

Summary
Die in diesem Artikel beschriebene indirekte Immunofluoreszenz-Protokoll ermöglicht den Nachweis und die Lokalisierung von Proteinen in der Milchdrüse der Maus. Eine vollständige Verfahren wird gegeben, um Brustdrüsenproben herzustellen, um die Immunhistochemie die Gewebeschnitte durch Fluoreszenzmikroskopie durchführen, um Bild und um Bilder zu rekonstruieren.
Abstract
Indirekte Immunfluoreszenz dient zum Erfassen und Lokalisieren von interessierenden Proteinen in einem Gewebe. Das hier vorgestellte Protokoll beschreibt ein vollständiges und einfaches Verfahren für die Immunerkennung von Proteinen, wobei die Maus laktierenden Milchdrüse als ein Beispiel genommen. Ein Protokoll für die Herstellung von Gewebeproben, insbesondere in Bezug auf die Präparation von Maus-Brustdrüsen, Gewebefixierung und gefrorenem Gewebe Schnitte, sind detailliert. Ein Standardprotokoll, um die indirekte Immunfluoreszenz durchführen, einschließlich eines optionalen Antigen-Retrieval-Schritt wird ebenfalls vorgestellt. Die Beobachtung der markierten Gewebeschnitte als auch die Bildaufnahme und Nachbehandlungen sind ebenfalls angegeben. Dieses Verfahren ergibt eine vollständige Übersicht, aus der Sammlung von Tiergewebe auf die zelluläre Lokalisation eines Proteins. Obwohl diese allgemeine Methode kann auf andere Gewebeproben angewendet werden, sollte es zu jedem Gewebe / primären Antikörper paar studierte angepasst werden.
Introduction
Die Brustdrüse ist ein atypisches Säugetier exokrinen Organ, dessen Hauptfunktion ist die Milch für Neugeborene zu ernähren. Die Entwicklung der Brustgewebe tritt vor allem nach der Geburt und wird durch ein einzigartiges Verfahren, in dem das Epithel dringt in die umgebenden Stroma ist. Dieses Gewebe viele Veränderungen (Wachstum, Differenzierung und Regression), insbesondere während des Erwachsenenlebens gleichzeitig mit Variationen in der Fortpflanzungszustand (Figur 1). Zusätzlich zu der Gesamtmorphologie des Gewebes, wobei die Anteile der verschiedenen Zelltypen sowie deren Anordnung innerhalb des Brustdrüsen dramatisch während der Entwicklung 1-5 ändern.
Während des embryonalen Lebens leitet das Brustepithel von Brustmilchleitungen, die durch eine geringfügige Verdickung und Schichtung des Ektoderms definiert ist, zwischen den Vorder- und Hinterbeinen an jeder Seite der Mittellinie herum Embryonaltag 10,5 (E10,5) (1A ).Auf E11.5 bricht der Milchleitung in einzelne Plakoden, die symmetrisch entlang der Brustmilchleitung an reproduzierbaren Stellen positioniert werden, und die umgebende Mesenchym beginnt zu kondensieren. Die Plakoden beginnen, tiefer in die Dermis Waschbecken und die Brust Mesenchym organisiert in konzentrischen Schichten rund um die Brustknospe (E12.5-E14.5). Ab E15.5 die Brustepithel, beginnt sich zu vermehren und zu verlängern, um die primäre sprießen, die durch den Brust Mesenchym gegen die Fettpolster drückt bilden. Der primäre Spross entwickelt ein hohles Lumen mit einer Öffnung an der Haut, durch die Ausbildung des Nippels Hülle gekennzeichnet. Auf E18.5 hat die Verlängerung Kanal in die Fettpolster gewachsen und hat sich zu einem kleinen arborized Gangsystem in das Fettpolster umgeben verzweigt. Im Wesentlichen wird festgenommen und die rudimentäre Brustdrüsen bleibt morphogenetisch Ruhe bis zur Pubertät. Beim männlichen Embryo, die Aktivierung des Androgen-Rezeptoren führt zur Degeneration der Knospen, die verschwindenvon E15.5. Ab E18, hört bis zur Pubertät 6-9 Brustentwicklung.
Bei der Geburt birgt die Brustdrüse ein rudimentäres Gangsystem, der und verlängert Niederlassungen langsam (isometrische Wachstum). Am Beginn der Pubertät, kugelförmige Gebilde an den Spitzen der Leitungen namens die terminalen Ende Knospen (TEBs), sind aus einer äußeren Schicht der Kappe Zellen und einer mehrschichtigen inneren Kern von Zellen (Körperzellen) gebildet. Diese Strukturen sind hoch proliferative und infiltrieren das umliegende Stroma-Gewebe als Reaktion auf hormonelle Signale. Proliferation innerhalb der TEBS Ergebnisse in duktalen Dehnung, mit Verzweigung Morphogenese gekoppelt. Dieser Prozess führt zur Einrichtung eines Grund epithelialen arborized Netzwerk von dem Nippel (1B Pubertät) ausgeht. Bei ~ 10-12 Wochen nach der Geburt, wenn das Epithel hat die ganze Fettpolster eingefallen, Haltestellen und die Expansion der TEBS verschwinden. Duktale Entwicklung durchläuft dann dynamischen Veränderungen, dh successive Proliferation und Regression von Epithelzellen nach Ovarialzyklen 10 (1B, Erwachsenen).
Vom Beginn der Schwangerschaft durchläuft das Brustgewebe wichtigen Wachstums und morphologische Änderungen für die Stillzeit vorzubereiten. Das Brustepithel ausgiebig proliferieren und differenzieren, was zu einem hochverzweigten tubulo-alveolären Netzwerk. Begleitend Brustepithelzellen (MEC) polarisiert und in der Lage, synthetisieren und sezernieren Milchprodukte. MEC organisieren in zahlreiche alveolären Strukturen (Acini), die von kontraktilen Myoepithelzellen umgeben sind und in einem Stroma der Binde- und Fettgewebe, Blutgefäße und Nervenenden (1B, Schwangerschaft) zusammengesetzt eingebaut. Weiterhin wird die basale Seite des MEC in engem Kontakt mit der Basalmembran (extrazelluläre Matrix) und Wechselwirkungen zwischen diesen beiden Einheiten eng regulieren sowohl die Morphogenese und sekretorische Funktion der Mammary Epithel 11-13.
Alle diese Verfahren beruhen auf der Wirkung verschiedener Umweltsignale, von denen die wichtigsten sind hormones14, parakrine Faktoren und der extrazellulären Matrix. Zum Beispiel, Progesteron induziert umfangreiche Seitenverzweigung 15 und alveologenesis, dass in Kombination mit Prolaktin (PRL) 16,17 fördert und pflegt die Differenzierung der Lungenbläschen. Zusätzlich zu Steroiden und PRL18, Zytokine und Signalwege mit Entwicklung verbunden (Wnt und Notch Signalwege) sind ebenfalls in Brustlinie Engagement und Entwicklung 19-21 beteiligt. Am Ende der Schwangerschaft, beginnen die luminale MEC um eine proteinreiche Milch Kolostrum im Lumen der Alveolen bekannt herzustellen. Außerdem fungiert Progesteron auf den epithelialen Permeabilität und da die Tight Junctions sind noch offen ist Kolostrum auch in den mütterlichen Blutstrom gefunden.
Nach der Geburt, die Mammary Epithel nimmt fast die gesamte Brustdrüsenvolumens und ist hoch organisierten (Abbildung 2, Brustepithel). Milch erzeugenden Einheiten, nämlich Alveolen (Abbildung 2, Alveole) werden von einer Monoschicht von polarisiertem Brustepithelzellen sekretorischen Zellen (MESCs) gebildet ist, mit ihren apikalen Plasmamembran, die das Lumen begrenzende. Alveolen ordnen sich in die Läppchen, die in Lappen, um Kanäle, die Milch an die Außenmilieu (Abbildung 2, Lappen) Drain gruppiert sind. Laktation auftritt, dh., MESCs beginnen reichliche Mengen von Milch, vor allem durch den Rückgang der Plazentahormone (hauptsächlich Progesteron) (1B, Laktation) ausgelöst absondern. Milchprotein-Gene sind in einer definierten zeitlichen zeitlichen Verlauf von Schwangerschaft Laktation 9,22,23 hauptsächlich in Reaktion auf Hypophysen PRL bei der Säugezeit Freigabe aktiviert. Begleitend Kontakte zwischen MESCs und der extrazellulären Matrix sowohl stimulieren Milchprotein synthesis durch Signale, die über die Wechselwirkungen zwischen zellulären Integrinen und Laminin 24,25 vermittelt werden, und die Apoptose in MESCs 26,27 zu unterdrücken. Diese Signalwege führen zu der Aktivierung von Milchprotein-Genpromotoren 28 durch die Aktivierung spezifischer Transkriptionsfaktoren 29. Zell-Zell-Kontakte sind ebenfalls wichtig für einige Aspekte der Differenzierung einschließlich der Einrichtung apikalen Polarität und vektoriellen Sekretion von Milchprodukten. Tight Junctions stark nahe nach dem Beginn der Laktation und MESCs fein orchestriert die Aufnahme von Molekülen aus dem Blut sowie die Synthese, den Transport und die Sekretion von Milchbestandteilen, in Reaktion auf die Ernährungsbedürfnisse von Neugeborenen. Zum Zeitpunkt der Säugezeit erfolgt die Kontraktion der Myoepithelzellen umgibt die Alveolen als Reaktion auf Oxytocin und führt zu Milchejektion durch die Kanäle und in die Nippel. Milch ist ein komplexes Fluid, das Proteine enthält (hauptsächlichCaseine), Zucker (hauptsächlich Lactose), Lipiden und Mineralien sowie bioaktiven Molekülen, wie Immunglobulinen A (IgA), Wachstumsfaktoren und Hormone. Caseine synthetisiert, in supramolekularen Strukturen zusammengesetzt, nämlich Caseinmicellen entlang des sekretorischen Wegs transportiert werden und dann durch Exozytose frei, also die Verschmelzung von caseinhaltigen sekretorischen Vesikel (SV) mit der apikalen Plasmamembran der MESC (Abbildung 2).
Intrazelluläre Verkehr stützt sich auf Material des Austauschs zwischen membranFächern und beinhaltet Lösliche N-Ethylmaleimid-Sensitive Fusion (NSF) Befestigung Protein (SNAP) Rezeptor (SNARE) 30,31. Die SNARE-Proteine der Familie ist in vesikulären SNAREs (v-SNAREs), in der Vesikelmembran Gegenwart und Ziel SNAREs (t-SNAREs), auf die Zielmembranen lokalisiert unterteilt. Durch Komprimieren durch ihre Coiled-Coil-Domänen, V- und T-SNARE versammeln, um eine hochstabile Vierhelixbündel-Komplex zu bilden, bezeichnet als the SNARE-Komplex. Dieser Komplex fördert die Fusion von zwei gegenüberliegenden Lipiddoppelschichten durch die schrittweise bringen sie in die Nähe 30,32. Danach werden SNARE-Komplexen durch die NSF Adenosintriphosphatase dissoziiert und seine Adapter Protein SNAP und SNARE-Proteine sind zurück in ihre Herkunftsfach 33 zurückgeführt. Interessanterweise liegt überwiegend jeweils SNARE-Protein in verschiedenen zellulären Kompartimenten und SNARE Paarung kann die Spezifität der intrazellulären Fusionsereignisse 34 beizutragen. Frühere Studien deuten darauf hin, dass zumindest Synaptosomale-Associated Protein 23 (SNAP23) und Vesikel-assoziiertes Membranprotein 8 (VAMP8) und Syntaxinen (STX) -7 und -12 eine Rolle bei Casein Exozytose 35,36. Diese Proteine wurden ebenfalls in Verbindung mit der Lipidfraktion von Milch, das heißt Milchfettkügelchen (MFG) 37 gefunden. Die aktuelle vorherrschende Modell postuliert, dass zytoplasmatischen Lipidtröpfchen (CLDs) sind durch die Akkumulation von neutralen l gebildetipids (hauptsächlich Triglyceride und Sterolester) und Cholesterin aus der Ernährung der Mutter zwischen den beiden Blättchen des endoplasmatischen Retikulums (ER) -Membran 38-41 abgeleitet ist. Große CLDs ausgebildet sind, zumindest teilweise durch die Fusion von kleineren CLDs während es an die apikalen Seite MESCs wo sie als MFG freigegeben werden (1-10 & mgr; m im Durchmesser) durch Knospung transportiert, wobei durch den MESC apikalen Plasmamembran umhüllt 40-42. Stillzeit aufhört, nachdem Welpen entwöhnt und die MESCs schrittweise sterben durch Apoptose, was zur Rückbildung des Brustgewebe wieder zu einem pubertären Zustand (1B, Rückbildung).
