

Summary
Le protocole d'immunofluorescence indirecte décrite dans cet article permet la détection et la localisation de protéines dans la glande mammaire de la souris. Procédé complète est donnée pour préparer des échantillons de la glande mammaire, pour effectuer l'immunohistochimie, l'image de coupes de tissus par microscopie à fluorescence, et de reconstruire des images.
Abstract
L'immunofluorescence indirecte est utilisé pour détecter et localiser des protéines d'intérêt dans un tissu. Le protocole présenté ici décrit un procédé simple et complet pour la détection de protéines immunitaire, la souris glande mammaire en lactation étant pris comme exemple. Un protocole de préparation des échantillons de tissu, en particulier en ce qui concerne la dissection de la glande mammaire de la souris, la fixation du tissu et la coupe de tissu congelé, sont décrites en détail. Un protocole standard pour effectuer immunofluorescence indirecte, y compris une étape de récupération de l'antigène en option, est également présenté. L'observation des coupes de tissus marqués ainsi que l'acquisition de l'image et post-traitements sont également indiqués. Cette procédure donne une vue d'ensemble, à partir de la collection de tissus d'origine animale pour la localisation cellulaire d'une protéine. Bien que ce procédé général peut être appliqué à d'autres échantillons de tissu, elle doit être adaptée à chaque couple d'anticorps de tissu / primaire étudié.
Introduction
La glande mammaire est un organe exocrine mammifère atypique dont la fonction principale est de produire du lait pour nourrir les nouveau-nés. Le développement du tissu mammaire survient principalement après la naissance et se caractérise par un processus unique dans lequel l'épithélium envahit le stroma environnant. Ce tissu subit de nombreux changements (croissance, la différenciation et de régression), en particulier au cours de la vie adulte, de manière concomitante avec des variations dans l'état reproducteur (Figure 1). En plus de la morphologie globale du tissu, les proportions des différents types de cellules, ainsi que leur disposition à l'intérieur de la glande mammaire changer de façon spectaculaire au cours du développement 5.1.
Au cours de la vie embryonnaire, l'épithélium mammaire dérive de lignes de lait mammaires, qui est défini par un léger épaississement et la stratification de l'ectoderme, entre les parties avant et arrière des membres de chaque côté de la ligne médiane autour jour embryonnaire 10,5 (de E10.5) (Figure 1A ).Sur E11.5, la ligne de lait se décompose en placodes individuels, qui sont positionnés de manière symétrique le long de la ligne de lait mammaire en des emplacements reproductibles, et le mésenchyme environnant commence à se condenser. Les placodes commencent à sombrer plus profondément dans le derme et le mésenchyme mammaire organise en couches concentriques autour du bourgeon mammaire (E12.5-E14.5). Comme de E15.5, l'épithélium mammaire, commence à proliférer et allongé pour former le germe primaire qui pousse à travers le mésenchyme mammaire vers le coussinet adipeux. Le germe primaire développe un lumen creux avec une ouverture sur la peau, caractérisée par la formation de la gaine du mamelon. Sur a 18,5, le conduit allongeant a grandi dans le coussinet de graisse et a ramifié dans un petit système canalaire arborisée englobé dans le coussinet adipeux. Développement est essentiellement arrêté et la glande mammaire reste rudimentaire morphogenetically repos jusqu'à la puberté. Dans l'embryon mâle, l'activation des récepteurs androgènes conduit à la dégénérescence des bourgeons, qui disparaissentpar E15.5. Comme de E18, le développement mammaire cesse jusqu'à la puberté 6-9.
À la naissance, la glande mammaire abrite un système canalaire rudimentaire qui allonge et les branches lentement (croissance isométrique). Au début de la puberté, les structures sphériques situés aux extrémités des conduits appelés les bourgeons terminaux (TEBS), sont formées d'une couche externe de cellules de la coiffe et une âme interne multicouche de cellules (cellules de l'organisme). Ces structures sont très proliférative et infiltrent le tissu stromal environnante en réponse aux signaux hormonaux. Prolifération dans les résultats TEBS d'allongement canalaire, couplé avec la morphogenèse de ramification. Ce processus conduit à la mise en place d'un réseau arborisée épithéliale de base émanant de la tétine (figure 1B, la puberté). A ~ 10-12 semaines après la naissance, lorsque l'épithélium envahi tout le a coussinet adipeux, son expansion et arrête le TEB disparaître. Le développement de Ductal subit alors des modifications dynamiques, à savoir, successive la prolifération et la régression des cellules épithéliales selon des cycles oestraux 10 (figure 1B, adultes).
Dès le début de la gestation, le tissu mammaire subit une croissance importante et des changements morphologiques pour se préparer à l'allaitement. L'épithélium mammaire largement proliférer et se différencier, ce qui conduit à un réseau tubulo-alvéolaire hautement ramifié. En même temps, les cellules epitheliales mammaires (CEM) deviennent polarisés et capable de synthétiser et sécréter des produits laitiers. CEM organisent en de nombreuses structures alvéolaires (de acini) qui sont entourés par des cellules myoépithéliales contractiles et incorporés dans un stroma composée de tissus conjonctifs et adipeux, les vaisseaux sanguins et les terminaisons nerveuses (figure 1B, la grossesse). En outre, le côté basal de CEM est en contact étroit avec la membrane basale (de la matrice extracellulaire), et les interactions entre ces deux entités réguler étroitement à la fois fonction de la morphogenèse et sécrétoire de la mamanry épithélium 11-13.
Tous ces procédés reposent sur l'action de divers signaux de l'environnement, dont les plus importants sont les facteurs paracrines hormones14, et la matrice extracellulaire. Par exemple, la progestérone induit étendue latérale de ramification 15 et alveologenesis qui, en combinaison avec la prolactine (PRL) 16,17, favorise et entretient la différenciation des alvéoles. En plus de stéroïdes et PRL18, les cytokines et les voies de signalisation associées au développement (Wnt et Notch voies de signalisation) sont également impliqués dans l'engagement de la lignée mammaire et le développement 19-21. À la fin de la grossesse, les CEM luminal commencent à produire un lait riche en protéines connu sous le nom de colostrum dans la lumière des alvéoles. En outre, la progestérone agit sur la perméabilité épithéliale et depuis les jonctions serrées sont encore ouverts, le colostrum est également trouvé dans la circulation sanguine maternelle.
Après l'accouchement, la mammarépithélium y occupe la quasi-totalité du volume de la glande mammaire et est hautement organisée (Figure 2, l'épithélium mammaire). Unités productrices de lait, à savoir les alvéoles (Figure 2, alvéole), sont formés par une monocouche de cellules épithéliales mammaires sécrétoires polarisées (de mESCs), avec leur membrane plasmique apicale délimitant la lumière. Alvéoles se disposent en lobules qui sont regroupés en lobes reliés à des conduits qui drainent le lait au milieu extérieur (Figure 2, lobe). Allaitement se produit, ie., MESCs commencent à sécréter des quantités abondantes de lait, déclenchées principalement par la baisse des hormones placentaires (principalement la progestérone) (figure 1B, lactation). Les gènes de protéines de lait sont activés dans un cours à temps temporelle définie allant de la grossesse à l'allaitement 9,22,23, principalement en réponse à PRL pituitaire publié au moment de la tétée. Parallèlement, les contacts entre mESCs et la matrice extracellulaire de protéines de lait à la fois stimuler Synthesis par des signaux qui sont médiés par les interactions entre les intégrines cellulaires et laminine 24,25, et suppriment l'apoptose dans mESCs 26,27. Ces voies de signalisation donnent lieu à l'activation des protéines de lait de 28 promoteurs de gènes par le biais de l'activation de facteurs de transcription spécifique 29. Contacts cellule-cellule sont également importants pour certains aspects de la différenciation, y compris l'établissement de la polarité apicale et la sécrétion vectoriel de produits laitiers. Jonctions serrées rapidement fermer après le début de la lactation et mESCs orchestrer finement l'absorption de molécules dans le sang ainsi que la synthèse, le transport et la sécrétion des composants du lait, en réponse aux besoins nutritionnels des nouveau-nés. Au moment de la tétée, la contraction des cellules myoépithéliales entourant les alvéoles se produit en réponse à l'ocytocine et conduit à l'éjection du lait à travers les conduits et dans le mamelon. Le lait est un liquide complexe qui contient des protéines (surtoutcaséines), des sucres (lactose principalement), des lipides et des minéraux, ainsi que des molécules bioactives telles que des immunoglobulines A (IgA), des facteurs de croissance et hormones. Les caséines sont synthétisés, assemblés en structures supramoléculaires, à savoir les micelles de caséine, transportés le long de la voie sécrétoire, et ensuite libérés par exocytose, à savoir la fusion des vésicules de sécrétion contenant de la caséine (SV) avec la membrane plasmique apicale des MESC (figure 2).
