCR and NoV GIB PCR), norovirus genogroup II (NoV GII PCR), and hepatitis G (HGV PCR) assays.

Figure 3. Mean Poliovirus and Murine Norovirus Recovery (%) from Ground and Reagent-Grade Water. The mean percent recovery is shown for poliovirus from ground ( ; n = 7) and from reagent grade (
; n = 7) and from reagent grade ( ; n = 12) water and for murine norovirus from ground (
; n = 12) water and for murine norovirus from ground ( ; n = 7) and from reagent grade (
; n = 7) and from reagent grade ( ; n=12) water (1), where “n” is the number of separate water samples processed. Error bars represent standard error.
; n=12) water (1), where “n” is the number of separate water samples processed. Error bars represent standard error.

Figure 4. Enterovirus and Norovirus GII Standard Curve. Typical standard curves for enterovirus and norovirus GII are shown. The formulas giving slope and R2 values for each curve are calculated by the thermal cycler.
Supplemental File 1. Please click here to download this file.
| Virus Group | Primer/Probe Name (1) | Sequence (2) | Reference | |
| Enterovirus | ||||
| EntF (EV-L) | CCTCCGGCCCCTGAATG | 20 | ||
| EntR (EV-R) | ACCGGATGGCCAATCCAA | 20 | ||
| EntP (Ev-probe) | 6FAM-CGGAACCGACTACTTTGGGTGTCCGT-TAMRA | 21 | ||
| Norovirus GIA | ||||
| NorGIAF (JJV1F) | GCCATGTTCCGITGGATG | 22 | ||
| NorGIAR (JJV1R) | TCCTTAGACGCCATCATCAT | 22 | ||
| NorGIAP (JJV1P) | 6FAM-TGTGGACAGGAGATCGCAATCTC-TAMRA | 22 | ||
| Norovirus GIB | ||||
| NorGIBF (QNIF4) | CGCTGGATGCGNTTCCAT | 23 | ||
| NorGIBR (NV1LCR) | CCTTAGACGCCATCATCATTTAC | 23 | ||
| NorGIBP (NV1LCpr) | 6FAM-TGGACAGGAGAYCGCRATCT-TAMRA | 23 | ||
| Norovirus GII | ||||
| NorGIIF (QNIF2d) | ATGTTCAGRTGGATGAGRTTCTCWGA | 25 | ||
| NorGIIR (COG2R) | TCGACGCCATCTTCATTCACA | 25 | ||
| NorGIIP (QNIFS) | 6FAM-AGCACGTGGGAGGGCGATCG-TAMRA | 25 | ||
| Norovirus GV | ||||
| MuNoVF1 | AGATCAGCTTAAGCCCTATTCAGAAC | 14 | ||
| MuNoVR1 | CAAGCTCTCACAAGCCTTCTTAAA | 14 | ||
| MuNoVP1 | VIC-TGGCCAGGGCTTCTGT-MGB | 14 | ||
| Hepatitis G | ||||
| HepF (5'-NCR forward primer) | CGGCCAAAAGGTGGTGGATG | 19 | ||
| HepR (5'-NCR reverse primer) | CGACGAGCCTGACGTCGGG | 19 | ||
| HepP (hepatitis G TaqMan Probe | 6FAM-AGGTCCCTCTGGCGCTTGTGGCGAG-TAMRA | 1 | ||
Table 1. Primers and TaqMan Probes for Virus Detection by RT-qPCR.
(1) Method 1615 primer and probe names are the first three letters of the virus name concatenated to F, R, or P for forward, reverse, and probe. The norovirus genogroup is designated by adding GI and GII to the names. The two norovirus GI primer sets also are distinguished using A and B. Primer and probe names from the primary references are given in parentheses.
(2) The orientation of primer and probe sequences is 5’ to 3’. The following degenerate base indicators are used: N–a mixture of all four nucleotides; R–A + G; Y–T + C; W–A + T; and I–inosine.
| Ingredient | Volume per reaction (μl) (2) | Final concentration | Volume per Master Mix (μl) (3) |
| RT Master Mix 1 | |||
| Random primer | 0.8 | 10 ng/μl (c. 5.6 μM) | 84 |
| Hepatitis G Armored RNA (4) | 1 | 105 | |
| PCR grade water | 14.7 | 1543.5 | |
| Total | 16.5 | 1732.5 | |
| RT Master Mix 2 | |||
| 10x PCR Buffer II | 4 | 10 mM tris, pH 8.3, 50 mM KCl | 420 |
| 25-mM MgCl2 | 4.8 | 3 mM | 504 |
| 10-mM dNTPs | 3.2 | 0.8 mM | 336 |
| 100-mM DTT | 4 | 10 mM | 420 |
| RNase Inhibitor | 0.5 | 0.5 units/μl | 52.5 |
| SuperScript II RT | 0.3 | 1.6 units/μl | 31.5 |
| Total | 16.8 | 1764 | |
Table 2. RT Master Mix 1 and 2 (1).