Immunfluoreszenz (IF) ist ein in fast allen Bereichen der Biologie verwendet gemeinsamen analytischen Labormethode, sowohl in der Forschung und in der klinischen Diagnostik. IF-Techniken können auf Gewebeschnitten durchgeführt werden (Immunhistochemie, IHC) oder Zelle (Immunzytochemie, ICC) Proben. Diese leistungsstarke Ansatz beruht auf der Verwendung von Leuchtstoff-markierte Antikörper, die spezifisch an die (direkt oder indirekt) mit dem Antigen von Interesse, wodurch die Visualisierung der Verteilung im Gewebe durch Fluoreszenzmikroskopie. Fluoreszenzsignale meist hängen von der Qualität und der Konzentration der Antikörper und sachgemäße Handhabung der Probe. Eine einfache indirekte Immunfluoreszenz (IIF) -Protokoll wird präsentiert, um Milcherzeugnisse (Kaseine und MFG) und Proteinen in Milchprodukt Sekretion beteiligt erkennen (butyrophilin (btn1), SNARE-Proteine) an Gefrierschnitten von Mausbrustgewebe (Abbildung 3). Während dieses Protokoll bietet eine komplette Übersicht IHC reichen von Gewebesammlung für Bild Nachbehandlung, kritische und optionale Schritte sowie einige technische Empfehlungen werden ebenfalls vorgestellt und diskutiert.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
CD1-Mäusen wurden bei INRA (UE0907 IERP, Jouy-en-Josas, Frankreich) gezüchtet. Alle ethischen Aspekte der Tierpflege mit den einschlägigen Richtlinien und Zulassungsanforderungen, die von der Französisch Ministerium für Landwirtschaft gelegt eingehalten werden. Die verwendeten Verfahren wurden von der Ethikkommission (Vereinbarung 12/097 vom Comethea Jouy-en-Josas / Agroparistech) zugelassen.
1. Milchdrüse Probenvorbereitung
- Maus-Brustdrüse Dissektion
- Euthanize Mäusen am Tag 10 der Laktation durch Genickbruch und stecken Sie das Tier zu mit seinen Unterleib nach oben zeigt.
- Befeuchten Sie die Bauchbereich mit Ethanol und trocknen Sie es mit einem Papiertuch.
- Mit einer Pinzette, ziehen Sie die Bauchhaut zwischen den beiden Hinterbeinen und einen Einschnitt (nur durch die Haut) von etwa 1 cm mit einer scharfen Schere. Ausgehend von diesem ersten Schnitt, dann mit einer Schere, um die Haut bis zum Hals auf der Maus zu schneiden. Ziehen Sie die Haut von dem Bauchfell und pin auf der einen Seite der Haut zu einer Zeit, Stretching gelehrt.
- Sammeln Sie die Bauch- und Leistenbrustdrüsen, indem Sie sie von der Haut mit einem Tupfer und schließlich Ziehen oder Schneiden sie weg von dem Bauchfell.
Anmerkung: In diesem Schritt Carmine Färbung kann, um das Brustepithel innerhalb der gesamten Drüse 43 visualisieren geführt werden. Dieser Ansatz kann nützlich sein, um die globale Morphologie der Brustdrüse unter verschiedenen Bedingungen (physiologische Entwicklungsstadien, Krankheiten in vivo Behandlungen) analysieren. - Entfernen der Lymphknoten an der Verbindungsstelle der Bauch- und Inguinaldrüsen 44 angeordnet.
- Brustgewebefixierung
- Schneiden das Brustgewebe in 3 mm 3 Fragmente mit einem Skalpell und unmittelbar spülen diese Fragmente in einer phosphatgepufferten Salzlösung (PBS), pH 7,4, um so viel Milch wie möglich zu entfernen.
- Schnell trocknen die Fragmente auf einem PapierHandtuch und legte sie in einem kalten PBS-Lösung, die 4% Paraformaldehyd (PFA, HCHO, 32% Formaldehydlösung, VORSICHT) für 10 bis 15 Minuten auf Eis.
Hinweis: Dies ist genug Zeit, um die nachfolgende Analyse auf Brustgewebeschnitt durch IIF36 und / oder in situ Hybridisierung 45 zu ermöglichen. Jedoch, wie Aldehyd-Fixierungsmittel eindringen eher langsam in Gewebestücke (~ 1-3 mm pro Stunde), kann diese Zeit verlängert werden, um eine optimale Fixierung der Gewebeprobe zu gewährleisten. Alternativ zu beheben Geweben in vivo durch Perfusion eines betäubten Tier mit einem Fixierungslösung (nicht in der vorliegenden Studie beschrieben).
- Saccharose Infusions
- Schnell Spülen der Milch Fragmente in kaltem PBS und tauche sie in kalte PBS-Lösung, enthaltend 40% Sucrose (D-Saccharose, C12H22O11, Mr 342,3 g / mol) für 16 bis 48 h bei 4 ° C unter leichtem Schütteln.
- Gewebeeinbettung
Hinweis: Bei diesem Schritt Brust Fragmente können, um kleinere Fragmente (2-3 mm 3 zu machen erneut geschnitten werden) Oder ihre Form ändern.- Richtig beschriften Sie die Kunststoffformen und füllen ein Drittel des Volumens der Form mit OCT-Verbindung, bei RT gehalten. Legen Sie ein Fragment (2-3 mm 3) von Brustgewebe pro Form und decken Sie es mit OCT-Verbindung.
- Platzieren der Formen an der Oberfläche des flüssigen Stickstoffs (auf einer Platte aus Aluminium oder mit einem metallischen Sieb) und damit das Produkt einfriert.
Hinweis: Es muss fest und weiß vor dem Eintauchen der Form in flüssigem Stickstoff zu werden.
- Bewahren Sie die gefrorenen Proben bei -80 ° C, bis Gewebeschnitten durchgeführt.
2. Gefrorene Gewebe Sectioning
Anmerkung: einen Kryostaten, der im wesentlichen ein Mikrotom in einer Kühltruhe, ist erforderlich, um gefrorene Gewebesektionen vorzunehmen. Eine niedrigere Temperatur wird oft für Fett oder lipidreiche Gewebe wie natives Brustdrüse erforderlich.
- Stellen Sie die Temperatur des Kryostaten auf -26 ° C und warten, bis es hat Stabisiert. Pflegen Sie die gefrorenen Gewebeblock bei -26 ° C über den gesamten Schneideverfahren. Absolut zu vermeiden Auftauen des Gewebes zu jedem Zeitpunkt während des Verfahrens.
- Kühlen Sie die Rasierklinge, die Schneidunterlage, die Wankstützeinrichtung und die Bürste bis -26 ° C, indem sie in den Kryostaten für mindestens 10 min. Außerdem legen Sie eine Dia-Box im Inneren des Kryostaten, um in der Lage, Glasobjektträger zu speichern, wie die Schnitte gemacht werden können.
- Richtig beschriften Sie die Glasobjektträger, die verwendet werden, um die Gewebeschnitte bei RT sammeln und diese beibehalten werden; sonst Gewebeschnitte werden nicht an sie halten. Entfernen Sie die Probe aus der Form im Inneren des Kryostaten.
Hinweis: Die Verwendung positiv geladenen Objektträgern wird die Haftung der frische Gefrierschnitte stark begünstigt durch höhere elektrostatische Anziehung. - Decken Sie die Oberfläche eines Metallgewebescheibe mit OCT-Verbindung (bei RT gehalten) und drücken Sie die gefrorene Probe darauf. Platzieren Sie das nasse Halterung im Inneren des Kryostaten und lassen Sie es cool mindestens 15 min.
- Setzen Sie den nassen Halterung in den Plattenhalter des Kryostaten. Stellen Sie die Schnittdicke von 5-6 & mgr; m und, wenn möglich, verwenden Sie eine neue scharfe Klinge oder zumindest ändern Sie den Bereich auf der Klinge verwendet, um jede Probe, da einige Gewebe werden schnell langweilig schneiden.
- Einstellen der Position der Wankstützeinrichtung über den Rasierklingen indem Schnitte der Befestigungsmedium, bis die Scheiben gleichmßig und korrekt gebildet wird. Idealerweise wird die Wankstützeinrichtung über der Rasierklinge um etwa 1 mm treten.
- Wenn die Einstellungen korrekt sind, führen Gewebeschnitte durch Drehen des Rades in einem kontinuierlichen gleichförmigen Bewegung. Es sei denn, die Temperatur ist ideal, ein Gewebeschnitt wird, von der Natur, versuchen Sie, machen Sie es sich.
- Verwenden Sie eine Bürste, um zu packen und zu manövrieren im Abschnitt über die Bühne, um sie zu legen, wie auf dem Objektträger gewünscht. Verwenden Sie den Pinsel zu bereinigen die Reste gegebenenfalls auf der gefrorenen Gewebeblock und / oder der Rasierklinge vorhanden.
- ziehender Gewebeschnitt in Richtung des Benutzers und zur Vermeidung von Eindrücken auf den Kryostaten Bühne. Nicht auf die Gewebeabschnitt auf den Kryostaten Stadium, da es zu der Adhäsion der Gewebescheibe auf der Bühne und damit die Unfähigkeit, mit dem Glasträger wiederherzustellen führen.
- Abrufen Gewebeschnitte einzeln durch abholen an der Oberfläche eines Glasobjektträger, indem Sie sie über dem Bereich und Angel es nach unten, um den Gewebeschnitt zu berühren.
Hinweis: Die Gewebeschnitte schnell auf die warme Glas haften durch statische Anziehungskraft. Wenn mehrere Gewebeschnitte werden auf demselben Objektträger gelegt, darauf achten, sie nicht zu überlappen und um Platz zu ihnen genug, um einzeln schließen Sie sie in einem hydrophoben Kreis sein (siehe Abschnitt 3.1.1.).
3. Die indirekte Immunfluoreszenz
- Lokalisierung Abschnitten
- Verwenden Sie eine hydrophobe Sperrstift, um eine hydrophobe Kreis um Objektträger angebrachten Gewebe ziehen. Lassen Sie den Kreis trocken für ca. 1 min bei RT. Zeichnen Sie eine Linie um die tThema Abschnitte mit einem feinen schwarzen Filzstift als gut, aber auf der Seite der Glasobjektträger gegenüber der einen, wo die Gewebeschnitte sind.
Hinweis: Dieser Kreis ist wasserabweisend und Aceton und Alkohol unlöslich. Es ist daher ein Hindernis für die während der IHC Verfahren verwendeten wässrigen Lösungen und verringert das Volumen der erforderlichen Reagenzien. - Rehydrieren Gewebeschnitte von ihnen bedeckt mit einem Tropfen von ~ 250 & mgr; l PBS für ein paar Minuten bei RT. Fix Gewebeschnitte von ihnen Abdecken mit ~ 250 & mgr; l einer frisch hergestellten 3% PFA-Lösung in PBS für 10 bis 15 min.
Hinweis: Wahlweise in diesem Fall verwenden Sie einen Aldehyd Abschrecken Lösung (50 mM Ammoniumchlorid (NH 4 Cl, Herr 53,5 g / mol) in PBS oder 0,1 M Glycin (C 2 H 5 NO 2, Mr 75,07 g / mol) in PBS ), um die Fixierungsreaktion zu stoppen. Einfache und reichlich PBS waschen ist in der Regel ausreichend, um nicht umgesetzte Aldehyd zu entfernen.
- Verwenden Sie eine hydrophobe Sperrstift, um eine hydrophobe Kreis um Objektträger angebrachten Gewebe ziehen. Lassen Sie den Kreis trocken für ca. 1 min bei RT. Zeichnen Sie eine Linie um die tThema Abschnitte mit einem feinen schwarzen Filzstift als gut, aber auf der Seite der Glasobjektträger gegenüber der einen, wo die Gewebeschnitte sind.
- Antigen-Retrieval (optional)
- Legen Sie das AR-Lösung (100 mM Tris (C 4 H 11 NO 3, Herr 121,14) 5% Harnstoff (NH 2 CONH 2, Mr 60,06) pH 9,6) in einem Becherglas. Das Volumen der AR-Lösung muss ausreichen, um die Glasobjektträger in einem Glashalter platziert vollständig zu decken.
- Heizen Sie den AR-Lösung auf 95 ° C durch die Überwachung der Temperatur mit einem Thermometer und dann die Glasobjektträger auf einem geeigneten Gestell, tauchen Sie das Rack in der heißen Puffer, Abdeckung, um Verdunstung zu begrenzen und Inkubation für 10 min bei 95 ° C.
- Das Becherglas aus dem Wasserbad und lassen Sie die Glasobjektträger weitere 10 Minuten im Puffer.
- Spülen Gewebeschnitte mit PBS (~ 250 & mgr; l / Schnitt) und sättigen sie mit einer Lösungvon 3% Rinderserumalbumin (BSA, ~ 250 & mgr; l / Schnitt) in PBS für mindestens 30 min bei RT.