Trafic intracellulaire repose sur les échanges de matériels entre les compartiments membranaires et implique Soluble N-éthylmaléimide sensible Fusion (NSF) Attachment Protein (SNAP) Receptor (SNARE) 30,31. La famille des protéines SNARE est subdivisé en SNAREs vésiculaires (v-SNARE), présents dans la membrane des vésicules, et des pièges cibles (t-SNARE), localisés sur les membranes cibles. Par zipping à travers leurs domaines coiled-coil, V- et t-SNARE assemblent pour former un complexe de faisceau à quatre hélices hautement stable, dénommé ecomplexe SNARE e. Ce complexe favorise la fusion de deux bicouches lipidiques opposées en les amenant progressivement à proximité immédiate 30,32. Ensuite, complexes SNARE sont dissociées par l'adénosine triphosphatase NSF et ses protéine adaptatrice protéines SNAP et le filet, sont recyclés à leur compartiment d'origine 33. Fait intéressant, chaque protéine SNARE réside principalement dans les compartiments cellulaires distincts et SNARE appariement peut contribuer à la spécificité des événements intracellulaires 34 de fusion. Des études antérieures suggèrent qu'au moins 23 protéines (SNAP23) et associée à la vésicule Membrane Protein 8 (VAMP8), et syntaxins (STX) Synaptosomal associée -7 et -12 jouent un rôle dans la caséine exocytose 35,36. Ces protéines ont également été trouvés en association avec la fraction lipidique du lait, à savoir, matière grasse du lait globules (MFG) 37. Le modèle qui prévaut courant postule que les gouttelettes lipidiques cytoplasmiques (CLD) sont formées par l'accumulation de l neutreipids (principalement des triglycérides et des esters de sterol) et cholestérol dérivé de l'alimentation maternelle entre les deux feuillets du réticulum endoplasmique (ER) de 38 à 41 membrane. Pour les CLD sont formées, au moins en partie, par la fusion de petites CLD pendant son transport vers le côté apical de mESCs où ils sont libérés en tant que MFG (1-10 um de diamètre) par bourgeonnement, étant enveloppé par la membrane plasmique apicale des MESC 40-42. Allaitement cesse après petits sont sevrés et les mESCs meurent progressivement par apoptose, conduisant à la régression du tissu mammaire à un état pubertaire (figure 1B, l'involution).
Immunofluorescence (IF) est une méthode de laboratoire analytique commun utilisé dans presque tous les aspects de la biologie, à la fois dans la recherche et en diagnostic clinique. Si les techniques peuvent être effectuées sur des coupes de tissus (immunohistochimie, IHC) ou cellulaires (immunocytochimie, CPI) échantillons. Cette approche puissante repose sur l'utilisation de fluorescent-des anticorps marqués qui se lient spécifiquement (directement ou indirectement) à l'antigène d'intérêt, permettant ainsi la visualisation de sa distribution dans les tissus par microscopie de fluorescence. Les signaux de fluorescence dépendent principalement de la qualité et la concentration des anticorps et la manipulation de l'échantillon. Un protocole simple immunofluorescence indirecte (IIF) est présenté pour détecter les produits de lait (caséines et MFG) et des protéines impliquées dans la sécrétion de lait produit (butyrophiline (BTN1), caisse claire protéines) sur des sections congelées de tissu mammaire de la souris (Figure 3). Bien que ce protocole fournit un aperçu complet IHC, allant de la collecte de tissus à l'image de post-traitement, critique et des étapes facultatives ainsi que quelques recommandations techniques sont également présentés et discutés.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Souris CD1 ont été élevés à l'INRA (UE0907 IERP, Jouy-en-Josas, France). Tous les aspects éthiques du soin des animaux conformes aux lignes directrices et aux exigences d'octroi de licences prévues par le ministère français de l'Agriculture. Les procédures utilisées ont été approuvées par le comité d'éthique local (accord 12/097 du Comethea Jouy-en-Josas / AgroParisTech).
1. glande mammaire Préparation d'échantillons
- Souris glande mammaire dissection
- Euthanasier les souris au jour 10 de la lactation par dislocation cervicale et épingler l'animal vers le bas avec son abdomen vers le haut.
- Mouiller la zone ventrale avec de l'éthanol et le sécher avec une serviette en papier.
- En utilisant des pinces, tirez la peau abdominale entre les deux pattes de derrière et faire une incision (à travers la peau seulement) d'environ 1 cm avec des ciseaux pointus. A partir de cette première incision, puis utilisez des ciseaux pour couper la peau jusqu'au cou sur la souris. Tirez la peau loin du péritoine et pin sur un côté de la peau à la fois, l'étirant enseigné.
- Recueillir les abdominaux et les glandes mammaires inguinales en les poussant loin de la peau avec un tampon et enfin en tirant ou en les coupant du péritoine.
Remarque: À cette étape Carmine coloration peut être effectuée afin de visualiser l'épithélium mammaire dans le totalité de la glande 43. Cette approche peut être utile d'analyser la morphologie globale de la glande mammaire dans diverses conditions (traitements physiologiques stades de développement, les maladies, in vivo). - Retirer le ganglion situé à la jonction de l'abdomen et les ganglions inguinaux 44.
- Fixation du tissu mammaire
- Coupez le tissu mammaire en 3 mm 3 fragments avec un scalpel et rincez immédiatement ces fragments dans un tampon phosphate salin (PBS), pH 7,4, afin d'éliminer autant de lait que possible.
- Sécher rapidement les fragments sur un papierserviette et mettez-les dans une solution froide PBS contenant 4% de paraformaldéhyde (PFA, HCHO, 32% de solution de formaldéhyde, ATTENTION) pour 10 à 15 min sur la glace.
Note: Ceci est assez de temps pour permettre une analyse ultérieure sur des tranches de tissu mammaire par IIF36 et / ou hybridation in situ 45. Cependant, comme fixateurs aldéhyde pénètrent lentement dans les morceaux de tissus (~ 1-3 mm par heure), ce délai peut être prolongé pour assurer une fixation optimale de l'échantillon de tissu. Alternativement, fixer des tissus in vivo par perfusion un animal anesthésié avec une solution de fixation (non détaillé dans la présente étude).
- Perfusion de saccharose
- Rincer rapidement les fragments mammaires dans du PBS froid et les plonger dans une solution PBS froid contenant 40% de saccharose (D-saccharose, C12H22O11, M. 342,3 g / mol) pendant 16 48 h à 4 ° C sous agitation douce.
- Intégration tissulaire
Note: À cette étape, les fragments mammaires peut être re-découpée afin de faire fragments plus petits (2-3 mm 3) Ou pour régler leur forme.- Étiqueter correctement les moules en plastique et de remplir un tiers du volume du moule avec du composé OCT, maintenu à la température ambiante. Placez un fragment (2-3 mm 3) du tissu mammaire par moule et couvrir avec le composé octobre
- Placer les moules sur la surface de l'azote liquide (sur une feuille d'aluminium ou en utilisant un tamis métallique) et laisser le produit à congeler.
Remarque: Il doit devenir solide et blanc avant d'immerger le moule dans de l'azote liquide.
- Stocker les échantillons congelés à -80 ° C jusqu'à ce que les coupes de tissus sont exécutées.
2. sectionnement de tissu congelé
Remarque: un cryostat, qui est essentiellement un microtome à l'intérieur d'un congélateur, qui est nécessaire pour faire des coupes de tissus congelés. Une température plus basse est souvent nécessaire pour les tissus adipeux ou riches en lipides tels que la glande mammaire vierge.
- Réglez la température du cryostat à -26 ° C et attendre jusqu'à ce qu'il ait Stabilisée. Maintenir le bloc de tissu congelé à -26 ° C pendant toute la procédure de découpe entière. Absolument éviter la décongélation du tissu à tout moment pendant la procédure.
- On refroidit la lame de rasoir, le support de coupe, le dispositif anti-roulis et la brosse à -26 ° C en les plaçant dans le cryostat pendant au moins 10 min. Également placer une boîte de glissement à l'intérieur du cryostat afin d'être en mesure de stocker les lames, les sections sont faites.
- Étiqueter correctement les lames de verre qui seront utilisés pour recueillir les coupes de tissus et de les maintenir à température ambiante; autrement coupes de tissus ne seront pas les respecter. Retirer l'échantillon du moule à l'intérieur du cryostat.
Note: Utilisation des lames de verre chargées positivement vont grandement favoriser l'adhérence des coupes de tissus congelés frais en raison de l'attraction électrostatique élevé. - Couvrez la surface d'un disque de tissu métallique avec le composé OCT (maintenu à RT) et pousser l'échantillon congelé sur elle. Placer le montage humide à l'intérieur du cryostat et laissez-cool pendant au moins 15 min.
- Placez le montage humide dans le support de disque du cryostat. Réglez l'épaisseur de coupe à 5-6 pm et, si possible, utiliser une nouvelle lame tranchante ou au moins modifier la zone sur la lame utilisée pour couper chaque échantillon, car il certains tissus suffit à émousser rapidement.
- Ajustez la position du dispositif anti-roulis sur la lame de rasoir en faisant des coupures du milieu de montage jusqu'à ce que les tranches sont formées uniformément et correctement. Idéalement, le dispositif anti-roulis va enjamber la lame de rasoir par environ 1 mm.