(1) Prepare RT Master Mixes in a clean room, i.e., a room where molecular and microbiological procedures are not performed.
(2) The final RT assay volume is 40-µl.
(3) The volumes show are based on 105 assays. This is sufficient for a 96-well PCR plate with the extra assays added to account for losses. The amount may be scaled up or down according to the number of samples and controls that will be analyzed.
(4) Determine the amount of hepatitis G reagent to include in the RT Master Mix 1 as described in supplemental materials Step S4.
| Ingredient | Volume per reaction (μl) (2) | Final concentration | Volume per Master Mix (μl) (3) |
| 2x LightCycler 480 Probes Master Mix | 10 | Proprietary | 1050 |
| ROX reference dye (4) | 0.4 | 0.5 mM | 42 |
| PCR grade water | 1 | 105 | |
| 10 μM EntF | 0.6 | 300 nM | 63 |
| 10 μM EntR | 1.8 | 900 nM | 189 |
| 10 μM EntP | 0.2 | 100 nM | 21 |
| Total | 14 | 1470 |
Table 3. PCR Master Mix for Enterovirus (EV) Assay(1).
(1) Prepare all PCR Master Mixes in a clean room.
(2) The final qPCR assay volume is 20 µl.
(3) The volumes show are based on 105 assays. This is sufficient for a 96-well PCR plate with the extra assays added to account for losses. The amount may be scaled up or down according to the number of samples and controls that will be analyzed.
(4) Substitute PCR grade water for this reagent when using instruments that do not require it.
| Ingredient | Volume per reaction (μl) (2) | Final concentration | Volume per Master Mix (μl) (3) |
| 2x LightCycler 480 Probes Master Mix | 10 | Proprietary | 1050 |
| ROX reference dye (4) | 0.4 | 0.5 mM | 42 |
| PCR grade water | 1.4 | 147 | |
| 10 μM NorGIAF | 1 | 500 nM | 105 |
| 10 μM NorGIAR | 1 | 500 nM | 105 |
| 10 μM NorGIAP | 0.2 | 100 nM | 21 |
| Total | 14 | 1470 |
Table 4. PCR Master Mix for Norovirus GIA (NoV GIA) Assay(1).
See Table 3 for footnotes (1)–(4).
| Ingredient | Volume per Reaction (μl) (2) | Final Concentration | Volume per Master Mix (μl) (3) |
| 2x LightCycler 480 Probes Master Mix | 10 | Proprietary | 1050 |
| ROX reference dye (4) | 0.4 | 0.5 mM | 42 |
| PCR grade water | 0.3 | 31.5 | |
| 10 μM NorGIBF | 1 | 500 nM | 105 |
| 10 μM NorGIBR | 1.8 | 900 nM | 189 |
| 10 μM NorGIBP | 0.5 | 250 nM | 52.5 |
| Total | 14 | 1470 |
Table 5. PCR Master Mix for Norovirus GIB (NoV GIB) Assay(1).
See Table 3 for footnotes (1)–(4).
| Ingredient | Volume per Reaction (μl) (2) | Final Concentration | Volume per Master Mix (μl) (3) |
| 2x LightCycler 480 Probes Master Mix | 10 | Proprietary | 1050 |
| ROX reference dye (4) | 0.4 | 0.5 mM | 42 |
| PCR grade water | 0.3 | 31.5 | |
| 10 μM NorGIIF | 1 | 500 nM | 105 |
| 10 μM NorGIIR | 1.8 | 900 nM | 189 |
| 10 μM NorGIIP | 0.5 | 250 nM | 52.5 |
| Total | 14 | 1470 |
Table 6. PCR Master Mix for Norovirus GII (NoV GII) Assay(1).