- Gesetzt 30-50 ul des primären Antikörpers in PBS mit 2% BSA für jedes Gewebeschnitt verdünnt.
Anmerkung: Dieser Band ist genug, um einen Tropfen, der den Gewebeschnitt vollständig bedeckt. - Stellen das gleiche Volumen des Verdünnungsmittels (2% BSA in PBS) alleine auf einem Gewebeschnitt, eine negative Kontrolle ohne primären Antikörper durchzuführen.
- Systematisch, um diesen Negativkontrolle in jedem IHC Experiment und führen für jeden sekundären Antikörper verwendet, um den Hintergrund des Experiments abzuschätzen (nicht-spezifische Markierung aufgrund des sekundären Antikörpers und / oder der Gewebeautofluoreszenz). Andere Arten von positiven oder negativen Kontrollen kann auch durchgeführt werden, um die Spezifität der Markierung zu gewährleisten (siehe Diskussion).
- Legen Sie die Glasobjektträger in einer feuchten Box O / N bei 4 ° C.
Hinweis: Primäre Antikörper waren monoklonale Maus-Anti-Cytokeratin8 (CK8, Verdünnung 1:50), monoklonalen Maus-anti-Cytokeratin 14 (CK14, Verdünnung 1:50), polyklonalen Kaninchen-Anti-Maus-Casein (# 7781, Verdünnung 1:50, großzügig von MC Neville, University of Colorado Health bereitgestellt Sciences Center, CO, USA), polyklonalen Kaninchen-anti-btn1 (1: 300 Verdünnung, großzügig von IH Mather, Abteilung Tier und Avian Sciences, University of Maryland, College Park, MD, USA), polyklonalen Kaninchen-anti-Stx6 (bereitgestellt Verdünnung 1:50, großzügig von S. Tooze, Cancer Research UK, London Research Institute, London, Großbritannien) und polyklonalen Kaninchen-anti-VAMP4 (1:50 Verdünnung) zur Verfügung gestellt. - Gründlich Gewebeschnitte mit PBS mindestens viermal für 10 min bei RT.
- Verdünne den geeigneten sekundären Antikörper (Rhodamin-konjugiertes Ziegen-Anti-Kaninchen-IgG (H + L), 1: 300 Verdünnung) in PBS, enthaltend 2% BSA, legen 30-50 ul dieser Lösung auf allen Gewebeschnitten, und Inkubation für 1,5 h bei RT.
- Seit Fluorochrome sind lichtempfindliche Moleküle, nichtaussetzen Gewebeschnitte, um Licht bis zu ihrer Analyse. Für IIF an Gewebeschnitten, begünstigen Sekundärantikörper auf einem roten Fluorophor gekoppelt seit Zellmembranen sind in der Regel eine grüne Autofluoreszenz, die mit geringer Kennzeichnungs stören können generieren. Außerdem die Wahl eines roten Fluoreszenzfarbstoff-gekoppelte sekundäre Antikörper ermöglicht die gleichzeitige Markierung von neutralen Lipiden (siehe unten).
- Gründlich die Gewebeschnitte mit PBS mindestens viermal waschen für 10 min bei RT.
- Für einige Experimente durchzuführen Postfixierung durch Inkubation der Proben mit 2% PFA in PBS für 10 min bei RT, verdünnt, um die Antigen / Antikörper-Gerüste zu stabilisieren. Allerdings kann dieser Schritt in den meisten Fällen verzichtet werden.
- Um CLDs und MFG, Farbe neutralen Lipiden durch Inkubation von Gewebeschnitten in 30-50 ul einer PBS-Lösung, enthaltend 3 ug / ml des Bodipy 493 visualisieren/ 503 10 min bei RT. Schnell Gewebeschnitte zweimal mit PBS spülen.
- Gegenfärbemittel nuklearen DNA mit 30-50 ul einer PBS-Lösung, die 3 uM DAPI (4-6-Diamidino-2-phenylindol, 5 mg / ml Stammlösung) 10 min bei RT. Die Gewebeschnitte vor der Montage der Folien für die Beobachtung zweimal mit PBS waschen.
- Entfernen PBS und setzen Sie einen Tropfen Eindeckmedium auf jedem Gewebeschnitt.
- Legen Sie eine Seite des Deckglases in einem Winkel gegen den Schieber, den Kontakt mit der Außenkante der Flüssigkeitstropfen und senken Sie dann langsam den Deckel, Luftblasen zu vermeiden. Lassen Sie die Flüssigkeit zwischen dem Objektträger und Deckglas für ein paar Minuten zu verbreiten und dann entfernen Sie das überschüssige Eindeckmedium mit einem Papiertuch.
- Dichten Sie das Deckglas auf den Objektträger mit Nagellack und speichern Gewebeschnitte bei 4 ° C auf ihre Belichtung bis Beobachtungs verhindern.
4. Fluoreszenzbeobachtung und Image Acquisition
Hinweis: Ein Fluoreszenzmikroskop mit einer Kamera durch eine Bildaufnahme-Software gesteuert wird, erforderlich, um die Ergebnisse zu beobachten IHC ausgestattet.
- Vor der Erfassung von Bildern, überprüfen Sie die Intensität der Kennzeichnung und Bewertung der Hintergrund des Experiments, indem man die negativen Kontrollen. Erwerben Sie Bilder von jedem Fluoreszenzmarkierung (Farbkanal) einzeln.
- Erwerben Sie alle Bilder, auch die der entsprechenden Kontrollen bei den gleichen Bedingungen (Belichtung und allgemeine Einstellungen) für jeden Farbkanal.
- Konventionelle Mikroskopie
- Führen Epifluoreszenzmikroskopie mit einem Mikroskop mit Standard-Filter für Fluoresceinisothiocyanat (FITC, grün), Rhodamin (rot) und DAPI (blau) Emissionen ausgestattet, × 20-63 (Öl-Immersion, NA 1.3) Ziele und eine DP50 Bildkamera ×.
- Konfokale Mikroskopie
- Führen konfokale Mikroskopie mit microsBewältigung der ZEN-Software ausgestattet, mit × 20-63 (Öl-Immersion, NA 1.4) Ziele und die 488- und 568-nm-Anregungswellenlängen des Laser ×.
5. Bild Behandlung
Hinweis: Alle Bild Nachbehandlungen werden mit dem ImageJ freie Software (http://imagej.nih.gov/ij/) durchgeführt.
- Bild überlagern (Merge)
- Öffnen Sie die in jedem Kanal, die kombiniert werden (Datei / Öffnen) erfassten Bilder. Wenn die Arbeit mit 8-Bit-Graustufenbilder, Attribut künstliche Farbe, die jedem Kanal unter Verwendung der Nachschlagtabelle (Bild / Lookup Tables).
- Generieren Sie das zusammengesetzte Bild von Graustufen- oder Farbbilder mit dem Befehl "Zusammenführen Kanäle" (Bild / Farbe / Merge-Kanäle) und dann Zuordnung einer Farbe zu jedem Kanal.
- Führen Sie Bildstapeln Überlagerung auf die gleiche Weise durch Öffnen Stapel in jedem Kanal, die kombiniert werden sollen (Datei / Öffnen) erworben und mit dem Befehl "Zusammenführen Kanäle &# 8221; (Foto / Farbe / Merge-Kanäle), um eine Farbe für jeden Kanal zuzuordnen. Speichern Sie die Verbundstapel als Bildsequenz oder als Film (siehe Abschnitt 5.4).
- Bildstapel Z Projektions
- Verwenden Sie die Z-Projektionsfunktion (Bild / Stapeln / Zproject, Max Intensity), um eine zweidimensionale Darstellung aller Bilder eines Bildstapels durch Projektion entlang der Achse senkrecht zur Bildebene (z-Achse) zu liefern. Die Option "Maximum Intensity" erstellt ein Bild, in dem jedes Pixel den Maximalwert über alle Bilder im Stapel enthält. Dies erzeugt ein Einzelbild ermöglicht die Visualisierung des gesamten durch den ganzen Stapel für einen bestimmten Kanal oder nach der Überlagerung von mehreren Kanälen beobachtet Färbung.
- Bildstapel 3D-Projektion
- Verwenden Sie die 3D-Projektion-Befehl (Bild / Stapeln / 3D-Projekt, hellste Punkt, y-Achse), eine Folge von Projektionen eines rotierenden Volumen auf eine Ebene zu erzeugen. Die visuelle Darstellung der surfaces und internen Strukturen hängt sowohl von der Projektionsverfahren (nächste Punkt, hellste Punkt (verwendet) oder Mittelwert) und die Visualisierungsparameter ausgewählt. Jeder Rahmen des Animationssequenz ist das Ergebnis von einem anderen Betrachtungswinkel vorsteht.
- Drehen Sie das erstellte 3D-Bild um jede der drei orthogonalen Achsen (hier wurde die y-Achse gewählt). Speichern Sie das als ein einzelnes Bild oder eine Filmsequenz hergestellt.
- Bildstapel, um Film Umwandlung
- Öffnen Sie ein Bild Stapel (Datei / Öffnen) und speichern Sie es als Film im AVI-Format mit dem Befehl "AVI" (Datei / Speichern unter / AVI).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Die Brustdrüse ist eine subkutane Drüse entlang der ventralen Struktur sowohl des Thorax und des Abdomens bei Nagern entfernt. Die Lage der fünf Paare von Drüsen der Maus während der Schwangerschaft ist in Abbildung 4 dargestellt. Die Morphologie der Brustdrüse drastisch ändert sich während seiner Entwicklung, was auf funktionelle Änderungen erforderlich, um für die vollständige Stillzeit (Abbildung 1B) vorzubereiten. In Jungfrau oder nulliparous Tieren, die Brustdrüse besteht aus einem wenig verzweigt Milchgangsepithel innerhalb einer dünnen Fett Stroma, die schwer zu finden sein kann, eingebettet. Vom Beginn der Schwangerschaft, der Milch Epithel wuchert und erweitert, was zu einer größeren Brustdrüsen, die leichter zu werden, um zu sehen und zu entfernen (Abbildung 4). Während der Stillzeit, der Brustgewebe dicker und erscheint weißer aufgrund der Anwesenheit von Milch. Nur Bauch- und Leistenbrustdrüsen sind, weil Hals- und Brustbrust glan gesammeltds weniger leicht durch ihre enge Verbindung mit Muskeln entfernt. Für einige Experimente Welpen aus der laktierenden weiblichen 4-6 h vor der Tötung, um die Milchsekretion von MESCs 46,47 begrenzen getrennt werden.
Bezeichnung des Brust myoepithelialen und Epithelzellen
Kontraktilen Myoepithelzellen die Alveolen umgeben kann von luminalen MESCs durch die Verwendung von Antikörpern, die gegen Marker spezifisch von jedem dieser Zelltypen exprimiert wird unterschieden werden. In der Brustdrüse, die aktuellen Marker werden Cytokeratine (CK). CKs sind eine große Familie von cytoplasmatischen Proteinen, die Zytoskelett-Intermediärfilamente (10 Nanometer im Durchmesser im Durchschnitt) in epithelialen Geweben polymerisieren. Die intermediären Filamente sind äußerst stabil und liefern eine mechanische Unterstützung für die Zellarchitektur und organisieren Gewebe durch einen Beitrag zur Zell-Zell-Adhäsion und Basalzell-Binde-Gewebe-Wechselwirkungen. Die Untermengen von CKS von Epithelzellen exprimiert hängen hauptsächlich von der Art von Epithel, dessen Entwicklungsstadium und deren Differenzierungsstatus. Ferner gilt dies auch für den malignen Pendants der Epithelien. Somit sind diese Marker sind einfach und wertvolle Werkzeuge, um Zellpopulationen in einem Gewebe unter physiologischen Bedingungen zu charakterisieren und für die Tumordiagnostik und Charakterisierung in chirurgischen Pathologie 48 verwendet.