- Une fois que les paramètres sont corrects, effectuer des coupes de tissus en tournant la roue dans un mouvement continu et uniforme. Sauf si la température est idéale, une coupe de tissu sera, par nature, essayer de se recroqueviller.
- Utilisez une brosse à saisir et manœuvrer la section sur la scène afin de le placer comme désiré sur la lame de verre. Utilisez la brosse pour nettoyer les restes éventuellement présents sur le bloc de tissu congelé et / ou la lame de rasoir.
- tirerla coupe de tissu vers l'utilisateur et éviter appuyant sur la scène du cryostat. Evitez d'appuyer sur la coupe de tissu sur la scène de cryostat elle peut conduire à l'adhésion de la tranche de tissu sur la scène et donc l'incapacité de le récupérer avec la lame de verre.
- Récupérer des coupes de tissus, un par un en les ramassant à la surface d'une lame de verre, en le tenant au-dessus de la section et l'inclinant vers le bas pour toucher la coupe de tissu.
Remarque: Les coupes de tissus adhèrent rapidement au verre chaud due à l'attraction statique. Si plusieurs sections de tissus sont placés sur la même lame, veiller à ne pas se chevaucher et de les espacer suffisamment pour être en mesure de les joindre individuellement dans un cercle hydrophobe (voir section 3.1.1.).
3. immunofluorescence indirecte
- Localisation sections
- Utilisez un stylo de barrière hydrophobe pour dessiner un cercle autour du tissu hydrophobe coulissant monté. Supposons que le cercle à sec pendant environ 1 min à température ambiante. Tracez une ligne autour de la tsections d'émission avec un marqueur permanent noir bien ainsi, mais sur le côté de la lame de verre opposée à celle où les coupes de tissus sont.
Remarque: Ce cercle est hydrofuge et acétone et insoluble dans l'alcool. Elle fournit donc un obstacle à des solutions aqueuses utilisées pendant la procédure IHC et réduit le volume des réactifs nécessaires. - Réhydrater les coupes de tissus en les recouvrant d'une baisse de ~ 250 pl de PBS pendant quelques minutes à la température ambiante. Correction des coupes de tissus en les recouvrant d'~ 250 ul d'une solution à 3% de PFA fraîchement préparée dans du PBS pendant 10 à 15 min.
Remarque: En option dans ce cas, utiliser une solution d'aldéhyde de trempe (chlorure d'ammonium 50 mM (NH 4 Cl, M. 53,5 g / mol) dans du PBS ou de glycine 0,1 M (C 2 H 5 NO 2, M. 75.07 g / mol) dans du PBS ) pour arrêter la réaction de fixation. Simple et abondante lavage PBS est généralement suffisante pour éliminer l'aldéhyde qui n'a pas réagi.
- Utilisez un stylo de barrière hydrophobe pour dessiner un cercle autour du tissu hydrophobe coulissant monté. Supposons que le cercle à sec pendant environ 1 min à température ambiante. Tracez une ligne autour de la tsections d'émission avec un marqueur permanent noir bien ainsi, mais sur le côté de la lame de verre opposée à celle où les coupes de tissus sont.
- La récupération de l'antigène (facultatif)
- Placer la solution de AR (Tris 100 mM (C 4 H 11 NO 3, MM 121,14) 5% d'urée (NH 2 CONH 2, M. 60,06) pH 9,6) dans un bêcher. Le volume de solution d'AR doit être suffisante pour recouvrir complètement les lames de verre placées dans un support en verre.
- Préchauffer la solution de AR à 95 ° C en contrôlant la température avec un thermomètre, puis placer les lames de verre sur un support approprié, immerger la crémaillère dans le tampon à chaud, couverture pour limiter l'évaporation et incuber pendant 10 min à 95 ° C.
- Retirer le bécher du bain d'eau et de laisser les lames de verre pendant 10 minutes dans le tampon.
- Rincer les sections de tissus avec du PBS (~ 250 pi / section) et les saturer avec une solutionde 3% d'albumine de sérum bovin (BSA, ~ 250 ul / section) dans du PBS pendant au moins 30 min à température ambiante.
- Mettre 30 à 50 pi de l'anticorps primaire dilué dans du PBS contenant 2% de BSA dans chaque section de tissu.
Remarque: Ce volume est suffisant pour former une goutte qui recouvre entièrement la section de tissu. - Placer le même volume de diluant (2% de BSA dans du PBS) seul sur une coupe de tissu pour effectuer un contrôle négatif sans anticorps primaire.
- Inclure systématiquement ce témoin négatif dans chaque expérience IHC et effectuer pour chaque anticorps secondaire utilisé pour estimer le fond de l'expérience (marquage non spécifique due à l'anticorps secondaire et / ou le tissu auto-fluorescence). D'autres types de contrôles positifs ou négatifs peuvent également être effectuées pour garantir la spécificité de l'étiquetage (voir la discussion).
- Placer les lames de verre dans une boîte humidifiée O / N à 4 ° C.
Remarque: Les anticorps primaires utilisés étaient monoclonal de souris anti-cytokératine8 (CK8, dilution 1:50), anticorps monoclonal de souris anti-cytokératine 14 (CK14, dilution 1:50), la caséine polyclonaux de lapin anti-souris (# 7781, dilution 1:50, généreusement offert par MC Neville, Université du Colorado Health Sciences Center, CO, USA), polyclonal de lapin anti-BTN1 (dilution 1: 300, généreusement fournis par IH Mather, Département des animaux et aviaires Sciences, Université du Maryland, College Park, MD, USA), polyclonal de lapin anti-STX6 ( dilution 1:50, généreusement offert par S. Tooze, Cancer Research UK, London Research Institute, Londres, Royaume-Uni) et polyclonaux de lapin anti-VAMP4 (dilution 1:50). - Laver soigneusement les coupes de tissus avec du PBS au moins quatre fois pendant 10 min à température ambiante.
- Diluer l'anticorps approprié secondaire (chèvre rhodamine conjugué anti-lapin IgG (H + L), dilution 1: 300) dans du PBS contenant 2% de BSA, placer 30 à 50 pl de cette solution sur toutes les sections de tissu, et incuber pendant 1,5 h à la température ambiante.
- Depuis fluorochromes sont des molécules sensibles à la lumière, ne pasexposer des coupes de tissus à la lumière jusqu'à ce que leur analyse. Par IIF sur des coupes de tissus, favoriser des anticorps secondaires couplés à un fluorochrome rouge depuis les membranes cellulaires ont tendance à produire une auto-fluorescence verte qui peut interférer avec le bas étiquetage. En outre, le choix d'un anticorps secondaire rouge fluorophore couplé permet l'étiquetage concomitante de lipides neutres (voir ci-dessous).
- Laver soigneusement les coupes de tissus avec du PBS au moins quatre fois pendant 10 min à température ambiante.
- Pour certaines expériences, effectuer post-fixation en incubant les échantillons contenant 2% de PFA dilué dans du PBS pendant 10 min à température ambiante, afin de stabiliser les échafaudages antigène / anticorps. Cependant, cette étape peut être omise dans la plupart des cas.
- Pour visualiser CLD et MFG, la couleur des lipides neutres en incubant les coupes de tissus dans 30-50 pi d'une solution de PBS contenant 3 ug / ml de BODIPY 493/ 503 pendant 10 min à température ambiante. Rincer rapidement des sections de tissu à deux reprises avec du PBS.
- Contre-ADN nucléaire avec 30-50 ul d'une solution de PBS contenant 3 M de DAPI (4-6 diamidino-2-phénylindole, 5 mg / ml solution mère) pendant 10 min à température ambiante. Laver les coupes de tissu avec du PBS avant de deux fois le montage des lames pour l'observation.
- Retirer du PBS et placer une goutte de milieu de montage sur chaque section de tissu.
- Placez un côté de la lamelle à un angle contre le tiroir, prendre contact avec le bord extérieur de la goutte de liquide, puis abaissez lentement le capot, éviter les bulles d'air. Laisser le liquide de se répandre entre la lame de verre et la lamelle pendant quelques minutes, puis retirez l'excès de milieu de montage avec une serviette en papier.
- Sceller la lamelle à la lame de verre avec des sections vernis à ongles et de tissus de conserver à 4 ° C pour éviter leur exposition à la lumière jusqu'à l'observation.
4. Observation fluorescence et Image Acquisition
Remarque: Un microscope à fluorescence équipé d'une caméra contrôlée par le logiciel d'acquisition d'image est nécessaire pour observer les résultats IHC.
- Avant l'acquisition d'images, de vérifier l'intensité de l'étiquetage et d'évaluer l'arrière-plan de l'expérience en regardant les contrôles négatifs. Acquérir des photos de chaque marqueur fluorescent (de canal de couleur) individuellement.
- Acquérir toutes les images, y compris celles des contrôles correspondants, dans les mêmes conditions (exposition et les paramètres généraux) pour chaque canal de couleur.