See Table 3 for footnotes (1)–(4).
| Ingredient | Volume per Reaction (μl) (2) | Final Concentration | Volume per Master Mix (μl) (3) |
| 2x LightCycler 480 Probes Master Mix | 10 | Proprietary | 1050 |
| ROX reference dye (4) | 0.4 | 0.5 mM | 42 |
| PCR grade water | 0.3 | 31.5 | |
| 10 μM MuNoVF1 | 1 | 500 nM | 105 |
| 10 μM MuNoVR1 | 1.8 | 900 nM | 189 |
| 10 μM MuNoVP1 | 0.5 | 250 nM | 52.5 |
| Total | 14 | 1470 |
Table 7. PCR Master Mix for Murine Norovirus Assay(1).
See Table 3 for footnotes (1)–(4).
| Ingredient | Volume per Reaction (μl) (2) | Final Concentration | Volume per Master Mix (μl)(3) |
| 2x LightCycler 480 Probes Master Mix | 10 | Proprietary | 1050 |
| ROX reference dye (4) | 0.4 | 0.5 mM | 42 |
| PCR grade water | 1.4 | 147 | |
| 10 μM HepF | 1 | 500 nM | 105 |
| 10 μM HepR | 1 | 500 nM | 105 |
| 10 μM HepP | 0.2 | 100 nM | 21 |
| Total | 14 | 1470 |
Table 8. PCR Master Mix for Hepatitis G (HGV) Assay(1).
See Table 3 for footnotes (1)–(4).
| Standard Curve Concentration | Genomic Copies per RT-qPCR Assay (1, 2) |
| 2.5 x 108 | 502,500 |
| 2.5 x 107 | 50,250 |
| 2.5 x 106 | 5,025 |
| 2.5 x 105 | 502.5 |
| 2.5 x 104 | 50.25 |
| 2.5 x 103 | 5.025 |
Table 9. Standard Curve Genomic Copies.
(1) Identify the standard curve wells as standards and place the genomic copies per RT-qPCR assay values in the appropriate place in the thermocycler software.
(2) An acceptable standard curve will have an efficiency of 70%–110%, an R2 value >0.97, and an overall standard deviation of <0.5 for norovirus and <1.0 for enterovirus.
| Criteria | Acceptable Value | |
| Norovirus | Enterovirus | |
| Overall Standard Deviation | <0.5 | <1.0 |
| R2 | >0.97 | >0.97 |
| Efficiency | 70% to 115% | 70% to 115% |
Table 10. Standard Curve Acceptance Criteria(1).
(1) Standard curves with % Efficiencies of 70%–110% are acceptable, but values in the 90%–115% range are ideal. Values less than 90% may indicate pipetting or dilution errors.
| QA Component | Mean Recovery Range (%) | Coefficient of Variation (%) |
| Lab Reagent Blank; negative PT or PE samples | 0 | N/A(1) |
| Lab Fortified Blank; Lab Fortified Sample Matrix | 5-200 | N/A |
| Positive PT and PE samples | 15-175 | ≤130 |
Table 11. Method 1615 Performance Criteria.
(1) Not applicable.
| Media | Composition |
| 0.15 M sodium phosphate, pH 7.0–7.5 | Prepare 0.15 M sodium phosphate by dissolving 40.2 g of sodium phosphate, dibasic (Na2HPO4 · 7H2O) in a final volume of 1 L dH2O. Adjust the pH to 7.0–7.5 with HCl. Autoclave at 121 °C, 15 psi for 15 min. Store sodium phosphate solution at RT for up to 12 months. |
| 5% BSA | Prepare by dissolving 5 g of BSA in 100 ml of dH2O. Sterilize by passing the solution through a 0.2-μm sterilizing filter. |
| PBS, 0.2% BSA | Prepare by adding 4 ml of 5% BSA to 96 ml of PBS. Sterilize by passing the solution through a 0.2-μm sterilizing filter. |
| TSM III buffer | Dissolve 1.21 g Trisma base, 5.84 g NaCl, 0.203 g MgCl2, 1 ml Prionex gelatin, and 3 ml Microcide III in 950 ml reagent grade water. Adjust the pH to 7.0 and then bring the final volume to 1 L. Sterilize by passing the solution through a 0.2-μm sterilizing filter. |
| 0.525% sodium hypochlorite (NaClO) | Prepare a 0.525% NaClO solution by diluting household bleach 1:10 in dH2O. Store 0.525% NaClO solutions for up to 1 week at RT. |
| 1-M sodium thiosulfate (Na2S2O3) pentahydrate | Prepare a 1 M solution by dissolving 248.2 g of Na2S2O3 in 1 L of dH2O. Store sodium thiosulfate for up to 6 months at RT. |
Table 12. Table of Media.