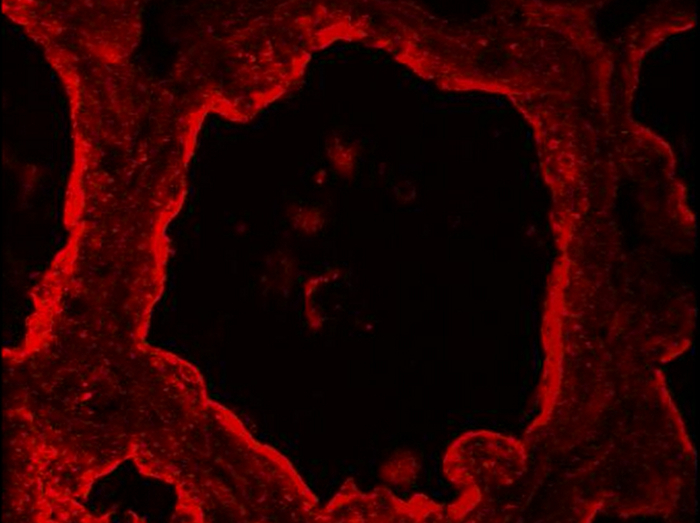
In der normalen Brustdrüse kann Myoepithelzellen und luminalen MESCs Zellen auf der Basis ihrer differentiellen Expression von CK14 und CK8, bzw. (5) unterscheiden. Diese zytoplasmatischen Marker in den Brustabschnitten laktierenden Mäusen nach PFA Fixation und AR detektiert. Bilder wurden mit einem herkömmlichen Epifluoreszenzmikroskop erworben. CK8 scheint über das Cytoplasma der luminalen MESCs (Abbildung 5, CK8) verteilt werden. Beachten Sie, dass die roten Hintergrund beobachtend für die negative Kontrolle ohne primären Antikörper (Abbildung 5, -Ig1) ist hauptsächlich auf dem Gewebeschnitt Faltung, wie durch die blaue DNA-Markierung, die mehrere Lagen von Kernen (5, -Ig1 Kerne) zeigt vorgeschlagen. CK14 wird speziell in flachen und länglichen Myoepithelzellen an der Basis der Alveolen (Abbildung 5, CK14) gelegen beobachtet. Weit verbreitet ist auch auf Myoepithelzellen identifizieren ist, alpha-Aktin der glatten Muskulatur zu erkennen (a - SMA) in diesen kontraktilen Zellen (siehe Abbildung 4 in 49) vor.
Der Nachweis von Maus-Milchprodukte
Nach der Geburt beginnen die ausdifferenzierten MESCs reichliche Mengen an Milch zu produzieren. Milchbestandteile werden durch verschiedene Wege 40,50 abgesondert. Caseinmicellen werden durch Exozytose von Golgi-abgeleitete SVs ausgeschieden, während Lipide werden als MFG von der Knospung der apikalen pl Freigabeasma Membran MESCs (Abbildung 2, Brustepithelzellen sekretorischen Zelle). Bei einigen Experimenten werden die Welpen von der weiblichen 4-6 Stunden vor dem Sammeln der Brustdrüsen, um die Milchsekretion 46,47 verlangsamen getrennt. Unter diesen Bedingungen kann der apikalen Plasmamembran der MESCs und der Inhalt des Lumens leicht beobachtet werden, was nicht der Fall bei säugenden da Alveolen kontrahiert und das Lumen geschlossen sind. Darüber hinaus verlangsamt die Sekretion ist auch wichtig, wenn das Studium Proteine in der Membranhandel beteiligt, wie SNAREs. In der Tat, Snares Zyklus zwischen Donor und Akzeptor Fächer und ihre subzelluläre Lokalisierung ist schwer zu bestimmen, da die Kennzeichnung oft diffus, wenn Membran Umsatz hoch ist, dh., Während der Säugezeit. Daher Verlangsamung der Milchsekretion, indem Sie die Welpen bietet den richtigen Bedingungen, um die intrazelluläre Lokalisation von SNAREs zu studieren, wenn die T- und v-SNAREs bevorzugt in der Spender aufzuhaltenund Akzeptorkompartiment Genauigkeit (siehe unten).
Abbildung 6 zeigt die Lokalisierung von Caseinen in der laktierenden Milchdrüse der Maus an Tag 10 der Laktation, in der Gegenwart (Figur 6 + p) oder in Abwesenheit (Abbildung 6, -p) von Jungtieren. Gewebeschnitte wurden sowohl durch konventionelle Epifluoreszenzmikroskopie (die drei Spalten auf der rechten Seite) und der konfokalen Mikroskopie (Abbildung 6, linke Spalte) beobachtet. Während der Säugezeit, erscheint Kaseine, um vor allem im apikalen Bereich angesammelt werden (Abbildung 6, + p, Pfeilspitzen). Konfokale Mikroskopie zeigt, daß Caseine sind ebenfalls vorhanden, wenn auch in geringerem Ausmaß, an der basalen Seite von MESCs in Gegenwart von Jungtieren (6, + p, Pfeile), die in konventioneller Mikroskopie (Abbildung 6, Caseine nicht klar beobachtet werden kann, Vergleichen linke und rechte Platten). In der Tat, im Weitfeld-Epifluoreszenz, die Fluoreszenz von der Probe (Hintergrund fluorescen emittiertce) durch das angeregte Volumen und verändert die Auflösung der in der Objektivbrennebene (out-of-focus Fluoreszenz) beobachteten Objekte. Dies gilt vor allem für dicke Proben (dicker als 2 & mgr; m). Konfokalmikroskopie ermöglicht, qualitativ hochwertige Bilder von Proben für Epifluoreszenz bereit zu erhalten, wie die Tiefe des Feldes gesteuert werden kann und die Hintergrundfluoreszenz von der Brennebene ausgeschlossen. Ferner wird in Gegenwart von Jungtieren (6, + p), wobei das Lumen der Alveolen sind ganz geschlossen und der apikalen Seite MESCs in Abwesenheit von Jungtieren (6, -p) besser beobachtet werden, wenn das Lumen des Alveolen geweitet aufgrund der Ansammlung von Milchprodukten. Wenn Milchsekretion verlangsamt auch Caseine werden unterhalb der apikalen Plasmamembran (6, -p, Pfeilspitzen) akkumuliert und deutlich in der basalen Seite MESCs (Abbildung 6, -p, Pfeile) beobachtet. Negativkontrollen ohne Primär ANTIBody fanden keine Kennzeichnung (Abbildung 6, -Ig1) zeigen.
Milchprodukte können leicht durch die Kombination von IHC für Kaseine und neutrales Lipid Gegenfärbung der CLDs und MFG (Abbildung 7) co-erkannt werden. Gewebeschnitte wurden als Z-Stapel durch konfokale Mikroskopie, die mit ImageJ nachbehandelt bis Z Vorsprünge oder 3D-Projektionen für jeden zu produzieren waren (Abbildung 7, Kaseine, Lipide) oder alle Farbkanäle (Abbildung 7, zusammenführen) abgebildet. Die erzeugten Bildsequenzen sind als Einzelbilder (7 und 8) oder Filme (siehe Ergänzungs Movies) gespeichert.
Obwohl einige Markierung wurde auf ihrer basalen Seite beobachtet wurden Caseine meist auf der apikalen Seite MESCs (Abbildung 7, + p) angesammelt hat, wie bereits beschrieben, wenn Weibchen wurden nicht zuvor von Jungtieren (6, + p) getrennt. CLDs werden auch vor allem im apikalen Bereich der MESCs lokalisiert, wohingegen größere Geheimnised MFG sind in dem Lumen der Alveolen vorhanden. Man beachte, daß Kasein und MFG sind leicht in das Lumen der Alveolen in der Abwesenheit von Welpen visualisiert (Abbildung 7, vergleichen + p und p). Kaseine nicht co-lokalisieren mit CLDs oder MFG in eine dieser Bedingungen, da die Überlagerung der beiden Farbkanäle nicht produzieren gelbe Markierung (Abbildung 7, fusionieren Bilder). Bildstapel Nachbehandlungen zeigen jedoch, dass Kaseine umgeben die sezerniert MFG im Lumen der Alveolen, was darauf hindeutet, dass diese Proteine mit der MFG zusammenwirken (Abbildung 7, fusionieren Bilder). Beachten Sie den Unterschied der von jedem Nachbehandlung verwendet erzeugten Bilder (Abbildung 7, vergleichen Zproj und 3D-proj für jeden Farbkanal).
Der Nachweis von butyrophilin, ein Protein Marker der MFG.
Btn1 ist eines der Hauptproteine mit MFG in Milch 51 verbunden. Diese Transmembranprotein ist mainly an der apikalen Plasmamembran der MESCs lokalisiert und wird folglich an der Oberfläche des MFG nach seiner Freigabe durch Knospung 52 gefunden. 8 zeigt, dass am Tag 10 der Laktation btn1 wird hauptsächlich an der apikalen Plasmamembran lokalisiert und in in geringerem Maße, im apikalen Bereich der MESCs. Btn1 umgibt auch die in dem Lumen des Alveolen sowie einige der apikalen CLDs (Abbildung 8, 3D proj merge, Pfeilspitzen) vorhanden MFG. Ergebnisse sind als ein einzelnes Bild aus dem erfassten Bild Z-Stapel (8, Bild) oder als 3D-Ansicht mit der 3D-Projektion Befehl ImageJ erzeugten extrahierten gezeigt, wie oben (Abbildung 8, 3D proj) beschrieben. Man beachte, dass ein einzelnes Bild kann ausreichen, um die apikale Verteilung des Proteins, sondern auch die räumliche Zuordnung der btn1 mit sezerniert MFG oder apikalen CLDs erst nach 3D-Rekonstruktion des Z-Stapel beobachtet beobachten (Figur 8 btn1 Bild zu vergleichen und 3D proj merge pictmen). Die Z-Stapel kann auch als ein Film, eine bessere räumliche Ansicht der Verteilung des Proteins geben rekonstruieren. Das Bild Z-Stapel allein btn1 erworben (Ergänzende Filme 1 und 3) oder mit den beiden anderen Farbkanäle überlagert (Merge, Ergänzender Filme 2 und 4) sind als Beispiele dargestellt. Die Z-Stapel Bild-für-Bild von oben nach unten gelesen werden (Supplementary Filme 1 und 2) oder als Drehansicht (Y-Achse) der 3D-Projektion des gesamten Bildstapel (Supplementary Filme 3 und 4 ).
Nachweis von zwei SNARE-Proteine: Stx6 und VAMP4
Wie bereits erwähnt, sind SNAREs membrangebundene Proteine, die Zyklus zwischen Donor und Akzeptor-Membranen. Es ist daher besser, Membran Umsatz mit der hohen sekretorischen Aktivität der MESCs durch die Trennung der Frauen von Jungtieren assoziiert vor dem Sammeln der Brustdrüse zu verlangsamen, wenn das Studiumdiese Proteine. Stx6 und VAMP4 haben sowohl als mit dem trans-Golgi-Netzwerk 53,54 assoziiert beschrieben worden. Jedoch können diese auch SNARE-Proteine spielen eine wichtige Rolle auf dem Niveau der anderen Zellkompartimenten wie der sekretorischen Granula (Stx6) 55,56 und den Golgi-Apparat (VAMP4) 57. Frühere Studien legen nahe, dass SNARE-Proteine spielen eine Rolle bei Casein Sekretion 35,36. Während der Stillzeit, Stx6 und VAMP4 sind im Unter Apikalbereich MESCs entfernt. Stx6 ist zwischen dem Kern und der apikalen Membran der MEC beobachtet, entsprechend dem Golgi und das Trans-Golgi-Netzwerk (9, Stx6), und ist auch vorhanden, wenn auch in geringerem Maße von caseinhaltigen SVs 36. VAMP4 ist auch in der Unter Apikalbereich MESCs lokalisiert, aber die Markierung scheint eher punktförmige und unterhalb der apikalen Plasmamembran akkumuliert (Abbildung 9, VAMP4) aufgrund seiner Verbindung mit den beiden CLDs und Casein-containing SVs 36. Negativkontrolle ohne primären Antikörper hat keinen Anlass zu jeder Etikettierung.