- Microscopie conventionnelle
- Effectuer microscopie à épifluorescence avec un microscope équipé de filtres standard pour l'isothiocyanate de fluorescéine (FITC, vert), la rhodamine (rouge) et DAPI (bleu) les émissions, × 20 à × 63 (immersion d'huile, NA 1.3) objectifs et une caméra d'imagerie DP50.
- La microscopie confocale
- Effectuer la microscopie confocale avec microsadaptation se trouve équipé du logiciel ZEN, en utilisant × 20 × 63 à (immersion d'huile, NA 1.4) et les objectifs 488- et 568-nm excitation longueurs d'onde du laser.
5. Traitement de l'image
Remarque: Toutes les images post-traitements sont effectués en utilisant le logiciel gratuit ImageJ (http://imagej.nih.gov/ij/).
- Superposer l'image (fusion)
- Ouvrez les images acquises dans chaque canal qui sera combinée (Fichier / Ouvrir). Si vous travaillez avec 8 bits images en niveaux de gris, couleur artificielle attribuer à chaque canal en utilisant la table de consultation (tableaux image / Lookup).
- Générer l'image composite à partir d'images en niveaux de gris ou de couleur en utilisant la commande «Fusionner les canaux" (Image / Couleur / fusionner des couches), puis attribuer une couleur à chaque canal.
- Effectuer l'image empile superposition de la même manière par des empilements acquises dans chaque canal qui sera combinée (Fichier / Ouvrir) ouvrant et en utilisant la commande «Fusionner canaux &# 8221; (Image / Couleur / fusionner des couches) d'attribuer une couleur à chaque canal. Enregistrer la pile composite comme une séquence d'images ou un film (voir section 5.4).
- Image pile Z projection
- Utilisez la fonction Z de projection (Image / Stack / Zproject, Intensité Max) pour fournir une vue en deux dimensions de toutes les images d'une pile d'images en les projetant sur l'axe perpendiculaire au plan de l'image (axe z). L'option "Maximum Intensity" crée une image dans laquelle chaque pixel contient la valeur maximale sur toutes les images de la pile. Cela génère une seule image permettant la visualisation de tous les coloration observée à travers l'ensemble de la pile d'image pour un canal particulier ou après la superposition de plusieurs canaux.
- Projection d'images 3D de pile
- Utilisez la commande de projection 3D (image projet / Stack / 3D, Brightest Point, l'axe Y) pour générer une séquence de projections d'un volume de rotation sur un plan. Le rendu visuel de surfaces et les structures internes dépend à la fois la méthode de projection (le plus proche point, point le plus lumineux (utilisé ici), ou valeur moyenne) et les paramètres de visualisation sélectionnés. Chaque trame de la séquence d'animation est le résultat de la projection à partir d'un angle de vue différent.
- Faire pivoter l'image 3D créée autour de chacun des trois axes orthogonaux (l'axe des y a été choisi ici). Enregistrer la séquence produite comme une seule image ou un film.
- Pile de l'image à la conversion de film
- Ouvrez une pile d'images (Fichier / Ouvrir) et l'enregistrer comme un film au format AVI en utilisant la commande "AVI" (Fichier / Enregistrer sous / AVI).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
La glande mammaire est une glande sous-cutané situé le long de la structure à la fois ventrale du thorax et de l'abdomen chez les rongeurs. L'emplacement des cinq paires de glandes de la souris pendant la gestation est montré dans la figure 4. La morphologie de la glande mammaire change radicalement au cours de son développement, reflétant des modifications fonctionnelles requises pour préparer la lactation complète (figure 1B). Chez les animaux vierges ou nullipares, la glande mammaire se compose d'un épithélium canalaire peu ramifiée intégré au sein d'un stroma gras mince qui peut être difficile à voir. Dès le début de la grossesse, les prolifère et se dilate épithélium mammaire, résultant dans les glandes mammaires grands qui deviennent plus faciles à voir et à retirer (Figure 4). Pendant la lactation, le tissu mammaire est plus épaisse et plus blanche apparaît en raison de la présence de lait. Seuls les glandes mammaires et inguinaux abdominaux sont collectées parce Glan mammaire cervicales et thoraciquesDS sont moins facilement retiré en raison de leur étroite association avec les muscles. Pour certaines expériences, les chiots peuvent être séparés de la femelle en lactation 4-6 h avant le sacrifice afin de limiter la sécrétion du lait par mESCs 46,47.
Identification des myoépithélial mammaires et les cellules épithéliales
Les cellules myoépithéliales contractiles entourant les alvéoles peuvent être distinguées des mESCs luminales grâce à l'utilisation d'anticorps dirigés contre des marqueurs spécifiquement exprimées par chacun de ces types de cellules. Dans la glande mammaire, les marqueurs actuellement utilisés sont les cytokératines (CK). CKs sont une grande famille de protéines cytoplasmiques qui se polymérisent pour former des filaments intermédiaires du cytosquelette (10 nanomètres de diamètre en moyenne) qui se trouvent dans les tissus épithéliaux. Les filaments intermédiaires sont extrêmement stables et fournissent un support mécanique pour l'architecture cellulaire, et organiser les tissus en contribuant à l'adhérence cellule-cellule et cellule-basale conjonctifinteractions tissulaires. Les sous-ensembles de CKs exprimées par les cellules épithéliales dépendent principalement du type de l'épithélium, son stade de développement et de son état de différenciation. En outre, cela vaut également pour les homologues malignes de l'épithélium. Par conséquent, ces marqueurs sont des outils simples et de grande valeur pour caractériser les populations de cellules dans un tissu dans des conditions physiologiques et sont utilisés pour le diagnostic et la caractérisation d'une tumeur en 48 pathologie chirurgicale.
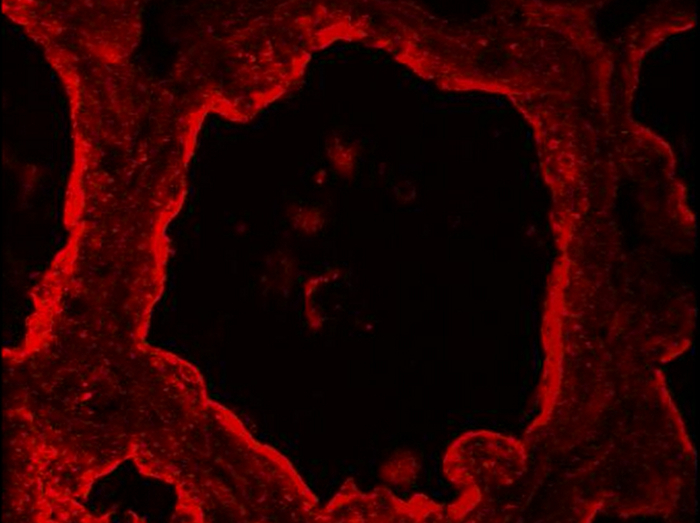
Dans la glande mammaire normale, les cellules myoépithéliales de mESCs luminales et peuvent être distinguées sur la base de leur expression différentielle de CK14 et CK8, respectivement (Figure 5). Ces marqueurs cytoplasmiques sont détectés dans les sections mammaires de souris allaitantes après PFA fixation et AR. Les images ont été acquises avec un microscope à épifluorescence classique. CK8 semble être distribué dans le cytoplasme des mESCs luminale (figure 5), CK8. Notez que le fond rouge observerD pour le contrôle négatif sans anticorps primaire (figure 5, -Ig1) est principalement due à la coupe de tissu pliage, comme suggéré par le marquage d'ADN bleu, qui affiche plusieurs couches de noyaux (Figure 5, -Ig1, noyaux). CK14 est spécifiquement observée dans les cellules myoépithéliales et allongées plates situées à la base des alvéoles (figure 5), CK14. Une autre manière courante pour identifier les cellules myoépithéliales est de détecter l'actine musculaire lisse alpha (a - SMA) présents dans ces cellules contractiles (voir Figure 4 à 49).
La détection de produits laitiers de la souris
Après l'accouchement, les mESCs complètement différenciées commencent à produire des quantités abondantes de lait. Composants laitiers sont sécrétées par des voies distinctes 40,50. Micelles de caséines sont sécrétées par exocytose de SV-Golgi dérivés, tandis que les lipides sont libérés comme MFG par le bourgeonnement de la pl apicaleasma membrane de mESCs (figure 2, une cellule épithéliale mammaire sécrétoire). Pour certaines expériences, les chiots sont séparés de la femme 4-6 heures avant la collecte des glandes mammaires, afin de ralentir la sécrétion du lait 46,47. Dans ces conditions, la membrane plasmique apicale des mESCs et le contenu de la lumière peuvent être facilement observés, ce qui est le cas au cours de lait depuis les alvéoles sont contractés et la lumière sont fermés. En outre, ce qui ralentit la sécrétion est également essentiel lors de l'étude des protéines impliquées dans le trafic membranaire comme SNAREs. En effet, le cycle pièges entre le donneur et accepteur compartiments et leur localisation subcellulaire est difficile à déterminer car l'étiquetage est souvent diffusé lorsque le chiffre d'affaires de la membrane est élevée, ie., Pendant l'allaitement. Par conséquent, le ralentissement de la sécrétion de lait en enlevant les chiots offre des conditions appropriées pour étudier la localisation intracellulaire de SNAREs lorsque le T et V-SNAREs résident préférentiellement chez le donneuret le compartiment accepteur, respectivement (voir ci-dessous).
La figure 6 montre la localisation des caséines dans la glande mammaire en lactation de la souris au jour 10 de la lactation, en présence (Figure 6, p +) ou en l'absence (Figure 6, -p) des chiots. Les coupes de tissus ont été observées à la fois par la microscopie conventionnelle à épifluorescence (les trois colonnes de droite) et la microscopie confocale (figure 6, colonne de gauche). Au cours de lait, les caséines semblent être principalement accumulée dans la région apicale (Figure 6, p +, des pointes de flèches). La microscopie confocale révèle que les caséines sont également présents, bien que dans une moindre mesure, sur le côté basal de mESCs en présence de petits (Figure 6, + p, flèches), qui ne peuvent être clairement observées en microscopie classique (Figure 6, les caséines, comparer les panneaux gauche et droit). En effet, en grand champ épifluorescence, la fluorescence émise par l'échantillon (fond fluorescence) passe à travers le volume excité et modifie la résolution des objets observés dans le plan focal objectif (out-of-focus fluorescence). Cela est particulièrement vrai pour les échantillons épais (épaisseur supérieure à 2 um). La microscopie confocale permet d'obtenir des images de haute qualité à partir d'échantillons préparés à épifluorescence, comme la profondeur de champ peut être contrôlée et la fluorescence de fond exclu du plan focal. En outre, en présence de petits (Figure 6, + p), la lumière des alvéoles sont tout à fait fermé et le côté apical de mESCs est mieux observé en l'absence de petits (Figure 6), -p, lorsque la lumière de la alvéoles est dilaté en raison de l'accumulation de produits laitiers. Lorsque la sécrétion de lait est ralenti, les caséines apparaissent également accumulés sous la membrane plasmique apicale (Figure 6, -p, des pointes de flèches), et sont clairement observés sur le côté basal de mESCs (Figure 6, -p, flèches). Les contrôles négatifs sans ANTIB primaireOdy n'a montré aucun étiquetage (Figure 6, -Ig1).
Les produits laitiers peuvent être facilement co-détectés en combinant IHC pour les caséines et lipide neutre contre-coloration des CLD et MFG (figure 7). Les coupes de tissus ont été imagées en Z-piles par microscopie confocale, qui étaient post-traités avec ImageJ pour produire des projections Z ou projections en 3D pour chaque (Figure 7, caséines, lipides) ou tous les canaux de couleur (Figure 7, fusionner). Les séquences d'images produites ont été sauvegardés comme des images simples (figures 7 et 8) ou des films (voir Films complémentaires).
Bien que certains marquage n'a été observé sur le côté de la base, les caséines sont principalement accumulées sur le côté apical de mESCs (figure 7, p +), comme déjà décrit, lorsque les femelles ont été jusqu'à présent pas été séparés des petits (Figure 6, + p). CLD sont aussi principalement localisée dans la région apicale de mESCs, alors que plus grand secret,MFG és sont présents dans la lumière des alvéoles. Notez que les caséines et les MFG sont facilement visualisées à la lumière d'alvéoles en l'absence de petits (Figure 7, comparer + p et -p). Caséines ne co-localisent pas avec les CLD ou MFG dans l'une de ces conditions depuis la superposition des deux canaux de couleur ne produit pas d'étiquetage jaune (figure 7, de fusionner des images). Cependant, post-traitements d'image pile montrent que les caséines entourent les MFG sécrétées dans la lumière des alvéoles, ce qui suggère que ces protéines peuvent interagir avec le MFG (figure 7, de fusionner des images). Notez la différence des images produites par chaque post-traitement utilisé (Figure 7, comparer Zproj et 3D proj pour chaque canal de couleur).
Détection de butyrophiline, un marqueur protéique de MFG.
BTN1 est l'une des principales protéines associées à MFG 51 dans le lait. Cette protéine transmembranaire est mainly localisée à la membrane plasmique apicale des mESCs et est par conséquent trouvé à la surface de la MFG après sa libération par bourgeonnement 52. La figure 8 montre qu'au jour 10 de la lactation, BTN1 est principalement localisée au niveau de la membrane plasmique apicale et, dans une moindre mesure, dans la région apicale de mESCs. BTN1 entoure également les MFG présents dans le lumen d'alvéoles, ainsi que certains des CLD apicaux (Figure 8, proj 3D fusion, des pointes de flèches). Les résultats sont présentés comme une seule image extraite de l'image Z-stack acquise (Figure 8, image) ou une vue 3D généré avec la commande de projection 3D d'ImageJ, comme décrit ci-dessus (Figure 8, proj 3D). Notez qu'une seule image peut être suffisante pour observer la distribution apical de la protéine, mais l'association spatiale des BTN1 avec MFG sécrétées ou apicale CLD est observée seulement après la reconstruction 3D de la Z-stack (Figure 8 Comparer image BTN1 et 3D de la fusion PICTUres). Le Z-pile peut également être reconstruit comme un film pour donner une meilleure vue de la distribution spatiale de la protéine. L'image Z-stack acquis pour BTN1 seul (des films supplémentaires 1 et 3) ou superposée avec les deux autres canaux de couleur (fusionner, films supplémentaires 2 et 4) sont présentés à titre d'exemples. Le Z-pile peut être lue image par image à partir du haut vers le bas (films complémentaires 1 et 2) ou comme une vue de rotation (axe y) de la projection 3D de l'ensemble de la pile d'images (films supplémentaires 3 et 4 ).
La détection de deux protéines SNARE: STX6 et VAMP4
Comme mentionné précédemment, des pièges sont des protéines membranaires ce cycle entre les membranes de donneur et accepteur. Il est donc préférable de ralentir le chiffre d'affaires de la membrane associée à l'activité sécrétoire élevé de mESCs en séparant les femelles de chiots avant de recueillir la glande mammaire lors de l'étudeces protéines. STX6 et VAMP4 ont tous deux été décrit comme étant associé avec le réseau trans-Golgi 53,54. Cependant, ces protéines SNARE peuvent également jouer un rôle important au niveau des autres compartiments cellulaires comme les granules sécrétoires (STX6) 55,56 et l'appareil de Golgi (VAMP4) 57. Des études antérieures suggèrent que les protéines SNARE jouent un rôle dans la sécrétion de caséine 35,36. Pendant la lactation, STX6 VAMP4 et sont situés dans la région sub-apicale de mESCs. STX6 est observée entre le noyau et la membrane apicale de CEM, correspondant à l'appareil de Golgi et le réseau trans Golgi (figure 9, STX6), et est également présent, bien que dans une moindre mesure, de SV-36 contenant de la caséine. VAMP4 est également localisée dans la région sub-apicale de mESCs, mais l'étiquetage semble être plus ponctuée et est accumulée sous la membrane plasmique apicale (Figure 9, VAMP4) en raison de son association avec les deux CLD et caséine-contaSV de INING 36. Contrôle négatif sans anticorps primaire n'a pas donné lieu à aucun étiquetage.