Abbildung 1. Maus-Brustdrüsenentwicklung während der embryonalen und adulten Lebens. (A) Die Maus Milchdrüsen beginnen um embryonalen Tag 10 (E10) aus den ektodermalen (hellblau) Milchleitungen (Rosa) zu entwickeln. Bei E11.5, bilden Plakoden symmetrisch entlang der Brustmilchleitung und dem umgebenden Mesenchym (dunkelblau) beginnt zu kondensieren. Die Plakoden invaginate Knospen (E12.5-E14.5) und zum E15.5 bilden, wobei das Brustepithel (pink), proliferieren und langgestreckt, um das primäre Spross, die durch den Brust Mesenchym Richtung der Fettpolster drückt bilden (hellgrün ). Einem hohlen Lumen bildet und öffnet den Aufstieg zum Nippel (lila) zu geben. Auf E18.5, eine Rudimenta bildet das Brustepithelry verzweigten Struktur an der Außenseite verbunden ist. 6 werden mit Genehmigung Angepasst von Macmillan Publishers Ltd: Nature Reviews Genetics, Copyright 2007. (B) Während der Pubertät, die Brustepithel (lila) tritt eine deutliche Wachstumsphase (umfangreiche Dehnung, Bifurkation und seitliche Verzweigung). Zu Beginn der Schwangerschaft, der häufig und schnell Proliferation sowie Seitenverzweigung auftreten, was zu der beträchtlichen Expansion des Brustepithel, die vollständig dringt das gesamte Brustfettpolster. Die Brustepithel eine hoch differenzierte Funktionszustand erreicht während der Stillzeit, wenn Lumen MESCs sezernieren große Mengen an Milch. Wenn der Stillzeit nicht mehr nach dem Absetzen der Brustdrüse Evolventen. MESCs durch Apoptose und Phagozytose entfernt, was zu dem Verschwinden der lobulo-alveolaren Strukturen, die von Fettgewebe ersetzt werden. Von Schema 1 von http://brisken-lab.epfl.ch/research und Kapitel 2.2 angepasst. http://tvmouse.ucdavis.edu/bcancercd/22/index.html. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
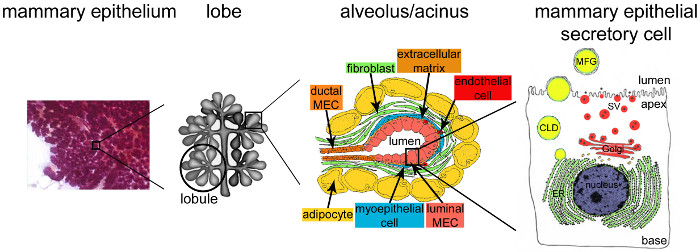
Abbildung 2. Architektur der Brustdrüse während der Stillzeit. Während der Stillzeit, die voll entwickelt und stark verzweigten Epithel (lila) Konten für die überwiegende Mehrheit der Brustgewebe. Das Epithelgewebe wird durch in einem Stroma, die verschiedenen Zelltypen (Fibroblasten, Fettzellen, Zellen der glatten Muskulatur, Blut- und Lymphgefäße sowie Nervenenden) enthält eingebettete Tubulointerstitielle Wabenstrukturen gebildet. MESCs in acinar Strukturen oder Alveolen, in Läppchen, die Lappen zusammengefügt organisiert. Jede Alveole ist ein Funktions Milch produzierenden Einheit, die zu einem hochverzweigten Netz von lobulären und interlobulären Kanälen verbunden ist, so dass die Milch wurde auf dregnen, zu der Außenseite. Jede Alveole ist von einer Monoschicht von polarisiertem MESCs begrenzt, der apikalen Seite davon grenzt an ein zentrales Lumen. Die basale Seite der MESCs steht in engem Kontakt mit einer extrazellulären Matrix und kontraktilen Myoepithelzellen. Milchprodukte werden an der apikalen Seite MESCs freigesetzt. Haupt Milch (Caseine) als Caseinmicellen (schwarze Punkte), die durch Exocytose von Golgi-abgeleitete sekretorische Vesikel (SVs) sezerniert, während Lipide werden als Milchfettkügelchen (MFG), durch Knospung der apikalen Plasmamembran der MESCs freigesetzt. CLD: zytoplasmatischen Lipidtröpfchen; ER: endoplasmatischen Retikulums; MEC: Mamma-Epithelzellen. Von Kapitel 2.2 angepasst. http://tvmouse.ucdavis.edu/bcancercd/22/index.html., Abb. 02 www.cellbiol.net/ste/alpHERCEPTIN1.php, Abb. 26-02 in 58 und 50. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
.within-page = "always"> 

Abbildung 3. Versuchsdurchführung, um die indirekte Immunfluoreszenz auf gefrorenen Schnitten von Mausbrustdrüse durchzuführen. Die Brustdrüse wird von einem CD1 weiblichen Maus am Tag 10 der Laktation gesammelt. Das Brustgewebe wird in kleine Fragmente, die mit Paraformaldehyd fixiert sind und bevor sie in OCT-Verbindung eingebettet und schockgefroren in Saccharose infundiert geschnitten. Die Brustdrüse Proben werden dann durch aufeinanderfolgende Inkubation mit primären und Fluorochrom konjugierte Sekundärantikörper in dünne Gefrierschnitte geschnitten und IIF verarbeitet sind. Nach der Montage werden die Proben mit einem Fluoreszenzmikroskop, so dass die Erfassung von Bildern, die anschließend nachbehandelt werden kann./53179/53179fig3large.jpg "Target =" _ blank "> Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.

. Abbildung 4. Anatomische Lage der Mausbrustdrüsen Links: ventralen Blick auf den Maus-Brustsystem am späten Schwangerschaftsphase. Rechts: Lokalisierung und Aspekt der Brustdrüse in der späten Schwangerschaftsphase in der Maus. Man beachte, dass während der Stillzeit, werden die Brustdrüsen dicker und weißer erscheinen aufgrund der Anwesenheit von Milch in den Alveolen. Von http://ctrgenpath.net/static/atlas/mousehistology/Windows/femaleu/mousemammgldiagram.html und http://www.pathbase.net/Necropsy_of_the_Mouse/index.php?file=Chapter_3.html angepasst. Bitte klicken Sie hier, um vergrößern Version dieser Figur.
-together.within-page = "always"> 
Figur 5. Identifizierung von luminalen Epithelzellen und Myoepithelzellen basal in der Maus-Brustdrüsen. Luminal MESCs und Myoepithelzellen durch IIF im Mausbrustdrüsen am Tag 10 der Laktation identifiziert, basierend auf ihrer Expression von CK-8 und CK-14 , beziehungsweise. Kern-DNA wurde mit DAPI (blau) gefärbt. Bilder wurden mit einem herkömmlichen Epifluoreszenzmikroskop erworben. Das zusammengesetzte Bild (merge) zeigt die Überlagerung der Kennzeichnung entsprechend Kaseine (rot) und Zellkerne (blau) auf. -Ig1, Negativkontrolle ohne primären Antikörper. Sternchen zeigen Lumen. Maßstabsbalken = 100 & mgr; m. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
E 6 "src =" / files / ftp_upload / 53179 / 53179fig6.jpg "/>
Abbildung 6. zelluläre Lokalisation von Casein in der Maus-Brustdrüse. Kaseine durch IIF in der Maus-Brustdrüse am Tag 10 der Laktation nachgewiesen. Die Brustdrüse wurde aus Weibchen in Gegenwart (+ p) oder in Abwesenheit (-p) von Jungtieren gesammelt. Die Bilder wurden mit einem herkömmlichen erworben (rechtes Bild, Caseine, Kerne und Zusammenführen) oder ein konfokales (Kaseine (rot), linke Tafel) Fluoreszenzmikroskop. In beiden Bedingungen werden Caseine (rot) in dem Scheitelbereich (Pfeilspitzen) und mehr oder weniger am basalen von MESCs (Pfeile) detektiert. Negativkontrollen ohne primären Antikörper keine Kennzeichnung (-Ig1) zeigen. Kern-DNA mit DAPI (blau) gefärbt. Das zusammengesetzte Bild (merge) zeigt die Überlagerung der Kennzeichnung entsprechend Kaseine (rot) und Zellkerne (blau) auf. Sternchen zeigen Lumen. Maßstabsbalken = 100 & mgr; m für Epifluoreszenz Bilder (rechtes Bild, Caseine, Kerne, fusionieren) und = 10 &# 181;. M für konfokale Bilder (linke Spalte) Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
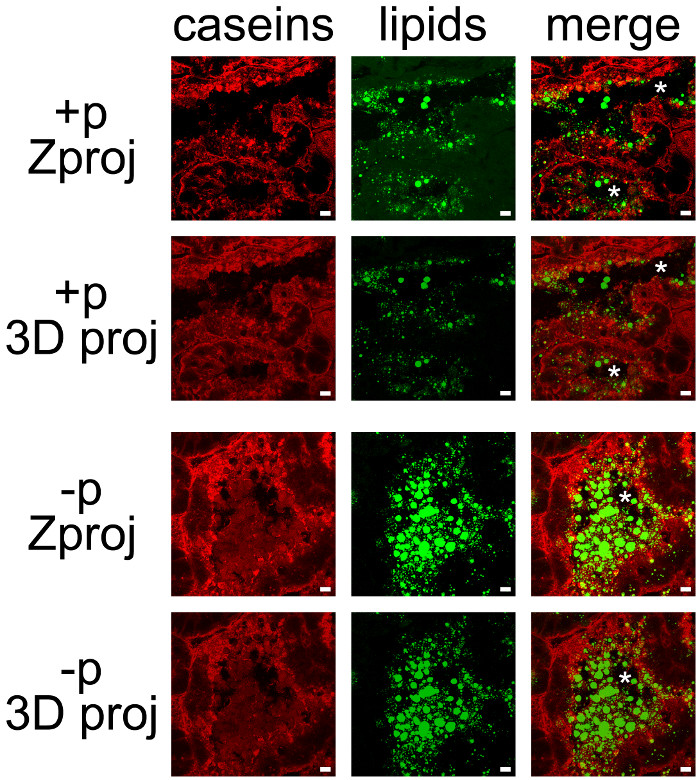
(+ P) Abbildung 7 zelluläre Lokalisation von Milchprodukten in den Maus-Brustdrüse. Kaseine (rot) durch IIF im Mausbrustdrüsen am Tag 10 der Laktation in Anwesenheit detektiert oder in Abwesenheit (-p) von Jungtieren. Neutralen Lipiden (CLDs und MFG) mit Bodipy 493/503 (grün) gegengefärbt. Die zusammengesetzten Bilder (merge) zeigen die Überlagerung der beiden Markierungen. Bilder wurden als Z-Stapel mit einem konfokalen Mikroskop aufgenommen. Z-Stapel wurden mit ImageJ nachbehandelt Z Vorsprünge (Zproj) oder 3D-Projektionen (y-Achse) (3D proj) der gesamte Stapel in jedem Kanal für beide (merge) zu erzeugen. Sternchen zeigen Lumen. Maßstabsleiste= 10 & mgr; m. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.

Abbildung 8. Zelluläre Lokalisation butyrophilin und Lipiden in der Maus-Brustdrüsen. Btn1 (rot) durch IIF in Maus-Brustdrüsen am Tag 10 der Laktation in Abwesenheit der Jungtiere nachgewiesen. Neutralen Lipiden (CLDs und MFG) und Kern-DNA mit Bodipy 493/503 (grün) und DAPI (blau) gegengefärbt sind. Bilder wurden mit einem konfokalen Mikroskop als Bild Z-Stapel erfasst. Die Ergebnisse werden als ein einzelnes Bild aus dem Bildstapel entnommen dargestellt (Bild, btn1, Lipide, Kerne und Zusammenführen) oder nach der Nachbehandlung mit ImageJ, um eine 3D-Ansicht (y-Achse) des gesamten Bildstapel zu erzeugen (3D proj, btn1 , Lipide, Kerne, zusammenführen). Die zusammengesetzten Bilder (merge) AnzeigeÜberlagerung der drei Farbkanäle. -Ig1, Negativkontrolle ohne primären Antikörper. Sternchen zeigen Lumen. Maßstabsbalken = 10 & mgr; m. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.

Abbildung 9. zelluläre Lokalisation von zwei SNARE-Proteine in der Maus-Brustdrüse. Syntaxin 6 (Stx6) und VAMP4 (V4) werden durch IIF in der Maus-Brustdrüse am Tag 10 der Laktation nachgewiesen. Bilder wurden mit einem herkömmlichen (CONV) Epifluoreszenz oder einem konfokalen (LSM) Mikroskop aufgenommen. Die zusammengesetzten Bilder (merge) zeigen die Überlagerung der für jede SNARE-Protein (rot) und für die Kern-DNA mit DAPI gegengefärbt (false grüne Farbe) bzw. beobachtet Kennzeichnung. -Ig1, Negativkontrolle ohne primären Antikörper. Sternchens zeigen Lumen. Maßstabsbalken = 10 & mgr; m für die konfokale Bilder und = 100 & mgr; m für Epifluoreszenz Bilder. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.


Tabelle 1 Immunhistochemie Anleitung zur Fehlerbehebung.

Ergänzende Film 1. Lokalisierung von butyrophilin in der Maus-Brustdrüse. Btn1 (rot) wird durch IIF in der Maus-Brustdrüse am Tag 10 der Laktation nachgewiesen. Bilder wurden mit einem Co erworben mit ImageJ nfocal Mikroskop als Z-Stapel und nachbehandelt, um einen Film zu erzeugen. Die Z-Stack wird von oben nach unten zu lesen. Bitte klicken Sie hier, um dieses Video anzusehen.

Ergänzende Film 2. Lokalisierung von butyrophilin und neutralen Lipiden in der Maus-Brustdrüse. Btn1 (rot) wird durch IIF in Maus-Brustdrüse am Tag 10 der Laktation nachgewiesen. Neutralen Lipiden (CLDs und MFG) und Kern-DNA mit Bodipy 493/503 (grün) und DAPI (blau) gegengefärbt sind. Bilder wurden mit einem konfokalen Mikroskop als Z-Stapel für jeden Farbkanal erworben und waren mit ImageJ nachbehandelt, um eine zusammengesetzte Z-Stapel, die die drei Farbkanäle überlagert zu erzeugen. Der resultierende Verbund Z-Stapel von oben nach unten gelesen.https://www.jove.com/files/ftp_upload/53179/supvid2.mp4 "target =" _ blank "> Bitte klicken Sie hier, um dieses Video anzusehen.