Figure 1. Souris de développement de la glande mammaire pendant la vie embryonnaire et adulte. (A) Les glandes mammaires de souris commencent à se développer autour de jour embryonnaire 10 (E10) des ectodermique (bleu clair) lignes de lait (rose). À E11.5, placodes forment symétriquement le long de la ligne de lait mammaire et le mésenchyme environnant (bleu foncé) commence à se condenser. Les placodes invaginent pour former des bourgeons (E12.5-E14.5) et, sur E15.5, l'épithélium mammaire (rose), prolifèrent et allongée pour former le germe primaire qui pousse à travers le mésenchyme mammaire vers le coussinet adipeux (vert clair ). Il se forme un lumen creux et ouvre de donner lieu à la tétine (violet). Sur a 18,5, l'épithélium mammaire forme un Rudimentary structure ramifiée reliée à l'extérieur. Adapté de 6 permission de Macmillan Publishers Ltd: Nature Reviews Genetics, copyright 2007. (B) Au cours de la puberté, l'épithélium mammaire (violet) entre dans une phase importante de la croissance (extensive allongement, bifurcation et les ramifications latérales). Au début de la gestation, la prolifération vaste et rapide ainsi que la ramification latérale se produire, conduisant à l'expansion considérable de l'épithélium mammaire, qui envahit entièrement toute la coussinet adipeux mammaire. L'épithélium mammaire atteint un état fonctionnel très différencié pendant la lactation lorsque mESCs luminal sécrètent de grandes quantités de lait. Lorsque l'allaitement cesse après le sevrage, les involue glande mammaire. MESCs sont éliminées par phagocytose et l'apoptose, ce qui conduit à la disparition de structures de lóbulo-alvéolaire qui sont remplacés par le tissu adipeux. Adapté du schéma 1 de http://brisken-lab.epfl.ch/research et le chapitre 2.2. http://tvmouse.ucdavis.edu/bcancercd/22/index.html. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
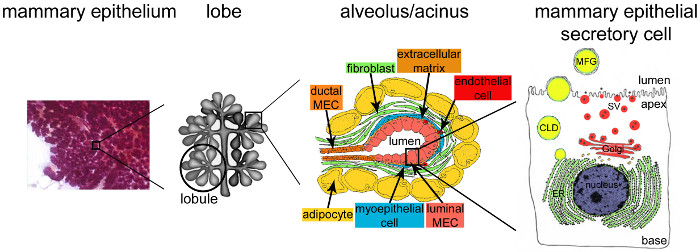
Figure 2. Architecture de la glande mammaire pendant la lactation. Pendant la lactation, l'épithélium entièrement développé et très ramifié (violet) représente la grande majorité du tissu mammaire. Le tissu épithélial est formée par des structures tubulo-alvéolaire noyés dans un stroma qui contient divers types de cellules (fibroblastes, les adipocytes, les cellules musculaires lisses, le sang et les vaisseaux lymphatiques et les terminaisons nerveuses). MESCs sont organisés en structures ou alvéoles acineuses, assemblés dans lobules qui forment lobes. Chaque alvéole est une unité produisant du lait fonctionnel qui est connecté à un réseau hautement ramifié de canaux interlobulaires et lobulaires, permettant ainsi le lait à Dplu vers l'extérieur. Chaque alvéole est délimitée par une monocouche de mESCs polarisées, le côté apical de qui borde un lumen central. Le côté basal de la mESCs est en contact étroit avec une matrice extracellulaire et les cellules myoépithéliales contractiles. Les produits laitiers sont libérés sur le côté apical de mESCs. Lait Major (de caséines) sont sécrétées sous forme de micelles de la caséine (points noirs) par exocytose des vésicules de sécrétion Golgi dérivés (SVS), tandis que les lipides sont libérés sous forme de globules de matière grasse du lait (de MFG) par bourgeonnement de la membrane plasmique apicale des mESCs. CLD: cytoplasmique gouttelette lipidique; ER: réticulum endoplasmique; MEC: cellules épithéliales mammaires. Adapté du chapitre 2.2. http://tvmouse.ucdavis.edu/bcancercd/22/index.html., Fig. Www.cellbiol.net/ste/alpHERCEPTIN1.php 02, Fig. 26-02 en 58, et de 50. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
.dans les pages = "always"> 

Figure 3. Procédure expérimentale pour effectuer immunofluorescence indirecte sur coupes congelées de la glande mammaire de la souris. La glande mammaire est recueilli à partir d'une souris femelle CD1 à 10 jours de lactation. Le tissu mammaire est coupé en petits fragments qui sont fixées avec du paraformaldehyde et infusées dans de saccharose avant d'être incorporé dans le composé PTOM et congélation rapide. Les échantillons de la glande mammaire sont ensuite découpés en coupes fines congelées et transformées pour IIF par incubation successive avec des anticorps primaires et secondaires conjugués à un fluorochrome, respectivement. Après le montage, les échantillons sont analysés avec un microscope à fluorescence, ce qui permet l'acquisition d'images pouvant ensuite être post-traitée./53179/53179fig3large.jpg "Target =" _ blank "> S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.

. Figure 4. anatomique emplacement des glandes mammaires de souris Gauche: vue ventrale du système mammaire de la souris à l'étape de fin de gestation. A droite: la localisation et l'aspect de la glande mammaire à la fin du stade de gestation chez la souris. On notera que pendant la lactation, la glande mammaire sont plus épaisses et plus blanches apparaissent en raison de la présence de lait dans les alvéoles. Adapté de http://ctrgenpath.net/static/atlas/mousehistology/Windows/femaleu/mousemammgldiagram.html et http://www.pathbase.net/Necropsy_of_the_Mouse/index.php?file=Chapter_3.html. S'il vous plaît cliquez ici pour voir une version plus grande de cette figure.
-together.within-page = "always"> 
Figure 5. Identification des cellules épithéliales luminales et les cellules myoépithéliales de base dans la glande mammaire de la souris. MESCs Luminal et les cellules myoépithéliales sont identifiés par IIF dans la glande mammaire de la souris au jour 10 de la lactation, sur la base de leur expression de la CK-8 et CK-14 , respectivement. L'ADN nucléaire a été coloré avec du DAPI (bleu). Les images ont été acquises avec un microscope à épifluorescence classique. L'image composite (fusionner) montre la superposition de l'étiquetage correspondant à caséines (rouge) et noyaux (bleu), respectivement. -Ig1, Contrôle négatif sans anticorps primaire. Les astérisques indiquent lumens. Barre d'échelle = 100 um. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
e 6 "src =" / files / ftp_upload / 53179 / 53179fig6.jpg "/>
Figure 6. Localisation cellulaire des caséines dans la glande mammaire de la souris. Les caséines sont détectés par IIF dans la glande mammaire de la souris au jour 10 de lactation. La glande mammaire a été recueilli à partir de femelles en présence (+ p) ou en l'absence (-p) des chiots. Les images ont été acquises avec un conventionnelles (panneau de droite, caséines, des noyaux et fusionnent) ou un microscope confocal (caséines (rouge), panneau de gauche) microscope à fluorescence. Dans les deux conditions, les caséines (rouge) sont détectés dans la région apicale (têtes de flèches) et plus ou moins à la base de mESCs (flèches). Les contrôles négatifs sans anticorps primaires ne montrent aucun étiquetage (-Ig1). L'ADN nucléaire est coloré avec du DAPI (bleu). L'image composite (fusionner) montre la superposition de l'étiquetage correspondant à caséines (rouge) et noyaux (bleu), respectivement. Les astérisques indiquent lumens. Barre d'échelle = 100 um pour les images à épifluorescence (panneau de droite, les caséines, noyaux, fusionner) et = 10 &# 181;. M pour les images confocal (colonne de gauche) S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
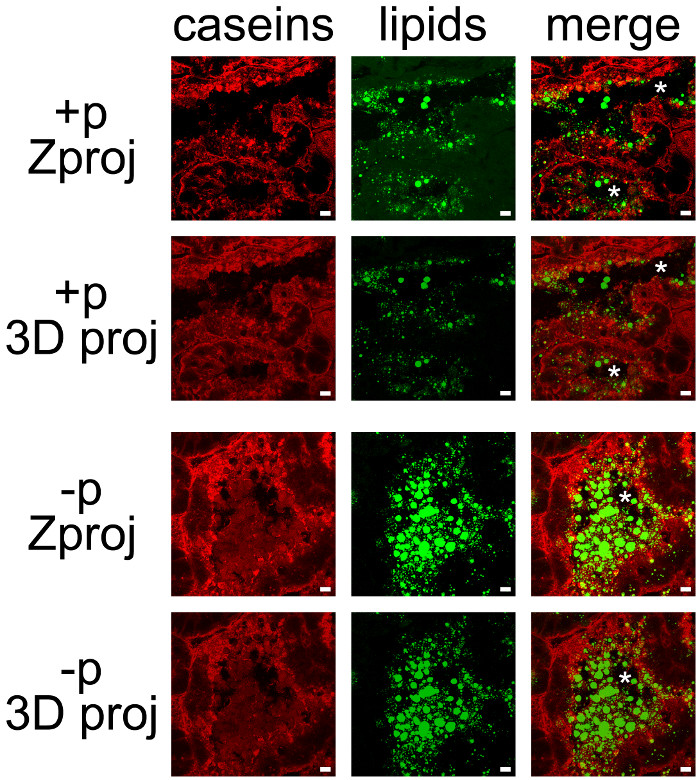
Figure 7. Localisation cellulaire des produits de lait dans la glande mammaire de la souris. Les caséines (rouge) sont détectés par IIF dans la glande mammaire de la souris au jour 10 de la lactation en présence (+ p) ou en l'absence (-p) des chiots. Les lipides neutres (CLD) et les MFG sont contre avec bodipy 493/503 (vert). Les images composites (de fusion) montrent la superposition des deux marquages. Les images ont été acquises dans le Z-piles avec un microscope confocal. Z-piles ont été post-traités avec ImageJ pour générer des projections de Z (Zproj) ou des projections 3D (axe des ordonnées) (proj 3D) de l'ensemble des piles dans chaque canal pour les deux (fusionner). Les astérisques indiquent lumens. La barre d'échelle= 10 um. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.

Figure 8. localisation cellulaire de butyrophiline et les lipides dans la glande mammaire de la souris. BTN1 (rouge) est détectée par IIF dans la glande mammaire de souris à 10 jours de lactation, en l'absence des chiots. Les lipides neutres (CLD et MFG) et l'ADN nucléaire sont de contraste avec bodipy 493/503 (vert) et DAPI (bleu), respectivement. Les images ont été acquises avec un microscope confocal comme images Z-piles. Les résultats sont présentés comme une seule image extraite de la pile d'images (images, BTN1, des lipides, des noyaux et fusionnent) ou après post-traitement avec ImageJ pour générer une vue 3D (axe des y) de l'ensemble de la pile d'image (proj 3D, BTN1 , des lipides, des noyaux, fusion). Les images composites (de fusion) montrent lessuperposition des trois canaux de couleur. -Ig1, Contrôle négatif sans anticorps primaire. Les astérisques indiquent lumens. La barre d'échelle = 10 um. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.

Figure 9. localisation cellulaire de deux protéines SNARE dans la glande mammaire de la souris. Syntaxine 6 (STX6) et VAMP4 (V4) sont détectés par IIF dans la glande mammaire de la souris au jour 10 de lactation. Les images ont été acquises avec un (conv) épifluorescence classique ou un microscope confocal (LSM) microscope. Les images composites (de fusion) montrent la superposition du marquage observé pour chaque protéine SNARE (rouge) et de l'ADN nucléaire DAPI (couleur vert faux), respectivement. -Ig1, Contrôle négatif sans anticorps primaire. Astérisques indiquent lumens. Barre d'échelle = 10 um pour les photos et confocale = 100 um pour épifluorescence images. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.


Tableau 1. L'immunohistochimie guide de dépannage.

Film supplémentaire 1. Localisation de butyrophiline dans la glande mammaire de la souris. BTN1 (rouge) est détectée par IIF dans la glande mammaire de la souris au jour 10 de lactation. Les images ont été acquises avec un co microscope nfocal comme un Z-stack et post-traité avec ImageJ pour générer un film. Le Z-stack est lu à partir du haut vers le bas. S'il vous plaît cliquez ici pour voir cette vidéo.

2. Localisation film supplémentaire et butyrophiline de lipides neutres dans la glande mammaire de la souris. BTN1 (rouge) est détectée par IIF dans la glande mammaire de souris à 10 jours de lactation. Les lipides neutres (CLD et MFG) et l'ADN nucléaire sont de contraste avec bodipy 493/503 (vert) et DAPI (bleu), respectivement. Les images ont été acquises avec un microscope confocal en tant que Z-pile pour chaque canal de couleur et sont post-traités avec ImageJ pour générer un Z-empilement composite qui superpose les trois canaux de couleur. Le composite Z-pile résultante est lue à partir du haut vers le bas.https://www.jove.com/files/ftp_upload/53179/supvid2.mp4 "target =" _ blank "> S'il vous plaît cliquer ici pour voir cette vidéo.
3. film supplémentaire de localisation de butyrophiline dans la glande mammaire de la souris. BTN1 (rouge) est détectée par IIF dans la glande mammaire de la souris au jour 10 de lactation. Les images ont été acquises avec un microscope confocal comme un Z-stack et post-traité avec ImageJ (projection 3D) pour générer un (axe y) tournant vue spatiale de l'étiquetage BTN1. S'il vous plaît cliquer ici pour voir cette vidéo.

Film supplémentaire 4. Localisation de butyrophiline et lipides neutres dans la MA de la sourismmary glande. BTN1 (rouge) est détectée par IIF dans la glande mammaire de la souris au jour 10 de lactation. Les lipides neutres (CLD et MFG) et l'ADN nucléaire sont de contraste avec bodipy 493/503 (vert) et DAPI (bleu), respectivement. Les images ont été acquises avec un microscope confocal en tant que Z-pile pour chaque canal de couleur et sont post-traités avec ImageJ pour générer un Z-empilement composite qui superpose les trois canaux de couleur. ImageJ (Projection 3D) a en outre été utilisé pour générer un (axe y) tournant vue spatiale de la Z-stack composite. S'il vous plaît cliquer ici pour voir cette vidéo.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
IHC est une méthode expérimentale relativement simple et directe pour localiser l'antigène dans des sections de tissus, qui dépend principalement sur les interactions épitope-anticorps spécifiques. Bien qu'un grand nombre de protocoles sont utilisés pour localiser une protéine par IIF, l'âme de ces procédures est presque toujours le même. Cependant, il ya certains aspects critiques qui peuvent fortement influencer le résultat et doivent donc être optimisés pour chaque étude IHC individu. L'aspect le plus difficile de cette approche est de déterminer les meilleures conditions expérimentales, ie., Ceux qui génèrent un signal fort et spécifique de l'antigène d'intérêt. Les variables qui doivent être considérés pour la conception expérimentale et optimisation sont les suivants: (1) le type d'antigène (espèces, les niveaux d'expression, localisation subcellulaire); (2) le type d'épitope (séquence, la conformation, modifications post-traductionnelles putatifs); (3) la préparation des échantillons (enrobage dans de la paraffine ou des sections gelées); (4) la meth de fixationod (immersion ou perfusion); (5) le fixateur utilisé (formaldéhyde, acétone ou alcool); (6) le réactif de blocage utilisé (sérum normal, BSA ou de lait non gras); (7) l'étape de AR; (8) la méthode de détection (directe ou indirecte); (8) le type d'anticorps primaire (monoclonaux ou polyclonaux); (9) l'anticorps secondaire (espèce et l'étiquette); (10) (contre-colorants nucléaire et / ou d'un autre marquage du compartiment cellulaire); et (11) le moyen de montage (voir le tableau 1 pour plus de détails). La fixation et les étapes de blocage, au moins, requièrent l'optimisation de facteurs tels que la concentration, le pH, la température, le temps d'incubation et de diluant.
Le premier aspect crucial concerne la préparation d'échantillons de tissus, qui est étroitement liée à la méthode de fixation, qui à son tour influence la qualité des résultats. Par exemple, les morceaux de tissu peuvent être fixés ou non avant l'enrobage. Cette étape peut également dépendre de la méthode d'enrobage choisi, à savoir, le composé octobre vs. inclusion en paraffine, Qui lui-même dépend parfois de l'anticorps primaire utilisé. Fixation de tissu peut être effectuée in vivo en perfusant un animal anesthésié avec une solution de fixation. Cette méthode est utile pour préserver antigènes lors de l'étude des tissus intacts, mais peut ne pas être suffisante pour fixer le tissu d'intérêt. Dans ce cas, les petits morceaux de tissus (pas plus épaisse que 10 mm) peuvent être immergées dans la solution de fixateur. Le tissu congelé peut être préparé par l'immersion du tissu dans de l'azote liquide ou l'isopentane, et enfichable gel est fortement recommandé pour la détection ultérieure de modifications post-traductionnelles telles que la phosphorylation. Cependant, à la différence de tissu inclus en paraffine, le gel ne convient pas pour une conservation à long terme des tissus due à la formation de cristaux de glace dans les cellules qui peuvent altérer la morphologie subcellulaire. Une fois coupé, des coupes de tissus congelés peuvent être conservés à -80 ° C pendant jusqu'à 1 an. Dans tous les cas, la préparation des échantillons de tissu est un compromis entre la préservation du tissu/ architecture cellulaire et en préservant l'intégrité de l'épitope.
Depuis il modifie la composition chimique des tissus, il est essentiel d'optimiser les conditions de fixation pour éviter à la fois incomplète (moins de fi xation) et excessive (overfixation) fi xation.
En effet, underfixation peut réduire le signal spécifique en favorisant la dégradation protéolytique de certains antigènes. D'autre part, overfixation peut modifier le marquage spécifique de l'épitope ou de masquage à générer un fort arrière-plan non spécifique. Ainsi, en plus du choix de la solution de fixateur, d'autres paramètres tels que le temps d'incubation, la température et le pH affecteront fixation de tissu. Bien que PFA est le fixateur le plus couramment utilisé pour IHC, il ne peut pas être considéré comme un fixateur "universelle". PFA induit protéine-protéine et acide des liaisons transversales des protéines nucléique et peut donc modifier artéfactuellement l'épitope (overfixation) puis empêcher sa recoreconnais- par l'anticorps primaire. Cependant, l'épitope peut encore être démasqué par des techniques AR (voir ci-dessous). PFA peut également être impropres à la détection de certains antigènes, comme il a été montré pour induire la translocation de certaines protéines phosphorylées de la membrane dans le cytoplasme. Dans de tels cas, PFA doit être remplacé par d'autres fixateurs appropriés, tels que l'alcool. Contrairement PFA, les alcools tels que le methanol ou l'éthanol ne masquent pas les epitopes car ils permettent la fixation des tissus par le remplacement des molécules d'eau dans les tissus. Cela peut conduire à la précipitation des protéines et ensuite empêcher une interaction anticorps / épitope de raison de changements de conformation. Il est largement considéré que les alcools ne pénètrent pas et donc ne conservent pas la morphologie des tissus ainsi que PFA. L'acétone est un autre autre fixateur, qui est couramment utilisé lorsque vous travaillez avec des coupes de tissus non fixées, snap-congelés. Toutefois, l'acétone est un puissant agent déshydratant et peut conduire à une précipitation irréversible des protéines tissulaires.