Ergänzende Film 3. Lokalisierung von butyrophilin in der Maus-Brustdrüse. Btn1 (rot) wird durch IIF in der Maus-Brustdrüse am Tag 10 der Laktation nachgewiesen. Die Bilder wurden mit einem konfokalen Mikroskop als Z-Stapel und nachbehandelt mit ImageJ (3D-Projektion), um eine rotierende (y-Achse) räumliche Ansicht des btn1 Kennzeichnung erzeugen erworben. Bitte klicken Sie hier, um dieses Video anzusehen.

Ergänzende Film 4. Lokalisierung von butyrophilin und neutralen Lipiden in der Maus mammary Drüse. btn1 (rot) wird durch IIF in der Maus-Brustdrüse am Tag 10 der Laktation nachgewiesen. Neutralen Lipiden (CLDs und MFG) und Kern-DNA mit Bodipy 493/503 (grün) und DAPI (blau) gegengefärbt sind. Bilder wurden mit einem konfokalen Mikroskop als Z-Stapel für jeden Farbkanal erworben und waren mit ImageJ nachbehandelt, um eine zusammengesetzte Z-Stapel, die die drei Farbkanäle überlagert zu erzeugen. ImageJ (3D-Projektion) wurde weiter verwendet werden, um eine sich drehende (y-Achse) räumliche Ansicht des zusammengesetzten Z-Stapel zu erzeugen. Bitte klicken Sie hier, um dieses Video anzusehen.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
IHC ist eine relativ einfache und direkte experimentelle Methode an Antigen in Gewebeschnitten, die in erster Linie auf spezifische Epitop-Antikörper-Wechselwirkungen abhängt lokalisieren. Obwohl eine große Anzahl von Protokollen verwendet werden, um ein Protein von IIF zu lokalisieren, ist der Kern dieser Verfahren fast immer gleich. Es gibt jedoch einige wichtige Aspekte, die einen starken Einfluss auf das Ergebnis und müssen daher für jede einzelne IHC Studie optimiert werden. Der schwierigste Aspekt dieses Ansatzes ist es, die besten experimentellen Bedingungen zu bestimmen, dh., Diejenigen, die ein starkes und spezifische Signal für das Antigen von Interesse. Die Variablen, die für experimentelle Gestaltung und Optimierung berücksichtigt werden müssen, sind: (1) die Art des Antigens (Spezies Expressionsniveaus, subzellulären Ort); (2) die Epitop-Typ (Sequenz Konformation putative posttranslationale Modifikationen); (3) Probenvorbereitung (Einbettung in Paraffin oder für Gefrierschnitte); (4) die Fixierung Method (Perfusion oder Eintauchen); (5) das Fixiermittel verwendet (Formaldehyd, Alkohol oder Aceton); (6) die Blockierungsmittel verwendet (Normalserum, BSA oder fettfreie Milch); (7) Der AR Schritt; (8) die Nachweisverfahren (direkt oder indirekt); (8) die Art der primären Antikörper (monoklonale oder polyklonale); (9), der sekundäre Antikörper (Arten und Aufkleber); (10) Gegenfärbungen (nukleare und / oder anderen Zellkompartiment Kennzeichnung); und (11) die Montagemedium (siehe Tabelle 1 für weitere Details). Die Fixierung und die Sperrschritten mindestens vor, dass die Optimierung von zusätzlichen Faktoren wie Konzentration, pH, Temperatur, Inkubationszeit und Verdünnungsmittel.
Der erste wichtige Aspekt betrifft die Herstellung von Gewebeproben, die eng mit dem Fixierungsverfahren, die wiederum Einfluss auf die Qualität der Ergebnisse verbunden ist. Beispielsweise können Gewebestücke befestigt werden oder nicht, vor dem Einbetten. Dieser Schritt kann auch von der gewählten Einbettungsverfahren, das heißt, OCT-Verbindung gegen Paraffineinbettung hängen, Die sich manchmal hängt von der primären Antikörpers verwendet. Gewebefixierung kann in vivo durch Perfusion eines betäubten Tier mit einem Fixierungslösung durchgeführt werden. Dieses Verfahren ist nützlich, um Antigene zu erhalten, wenn das Studium intakten Geweben aber nicht ausreicht, um das Gewebe von Interesse zu beheben. In diesem Fall können kleine Gewebestücke (nicht stärker als 10 mm) in die Fixierungslösung eingetaucht werden. Gefrorene Gewebe kann durch Eintauchen des Gewebes in flüssigem Stickstoff oder Isopentan hergestellt werden und das Schnellgefrieren ist hoch für die nachfolgende Detektion von post-translationalen Modifikationen wie Phosphorylierung empfohlen. , Im Gegensatz zu Paraffin eingebettetem Gewebe ist jedoch Einfrieren nicht ausreichend für langfristige Erhaltung der Gewebe aufgrund der Bildung von Eiskristallen in den Zellen, die subzellulären Morphologie verändern können. Nach dem Schneiden können gefrorene Gewebeschnitte bei -80 ° C für bis zu 1 Jahr gelagert werden. In jedem Fall ist die Herstellung der Gewebeproben ein Kompromiß zwischen der Erhaltung der Gewebe/ Zellarchitektur und die Erhaltung der Integrität Epitop.
Da es die chemische Zusammensetzung der Gewebe verändert, ist es entscheidend, um die Fixierung Bedingungen zu optimieren, um sowohl unvollständig (unter Fixierung) und übermäßige (overfixation) Fixierung zu vermeiden.
Tatsächlich kann underfixation das spezifische Signal durch die Förderung der proteolytischen Abbau bestimmte Antigene zu reduzieren. Auf der anderen Seite, können overfixation die spezifische Markierung durch Maskierung des Epitops oder Erzeugung eines starken unspezifischen Hintergrund zu ändern. Somit kann zusätzlich zu der Wahl der Fixierungslösung, andere Parameter, wie die Inkubationszeit, die Temperatur und der pH-Wert der Gewebefixierung beeinträchtigen. Obwohl PFA ist die häufigste Fixiermittel für IHC eingesetzt wird, kann es nicht als eine "universelle" Fixativ berücksichtigen. PFA induziert Protein-Protein- und Protein-Nukleinsäure-Querverbindungen und kann daher artefactually das Epitop (overfixation) ändern und dann seine reco verhindernkennung durch den primären Antikörper. Allerdings kann das Epitop durch AR-Techniken (siehe unten) entlarvt werden. PFA kann auch für den Nachweis von Antigenen geeignet bestimmte, wie es gezeigt worden ist, um die Translokation von einigen phosphorylierten Proteine von der Membran in das Cytoplasma zu induzieren. In solchen Fällen muss PFA durch geeignete alternative Fixativen wie Alkohol ersetzt werden. Anders als PFA, liegen Alkohole wie Methanol oder Ethanol keine Epitope maskieren, da sie eine Fixierung des Gewebes durch Austauschen Wassermoleküle im Gewebe. Dies kann zur Ausfällung der Proteine führen und dann zu verhindern Antikörper / Epitop Wechselwirkung aufgrund von Konformationsänderungen. Es wird allgemein angenommen, dass Alkohole nicht durchdringen und somit nicht Gewebemorphologie als auch zu erhalten, wie PFA. Aceton ist eine weitere Alternative Fixiermittel, die üblicherweise verwendet wird, bei der Arbeit mit nicht fixierten, schockgefroren Gewebeschnitten. Allerdings ist Aceton ein starkes Dehydratisierungsmittel und kann zu irreversiblen Ausfällung von Gewebeproteinen führen.
Bei einigen Antigenen kann ein zusätzlicher Schritt des AR erforderlich sein, um ein gutes Signal zu erhalten, vor allem, wenn das Fixiermittel induziert die Konformationsänderung oder ändert die elektrostatische Ladung des Epitops (Maskierung des Epitops). AR Methoden zielen darauf ab, diese Prozesse umzukehren, um die Immunoreaktivität des Epitops und die anschließende Wechselwirkung mit dem primären Antikörper wiederherzustellen. AR Verfahren auf zwei Ansätze beruhen hauptsächlich: (1) Protease-induzierte Epitopdemaskierungslösung, dh, mit Enzymen, wie Proteinase K, Trypsin oder Pepsin, die Peptide, die das Epitop zu maskieren spalten; und (2) hitzeinduzierte Epitopdemaskierung, das heißt, mit einem Mikrowellenherd, Schnellkochtöpfe, Pflanzen Dampfer, Autoklaven oder Wasserbädern. Dieser letztere Ansatz ist besonders zeit-, temperatur-, Puffer- und pH-sensitive und die optimalen Bedingungen müssen empirisch ermittelt werden (ein Beispiel ist in dem Protokoll Abschnitt). Alternativ kann die Affinität eines Antikörpers für sein Antigen verstärkt werdendurch Änderung des pH-Wertes oder der Kationenkonzentration der Antikörperverdünnung.
Eine Permeabilisierung Schritt wird manchmal benötigt, um ein gutes Signal für ein intrazelluläres Epitop in dicke Gewebeschnitte vor allem für nukleäre Antigen-Färbung zu erhalten. Dies kann auf verschiedene Weise erreicht werden, indem: (1) Alkoholen oder Aceton als Haftmittel; oder (2) Detergenzien, wie Triton, NP-40 (0,1-0,2% in PBS, 10 min), Digitonin, Saponin oder Tween 20 (0,2-0,5% für 10 bis 30 min) nach PFA Fixation. Die Wahl des Reinigungsmittels hängt jedoch von der Lokalisierung des erkannten Epitop. In der Tat sind scharfen Reinigungsmittel wie Triton-X100, die zelluläre Membranen löslich zu machen, geeignet für Kern Epitop Erkennung kann aber, um zu signalisieren Veränderung bei der Gewinnung von einigen membranProteinen führen. Die Verwendung von milder Detergentien (Saponin und Tween 20) besser geeignet sind für den Nachweis von zytoplasmatische Epitope.
Der zweite entscheidende Schritt ist die blocking unspezifische Färbung. Die Bindung eines Antikörpers an sein Target-Epitop durch zwischenmolekulare Kräfte (zB hydrophobe und ionische Wechselwirkungen, Wasserstoffbindung) geregelt. Somit können Wechselwirkungen von primären und / oder sekundären Antikörpern, die mit anderen Proteinen als ihre Zielantigene in unspezifische Färbung führen. Das schafft hohe Fluoreszenzhintergrund, die die Visualisierung des Proteins von Interesse (Low-Signal / Rausch-Verhältnis) verhindert. Blockierungsreagenzien vermindert unspezifische Wechselwirkungen ohne spezifische Antikörper / Epitop-Interaktion beeinträchtigen. Ein gemeinsames Verfahren besteht aus Inkubation Gewebeschnitte mit hitzeinaktiviertem normalem Serum oder BSA. Bei Verwendung eines normalen Serums, muss sie von der gleichen Spezies wie die Wirtstier des sekundären Antikörpers oder eines nicht verwandten Arten sein. In allen Fällen muss der ausgewählte Blockierungsreagenz auch die Verdünnungsmittel für die primären und sekundären Antikörper zugegeben werden. Weiterhin ist die Verwendung von nicht-ionischen Detergenzien, wie Triton X-100, Tween 20 oder Saponin hilft, um unspezifische Wechselwirkungen zu reduzieren.
Der dritte und wahrscheinlich wichtigste Parameter ist der primäre Antikörper die Auswahl und Optimierung. Offensichtlich ist die beste Wahl, ein hochwertiges Antikörper mit minimaler Kreuzreaktion. Als monoklonaler Antikörper weisen in der Regel hohe Affinität und Spezifität für ein einzelnes Epitop, sie sind die besten Werkzeuge, um ein bestimmtes Mitglied einer Proteinfamilie mit hoher Sequenzidentität zu unterscheiden. Jedoch kann der Antikörper / Epitop Wechselwirkung beeinträchtigt sein, wenn das Ziel-Epitop hat seine native Konformation verloren, oder wenn der Zugriff auf das Epitop durch Wechselwirkungen mit anderen Proteinen, post-translationale Modifikationen, Temperatur, pH, Fixierung und Salzkonzentration verhindert. In solchen Fällen, polyklonale Antikörper sind besser geeignet, da sie mehrere Epitope des gleichen Proteins erkennt. Außerdem sind sie oftmals stabiler als monoklonale Antikörper über einen weiten Bereich von pH und Salzkonzentration.Voruntersuchungen haben, die geeigneten Inkubationsbedingungen, dh die Arbeitslösung (monoklonaler Antikörper: 5-25 mg / ml, polyklonaler Antikörper: 1,7 bis 15 mg / ml) zu definieren, Inkubationszeit, Verdünnungsmittel und Temperatur, die sich als für die empirisch ermittelten jeder primären Antikörper. Diese Parameter müssen optimiert werden, um die Bedingungen, die die optimale Signal mit geringem Hintergrundrauschen erzeugen zu bestimmen. Die Spezifität der Markierung wird durch längere Inkubationszeiten mit niedrigeren Temperaturen begünstigt (dh., 4 ° C gegen RT).
Die Wahl, direkter oder indirekter Nachweis durchzuführen, hängt oft von der Ebene der Antigen-Expression. Beispielsweise kann ein stark exprimiertes Epitop einfach mit einem Fluorochrom konjugierte primäre Antikörper nachgewiesen werden, wodurch eine schnelle und einfache Mehrfarben-Färbung unter Vermeidung mögliche unspezifische Hintergrund aufgrund der Verwendung eines sekundären Antikörpers. Allerdings richten IF kann ein Low-Signal zu höheren Kosten erzeugt und kann sanchmal schwierig sein, sind, wenn markierte Antikörper nicht im Handel erhältlich. Umgekehrt ist IIF empfindlicher unteren exprimierten Epitope erkennen als das erzeugte Signal stärker durch die Wechselwirkung von mindestens zwei markierten sekundären Antikörper (gegen die primären Antikörper Wirtsart angehoben) mit dem primären Antikörper (Amplifikation). Ferner ist eine Vielzahl von sekundären Antikörpern gegen verschiedene Fluorophoren konjugiert sind im Handel erhältlich und relativ preiswert, und die Qualität kontrolliert. Jedoch kann dieser Ansatz Kreuzreaktivität induzieren und so erfordert sorgfältig auswählen primäre Antikörper, die nicht in der gleichen Art oder von unterschiedlichen Isotypen bei der Durchführung von Mehrfachmarkierungsexperimenten hergestellt werden. IIF auch manchmal erfordert zusätzliche Sperr Schritte und muss systematische negative Kontrollen (siehe unten) enthalten. Verstärkung kann weiter durch Verwendung eines Biotin-konjugierten sekundären Antikörpers erreicht werden und markiertem Avidin oder Streptavidin (vier Biotinmoleküle fluoreszierend pro mo gebundenenlecule). Dennoch ist diese Verstärkungsverfahren erfordert zusätzliche Schritte, um zu verhindern, nicht-spezifische Bindung und nicht für die Färbung von einigen Geweben (Leber, Niere, Herz, Gehirn, Lunge und laktierenden Brustdrüse) aufgrund der Anwesenheit von hohen Konzentrationen von endogenem Biotin angepasst werden . Jedoch kann endogenes Biotin durch Vorinkubation der Probe mit Avidin und anschließend mit Biotin vor der Inkubation mit dem primären Antikörper blockiert werden. Die Wahl der konjugierten Fluorochrome, die kleine chemische Moleküle mit der Eigenschaft, Licht zu emittieren, wenn mit Licht einer kürzeren Wellenlänge angeregt werden, hängt in erster Linie von der Art des Mikroskops Ausrüstung vorhanden.
Wenn richtig ausgelegt, um sowohl Kreuzreaktivität zwischen den Antikörpern und den Übergang zwischen den spektralen Eigenschaften der verwendeten Fluorochromen binden, erlaubt Immunofluoreszenz basierende IHC die gleichzeitige Visualisierung von mehreren zellulären Ziele.
Die letzte kritischePunkt in Bezug IHC-Experimente betrifft die positiven und negativen Kontrollen, die durchgeführt werden müssen, um die Gültigkeit der Färbung zu unterstützen, die experimentelle Artefakte erkennen und für eine genaue Interpretation der Ergebnisse. Einige Gewebe weisen eine hohe Fluoreszenzhintergrund (als Autofluoreszenz bezeichnet), die auf einer Fehlinterpretation der Ergebnisse führen könnten. So haben Gewebeschnitten unter sowohl Fluoreszenz als auch Hellfeldbeleuchtung vor dem Starten des IHC Experiment beobachtet werden. Eine negative Kontrolle, die den primären Antikörper weggelassen sind systematisch in jedem IHC Experiment, um zu gewährleisten, dass eine mögliche unspezifische Bindung des sekundären Antikörpers ist vernachlässigbar und nicht verdeckt oder ähneln den spezifischen Färbungsmuster aufgenommen werden. Eine Isotyp-Kontrolle bei der Arbeit mit einem monoklonalen primären Antikörper, der durch mit einem Nicht-Immun-Antikörper des gleichen Isotyps Ersetzen durchgeführt werden (zB IgG1, IgG2a, IgG2b IgM) mit der gleichen Konzentration. Diese Kontrolle hilft, esschätzt werden unspezifische Färbung, die durch die Wechselwirkung von Antikörper mit der Probe sein kann. (1) mit seiner löslichen Immunogen (10: 1 Molverhältnis) O / N bei 4 ° C, um die spezifische Bindung eines Antikörpers an sein Antigen zu zeigen, ein Absorptionssteuerung kann auf zwei Arten durch Vorinkubieren des primären Antikörpers erreicht werden, ; und (2) mit Zellen oder Gewebeschnitten, die das Epitop von Interesse exprimieren, aber das aus dem untersuchten Gewebe unterscheiden (siehe beispielsweise 4B in 59). In beiden Fällen sollten die daraus Abreicherung des primären Antikörpers an wenig oder keine Färbung führen. Eine andere Art der Steuerung kann mit einem irrelevanten primären Antikörpers durchgeführt werden, dh., Der gegen ein Epitop, das eine zelluläre Lokalisation, die sich von dem Epitop von Interesse ist (Kern gegen zytoplasmatische) aufweist gerichtet. Die irrelevanten Antikörper müssen vom gleichen Isotyp und Spezies wie der primäre Antikörper von Interesse sein. Zusätzliche Kontrollen für IHC-Experimente können die Verwendung von Proben aus tiss schließenues bekannt, zum Ausdruck bringen (transgene Tiere) oder nicht (Knock-out-Tiere) das Epitop von Interesse. Dies kann eine nützliche Referenz darstellen und dazu beitragen, die IHC Verfahren zu optimieren.
Eine Haupteinschränkung der IF-Techniken ist, dass sie nur auf feste (tote) angewendet werden und / oder permeabilisierten Zellen, die beide Verfahren potenziell induzieren Artefakte. Andere Grenzen dieses Ansatzes sind durch die Verwendung eines Mikroskops für die Beobachtung der Proben. Zunächst wird, wie die optische Auflösung des Epifluoreszenz und konfokalen Mikroskopen begrenzt, Lage oder Co-Location der detektierten Proteine sollten nicht überinterpretiert werden. Zweitens Photobleaching, das heißt. Ausbleichen der Fluoreszenzintensität über die Zeit, wenn sie Licht ausgesetzt wird, ist im Wesentlichen aufgrund der Erzeugung von reaktiven Sauerstoffspezies in der Probe auf der Fluoreszenzanregung, das wiederum führt zu photochemischen Zerstörung des Fluorophors. Photobleaching kann reduziert werden: a) Halten der Proben von geschütztenLicht während des IF-Experiment und Lagerung, bis ihre Beobachtung; b) unter Verwendung eines Antifade Mittel (reaktive Sauerstoffspezies Fänger) in der Montageträger; c) Verringern der Intensität und / oder Dauer des Anregungslichts; d) Erhöhung der Konzentration von Fluorophoren oder mit einer niedrigen Konzentration eines Fluorochroms mit hoher Quantenausbeute; und e) unter Verwendung von robusten Fluorophore, die weniger anfällig für Photobleaching (dh. Alexa Fluors, Seta Fluors oder DyLightFluors) sind. Drittens ist die Autofluoreszenz oft aufgrund der Anwesenheit von Flavin-Coenzyme (FAD und FMN: Absorption, 450 nm; Emission, 515 nm) und reduzierte Pyridinnucleotide (NADH: Absorption, 340 nm; Emission, 460 nm) in Säugerzellen. Weiterhin ist die Verwendung von Aldehyden, insbesondere Glutaraldehyd, um die Proben zu fixieren, können für einen hohen Autofluoreszenz führt. Dies kann durch Waschen der Proben mit 0,1% Natriumborhydrid in PBS vor der Antikörper-Inkubation und / oder durch Auswahl von Sonden und optische Filter, m minimiert werden,aximize das Fluoreszenzsignal gegenüber der Autofluoreszenz. Viertens Fluoreszenz Überlappung (auch als Durchschlag, Crossover- bzw. Übersprechen) ist vor allem auf die Emissions Spektraleigenschaften der Fluorophore, da sie oftmals eine sehr breite Bandbreiten unterschiedlich, asymmetrisch Spektralprofile, sowie verschiedene Peak-Emissionswellenlängen und die Anzahl der Maxima. Fluoreszenz Überlappung auftritt, wenn mit mehreren Fluorophoren (Farbenmarkierung) und wird durch die Emission eines Fluorophors in dem Kanal (Filter), gekennzeichnet von einem anderen Fluorophor. Durchschlagen Artefakte minimiert werden muß, da sie häufig erschweren die Interpretation, ob die Ergebnisse, insbesondere im Falle von Co-Lokalisierung oder quantitativen Untersuchungen. Als Ausgleich des Fluorophore Emission kann nur geringfügig durch die IF-Verfahren verbessert werden, Durchschlagen kann vor allem bei der Bildaufnahme durch die Verwendung eines optimierten Fluoreszenzfiltersätze und / oder Photomultiplier-Detektor, um richtig zu separ reduziert werdenaßen die spektralen Profile der Fluorophore. In dieser Hinsicht ist die konfokale Mikroskopie gut geeignet, um Mehrfarben-Abbildungs da sie die Differenzierung Fluoreszenzemissionsspektren der einzelnen Fluorophore, indem jedes Signal auf einen bestimmten Erfassungskanals. Darüber hinaus ermöglicht die konfokale Mikroskopie, die Verstärkung, Photomultiplierspannung oder Laserleistung für die einzelnen Detektionskanäle zum sequentiellen Übernahme (nur ein Fluorophor zu einem Zeitpunkt) der Kennzeichnung anpassen. Im Idealfall muss einfacher Bezeichnung Kontrollen durchgeführt werden, um den Durchschlag zu quantifizieren und schließlich entfernen Sie es rechnerisch. Eine Kontrolle ohne Sekundärantikörper (Hintergrundkontrolle) hergestellt werden können, um die Grenzen der Signalverstärkung und Offset jedes Kanals für eine optimale Bildaufnahme. Es kann auch für Post-Erfassungsverarbeitung, um eine korrekte Bildhintergrund (Autofluoreszenz) verwendet werden.
Im Ergebnis der beschriebenen Methode ist eine einfache Standard-Protokoll für die einfache realizatiAuf der Immunfärbung auf die Brustdrüse Abschnitte. Trotzdem muss die Hauptschritte einer IHC Experiment für jedes Antigen / Antikörper-Paares, um eine spezifische Färbung sichtbar zu machen und um unspezifische Hintergrundsignale zu minimieren, optimiert werden. Das beschriebene Verfahren umfasst auch einige grundlegende Methoden zur Nachbehandlung der meisten der erhaltenen Bilder. Fluoreszenz-basierte Immundetektion ist eine leistungsfähige Methode mit einem breiten Anwendungsspektrum von der zellulären Lokalisation eines Antigens zur Diagnose. Neue Fortschritte in diesen Ansätzen wird bei der zukünftigen Entwicklung von neuen Fluorophore, Erfassungsgeräte und Mikroskopietechniken bisher nicht beobachteten Details von biologischen Strukturen und Prozesse erreicht werden, die dem Bild.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren erklären, dass sie keine konkurrierenden finanziellen Interessen.
Acknowledgments
Die Autoren danken dem INRA MIMA2 Imaging Core Facility (INRA, UMR1198, Jouy-en-Josas) und an die Mitarbeiter des IERP Einheit (UE 0907, INRA, Jouy-en-Josas) für die Tierpflege und Einrichtungen. Wir möchten auch IH Mather, MC Neville und S. Tooze für die Bereitstellung sehr nützlich antibodie danken.
Materials
| Name | Company | Catalog Number | Comments |
| Dissection | |||
| Pins | |||
| Ethanol | |||
| Scissors | |||
| Scalpel and adapted blades | |||
| Ice | |||
| Towel paper | |||
| Tissue sample preparation | Company | Catalog Number | Comments/Description |
| Phosphate Buffered Saline (pH7.4) | Sigma | P-3813 | |
| Paraformaldehyde (PFA, 32% EM grade, 100 ml) | Electron Microscopy Sciences | 15714-S | personnal protection equipment required WARNING: this product will expose you to Formaldehyde Gas, a chemical known to cause cancer |
| OCT compound/Tissue Tek | Sakura | 4583 | |
| Sucrose (D-saccharose) | VWR | 27480.294 | |
| Plastic molds | Dominique Dutscher | 39910 | |
| Liquid nitrogen | |||
| Cryostat/sample support | Leica | CM3050S | |
| Razor blades (SEC35) | Thermo Scientific | 152200 | |
| Slide box | |||
| Glass slides Superfrost/Superfrost Ultra Plus | Thermo Scientific | 10143560W90/1014356190 | |
| Brushes | |||
| IHC | Company | Catalog Number | Comments/Description |
| Super Pap Pen | Sigma | Z377821-1EA | |
| Permanent marker (black) | |||
| 50 mM NH4Cl in PBS | Sigma | A-0171 | |
| 0.1 M glycine in PBS | VWR | 24403.367 | |
| Antigen Retrieval solution: Tris 100 mM 5% urea pH9.6 | |||
| Heater (up to 100°C) | |||
| Bovine Serum Albumin (BSA) | Sigma | A7906-100G | |
| Vectashield (anti-fading mounting medium) without DAPI/with DAPI | Vector Laboratories | H-1000/H-1200 | |
| Glass coverslips 22x50mm (microscopy grade) | VWR | CORN2980-225 | |
| Nail polish | |||
| Primary antibodies | Company | Catalog Number | Comments/Description |
| Rabbit anti-mouse caseins (#7781; 1:50 dilution) | generously gifted by M.C. Neville (University of Colorado Health Sciences Center, USA) |
||
| Mouse anti-cytokeratin 8 (CK8, clone 1E8, 1:50 dilution) | Biolegend (Covance) | MMS-162P | |
| Mouse anti-cytokeratin 14 (CK14, cloneLL002, 1:50 dilution) | Thermo Scientific | MS-115-P0/P1 | |
| Rabbit anti-butyrophilin (1:300 dilution) | generously gifted by I.H. Mather (Department of Animal and Avian Sciences University of Maryland College Park, USA) | ||
| Rabbit anti-Stx6 (1:50 dilution) | generously gifted S. Tooze (Cancer Research UK, London Research Institute, London, UK) |
||
| Rabbit anti-VAMP4 (1:50 dilution) | Abcam | ab3348 | |
| Secondary antibodies | Company | Catalog Number | Comments/Description |
| Rhodamine-conjugated goat anti-rabbit IgG (H + L) (1:300 dilution) | Jackson ImmunoResearch Laboratories | 111-025-003 | |
| Counterstains | Company | Catalog Number | Comments/Description |
| Bodipy 493/503 | Life Technologies (Molecular Probes) | D-3922 | |
| DAPI (4-6-diamidino-2-phenylindole) | Life Technologies (Molecular Probes) | D-1306 | |
| Observation/Image capture | Company | Catalog Number | Comments/Description |
| conventional fluorescence microscope | Leica Leitz DMRB microscope |
Standard filters for FITC, Rhodamine and DAPI emissions, ×63 oil-immersion objective (NA 1.3), DP50 imaging camera (Olympus), CellˆF software (Olympus) |
|
| Laser Scanning Microscope (confocal microscopy) | Zeiss LSM 510 microscope |
Plan-Apochromat ×63 oil-immersion objective (NA 1.4), CLSM 510 software, Confocal facilities, MIMA2 Platform, INRA Jouy-en-Josas, France, http://mima2.jouy.inra.fr/mima2) | |
| Image treatment | Company | Catalog Number | Comments/Description |
| ImageJ 1.49k software | Free software |
References
- Watson, C. J., Khaled, W. T. Mammary development in the embryo and adult: a journey of morphogenesis and commitment. Development. , 135-995 (2008).
- Smith, G. H. Experimental mammary epithelial morphogenesis in an in vivo model: evidence for distinct cellular progenitors of the ductal and lobular phenotype. Breast Cancer Res Treat. 39, 21-31 (1996).
- Van Keymeulen, A., et al. Distinct stem cells contribute to mammary gland development and maintenance. Nature. 479, 189-193 (2011).
- Oakes, S. R., Gallego-Ortega, D., Ormandy, C. J. The mammary cellular hierarchy and breast cancer. Cell Mol Life Sci. 71, 4301-4324 (2014).
- Visvader, J. E., Stingl, J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes & development. 28, 1143-1158 (2014).
- Robinson, G. W. Cooperation of signalling pathways in embryonic mammary gland development. Nat Rev Genet. 8, 963-972 (2007).
- Cowin, P., Wysolmerski, J. Molecular mechanisms guiding embryonic mammary gland development. Cold Spring Harb Perspect Biol. 2, a003251 (2010).
- Brisken, C., O'Malley, B. Hormone action in the mammary gland. Cold Spring Harb Perspect Biol. 2, a003178 (2010).
- Gjorevski, N., Nelson, C. M. Integrated morphodynamic signalling of the mammary gland. Nat Rev Mol Cell Biol. 12, 581-593 (2011).
- Daniel, C. W., Smith, G. H. The mammary gland: a model for development. J Mammary Gland Biol Neoplasia. 4, 3-8 (1999).
- Howlett, A. R., Bissell, M. J. The influence of tissue microenvironment (stroma and extracellular matrix) on the development and function of mammary epithelium. Epithelial Cell Biol. 2, 79-89 (1993).
- Edwards, G., Streuli, C. Signalling in extracellular-matrix-mediated control of epithelial cell phenotype. Biochem Soc Trans. 23, 464-468 (1995).
- Hennighausen, L., Robinson, G. W. Think globally, act locally: the making of a mouse mammary gland. Genes & development. 12, 449-455 (1998).
- Topper, Y. J., Freeman, C. S. Multiple hormone interactions in the developmental biology of the mammary gland. Physiol Rev. 60, 1049-1106 (1980).
- Brisken, C., et al. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc Natl Acad Sci U S A. 95, 5076-5081 (1998).
- Ormandy, C. J., Binart, N., Kelly, P. A. Mammary gland development in prolactin receptor knockout mice. J Mammary Gland Biol Neoplasia. 2, 355-364 (1997).
- Oakes, S. R., Rogers, R. L., Naylor, M. J., Ormandy, C. J. Prolactin regulation of mammary gland development. J Mammary Gland Biol Neoplasia. 13, 13-28 (2008).
- Hennighausen, L., Robinson, G. W. Information networks in the mammary gland. Nat Rev Mol Cell Biol. 6, 715-725 (2005).
- Kouros-Mehr, H., Werb, Z. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev Dyn. 235, 3404-3412 (2006).
- Khaled, W. T., et al. The IL-4/IL-13/Stat6 signalling pathway promotes luminal mammary epithelial cell development. Development. 134, 2739-2750 (2007).
- Asselin-Labat, M. L., et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol. 9, 201-209 (2007).
- Barcellos-Hoff, M. H., Aggeler, J., Ram, T. G., Bissell, M. J. Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development. 105, 223-235 (1989).
- Robinson, G. W., McKnight, R. A., Smith, G. H., Hennighausen, L. Mammary epithelial cells undergo secretory differentiation in cycling virgins but require pregnancy for the establishment of terminal differentiation. Development. 121, 2079-2090 (1995).
- Streuli, C. H., Bissell, M. J. Mammary epithelial cells, extracellular matrix, and gene expression. Cancer Treat Res. 53, 365-381 (1991).
- Streuli, C. H., et al. Laminin mediates tissue-specific gene expression in mammary epithelia. J Cell Biol. 129, 591-603 (1995).
- Boudreau, N., Sympson, C. J., Werb, Z., Bissell, M. J. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science. 267, 891-893 (1995).
- Pullan, S., et al. Requirement of basement membrane for the suppression of programmed cell death in mammary epithelium. J Cell Sci. 109 (Pt 3), 631-642 (1996).
- Schmidhauser, C., Bissell, M. J., Myers, C. A., Casperson, G. F. Extracellular matrix and hormones transcriptionally regulate bovine beta-casein 5' sequences in stably transfected mouse mammary cells. Proc Natl Acad Sci U S A. 87, 9118-9122 (1990).
- Streuli, C. H., et al. Stat5 as a target for regulation by extracellular matrix. J Biol Chem. 270, 21639-21644 (1995).
- Sollner, T., et al. SNAP receptors implicated in vesicle targeting and fusion. Nature. 362, 318-324 (1993).
- Jahn, R., Scheller, R. H. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 7, 631-643 (2006).
- Weber, T., et al. SNAREpins: minimal machinery for membrane fusion. Cell. 92, 759-772 (1998).
- Sollner, T., Bennett, M. K., Whiteheart, S. W., Scheller, R. H., Rothman, J. E. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 75, 409-418 (1993).
- McNew, J. A. Regulation of SNARE-mediated membrane fusion during exocytosis. Chem Rev. 108, 1669-1686 (2008).
- Wang, C. C., et al. VAMP8/endobrevin as a general vesicular SNARE for regulated exocytosis of the exocrine system. Mol Biol Cell. 18, 1056-1063 (2007).
- Chat, S., et al. Characterisation of the potential SNARE proteins relevant to milk product release by mouse mammary epithelial cells. Eur J Cell Biol. 90, 401-413 (2011).
- Reinhardt, T. A., Lippolis, J. D. Bovine milk fat globule membrane proteome. J Dairy Res. 73, 406-416 (2006).
- Robenek, H., et al. Butyrophilin controls milk fat globule secretion. Proc Natl Acad Sci U S A. 103, 10385-10390 (2006).
- Fujimoto, T., Ohsaki, Y., Cheng, J., Suzuki, M., Shinohara, Y. Lipid droplets: a classic organelle with new outfits. Histochem Cell Biol. 130, 263-279 (2008).
- Mather, I. H., Keenan, T. W. Origin and secretion of milk lipids. J Mammary Gland Biol Neoplasia. 3, 259-273 (1998).
- Heid, H. W., Keenan, T. W. Intracellular origin and secretion of milk fat globules. Eur J Cell Biol. 84, 245-258 (2005).
- McManaman, J. L., Russell, T. D., Schaack, J., Orlicky, D. J., Robenek, H. Molecular determinants of milk lipid secretion. J Mammary Gland Biol Neoplasia. 12, 259-268 (2007).
- de Assis, S., Warri, A., Cruz, M. I., Hilakivi-Clarke, L. Changes in mammary gland morphology and breast cancer risk in rats. Journal of visualized experiments : JoVE. , (2010).
- Plante, I., Stewart, M. K., Laird, D. W. Evaluation of mammary gland development and function in mouse models. Journal of visualized experiments : JoVE. , (2011).
- Galio, L., et al. MicroRNA in the ovine mammary gland during early pregnancy: spatial and temporal expression of miR-21, miR-205, and miR-200. Physiol Genomics. 45, 151-161 (2013).
- Linzell, J. L., Peaker, M. The effects of oxytocin and milk removal on milk secretion in the goat. J Physiol. 216, 717-734 (1971).
- Knight, C. H., Peaker, M., Wilde, C. J. Local control of mammary development and function. Rev Reprod. 3, 104-112 (1998).
- Walid, M. S., Osborne, T. J., Robinson, J. S. Primary brain sarcoma or metastatic carcinoma? Indian J Cancer. 46, 174-175 (2009).
- Hue-Beauvais, C., et al. Localisation of caveolin in mammary tissue depends on cell type. Cell Tissue Res. 328, 521-536 (2007).
- McManaman, J. L., Neville, M. C. Mammary physiology and milk secretion. Adv Drug Deliv Rev. 55, 629-641 (2003).
- Mather, I. H., Jack, L. J. A review of the molecular and cellular biology of butyrophilin, the major protein of bovine milk fat globule membrane. J Dairy Sci. 76, 3832-3850 (1993).
- Heid, H. W., Winter, S., Bruder, G., Keenan, T. W., Jarasch, E. D. Butyrophilin, an apical plasma membrane-associated glycoprotein characteristic of lactating mammary glands of diverse species. Biochim Biophys Acta. 728, 228-238 (1983).
- Bock, J. B., Klumperman, J., Davanger, S., Scheller, R. H. Syntaxin 6 functions in trans-Golgi network vesicle trafficking. Mol Biol Cell. 8, 1261-1271 (1997).
- Tran, T. H., Zeng, Q., Hong, W. VAMP4 cycles from the cell surface to the trans-Golgi network via sorting and recycling endosomes. J Cell Sci. 120, 1028-1041 (2007).
- Wendler, F., Page, L., Urbe, S., Tooze, S. A. Homotypic fusion of immature secretory granules during maturation requires syntaxin 6. Mol Biol Cell. 12, 1699-1709 (2001).
- Wendler, F., Tooze, S. Syntaxin 6: the promiscuous behaviour of a SNARE protein. Traffic. 2, 606-611 (2001).
- Shitara, A., et al. VAMP4 is required to maintain the ribbon structure of the Golgi apparatus. Mol Cell Biochem. 380, 11-21 (2013).
- Thibault, C., Levasseur, M. C. La reproduction chez les mammifères et l'homme. , INRA Editions. 928 (2001).
- Truchet, S., Wietzerbin, J., Debey, P. Mouse oocytes and preimplantation embryos bear the two sub-units of interferon-gamma receptor. Mol Reprod Dev. 60, 319-330 (2001).