Pour certains antigènes, une étape supplémentaire de AR peut être nécessaire pour obtenir un bon signal, surtout si le fixateur induit le changement de conformation ou modifie la charge électrostatique de l'épitope (masquage de l'épitope). AR méthodes visent à inverser ces processus de restauration de l'immunoréactivité de l'épitope et son interaction ultérieure avec l'anticorps primaire. Modes de AR reposent essentiellement sur deux approches: (1) épitope par la protease, soit avec des enzymes telles que la proteinase K, la trypsine ou la pepsine, qui clive les peptides qui masquent l'épitope; et (2) épitope induite par la chaleur, par exemple en utilisant un four à micro-ondes, autocuiseurs, cuiseurs à vapeur de légumes, des autoclaves ou des bains d'eau. Cette dernière approche est particulièrement imparti, en température, Tampon-, et sensible au pH, et les conditions optimales doit être déterminée de manière empirique (un exemple est fourni dans la section Protocole). En variante, l'affinité d'un anticorps pour son antigène peut être renforcéeen modifiant le pH ou la concentration en cation du diluant d'anticorps.
Une étape de perméabilisation est parfois nécessaire pour obtenir un bon signal pour un epitope intracellulaire dans des coupes de tissus épais, en particulier pour antigène coloration nucléaire. Ceci peut être réalisé de diverses manières en utilisant: (1) des alcools ou de l'acétone comme fixateur; ou (2) des détergents tels que Triton, NP-40 (0,1-0,2% dans du PBS, 10 min), la digitonine, la saponine ou de Tween 20 (0,2 à 0,5% pendant 10 à 30 min) après fixation PFA. Cependant, le choix du détergent dépend de la localisation cellulaire de l'épitope détecté. En effet, de détergents agressifs tels que le Triton-X100, qui solubilisent les membranes cellulaires, sont appropriés pour la détection de l'épitope nucléaire, mais peuvent conduire à signaler la modification sur l'extraction de certaines protéines membranaires. L'utilisation de détergents doux (de saponine et de Tween 20) sont plus appropriés pour la détection d'épitopes cytoplasmiques.
La deuxième étape critique est la Blocking de coloration non spécifique. La liaison d'un anticorps à son epitope cible est régi par des forces intermoléculaires (par exemple, les interactions, hydrophobes et ioniques, des liaisons hydrogène). Ainsi, les interactions des anticorps primaires et / ou secondaires avec d'autres protéines que leurs antigènes cibles peuvent donner lieu à une coloration non spécifique. Cela génère une forte fluorescence de fond, ce qui empêche la visualisation de la protéine d'intérêt (faible rapport signal / bruit). Blocage de réactifs réduit les interactions non spécifiques sans nuire à l'interaction spécifique anticorps / épitope de. Une procédure commune consiste à incuber des coupes de tissus avec du sérum normal inactivé par la chaleur ou de BSA. Lors de l'utilisation d'un sérum normal, il doit être de la même espèce que celle de l'animal hôte de l'anticorps secondaire ou d'une espèce non apparentées. Dans tous les cas, le réactif bloquant choisi doit également être ajouté à des diluants pour les anticorps primaires et secondaires. En outre, l'utilisation de détergents non-ioniques tels que le Triton X-100, TWeen 20 ou saponine aide à réduire les interactions non-spécifiques.
Le troisième et probablement le plus important paramètre est la sélection d'anticorps primaire et l'optimisation. De toute évidence, le meilleur choix est un anticorps de haute qualité avec un minimum de réactivité croisée. Comme anticorps monoclonaux présentent généralement une forte affinité et spécificité pour un épitope unique, ils sont les meilleurs outils pour discriminer un membre particulier d'une famille de protéines avec l'identité de séquence élevée. Cependant, l'interaction anticorps / épitope peut être compromise si l'épitope cible a perdu son état conformationnel natif ou lorsque l'accès à l'épitope est empêchée par des interactions avec d'autres protéines, des modifications post-traductionnelles, la température, le pH, la fixation et la concentration en sel. Dans de tels cas, les anticorps polyclonaux sont plus appropriés car ils reconnaissent des epitopes multiples de la même protéine. En outre, ils sont souvent plus stables que les anticorps monoclonaux sur une large gamme de pH et de concentration de sel.Des études préliminaires ont à définir les conditions d'incubation appropriées, à savoir, travailler dilution (anticorps monoclonal: 5-25 mg / ml, anticorps polyclonaux: de 1,7 à 15 mg / ml), temps d'incubation, diluants et de température, qui doivent être déterminées empiriquement pour chaque anticorps primaire. Ces paramètres doivent être optimisés afin de déterminer les conditions qui produisent le signal optimale avec un faible bruit de fond. La spécificité du marquage est favorisé par des temps d'incubation plus longs à des températures plus basses (par exemple., 4 ° C par rapport à la température ambiante).
Le choix à effectuer une détection directe ou indirecte dépend souvent du niveau d'expression d'antigène. Par exemple, un epitope hautement exprimé peut simplement être détectée avec un anticorps primaire conjugué à un fluorochrome, permettant ainsi une coloration multicolore simple et rapide possible tout en évitant fond non spécifique en raison de l'utilisation d'un anticorps secondaire. Cependant, directe si mai génère un signal faible, à un coût plus élevé, et peut sarfois être difficile, anticorps lorsqu'ils sont marqués ne sont pas disponibles dans le commerce. Inversement, IIF est plus sensible pour détecter des épitopes exprimés inférieurs tels que le signal généré est plus intense en raison de l'interaction d'au moins deux anticorps secondaires marqués (dirigé contre l'espèce de l'hôte l'anticorps primaire) avec l'anticorps primaire (amplification). En outre, une large gamme d'anticorps secondaires conjugués à des fluorophores différents sont disponibles dans le commerce, relativement peu coûteux et de qualité contrôlée. Cependant, cette approche peut induire une réactivité croisée et nécessite donc de choisir avec soin les anticorps primaires qui ne sont pas produites dans les mêmes espèces ou des différents isotypes lors de l'exécution des expériences multi-étiquetage. IIF également nécessite parfois des mesures de blocage supplémentaires et doit inclure des contrôles négatifs systématiques (voir ci-dessous). L'amplification peut être en outre réalisé en utilisant un anticorps secondaire conjugué à la biotine et marqué par fluorescence avidine ou la streptavidine (quatre biotines lié par molecule). Néanmoins, cette méthode d'amplification nécessite des mesures supplémentaires pour éviter la liaison non spécifique et ne peut être adapté pour la coloration de certains tissus (foie, reins, le cœur, le cerveau, les poumons et allaitantes glande mammaire) en raison de la présence de niveaux élevés de biotine endogène . Cependant, la biotine endogène peut être bloquée par pré-incubation de l'échantillon avec de l'avidine avec la biotine et par la suite avant l'incubation avec l'anticorps primaire. Le choix des fluorochromes conjugués, qui sont de petites molécules chimiques ayant la propriété d'émettre de la lumière lorsqu'il est excité par la lumière d'une longueur d'onde plus courte, dépend principalement du type d'équipement de microscope disponibles.
Lorsqu'il est correctement conçu pour limiter à la fois la réactivité croisée entre les anticorps et croisement entre les propriétés spectrales des fluorochromes utilisés, IHC base-immunofluorescence permet la visualisation simultanée de plusieurs cibles cellulaires.
La dernière critiquepoint concernant expériences IHC concerne les contrôles positifs et négatifs qui doivent être effectuées pour soutenir la validité de la coloration, d'identifier des artefacts expérimentaux et pour l'interprétation précise des résultats. Certains tissus présentent une forte fond fluorescent (dénommé autofluorescence) qui pourrait conduire à une mauvaise interprétation des résultats. Ainsi, des coupes de tissus doivent être observées sous deux fluorescence et champ lumineux d'éclairage avant de commencer l'expérience IHC. Un contrôle négatif qui omet l'anticorps primaire doit être systématiquement inclus dans chaque expérience IHC afin de veiller à ce qu'un non-spécifique potentiel de liaison de l'anticorps secondaire est négligeable et ne masque pas ou ressembler au motif de coloration spécifique. Un contrôle isotypique peut être effectuée lorsque l'on travaille avec un anticorps monoclonal primaire en le remplaçant par un anticorps non-immun du même isotype (par exemple, IgG1, IgG2a, IgG2b, IgM) à la même concentration. Ce contrôle contribue à es-estimer la coloration non spécifique, ce qui peut être dû à l'interaction des anticorps avec l'échantillon. Pour mettre en évidence la liaison d'un anticorps à son antigène spécifique, un contrôle de l'absorption peut être réalisée de deux manières par pré-incubation de l'anticorps primaire (1) avec son immunogène soluble (10: 1 rapport molaire) O / N à 4 ° C ; et (2) avec des cellules ou des coupes de tissus qui expriment l'épitope intéressant mais qui diffèrent du tissu étudié (par exemple, voir la figure 4B en 59). Dans les deux cas, l'épuisement conséquente de l'anticorps primaire devrait conduire à peu ou pas de coloration. Un autre type de commande peut être réalisé en utilisant un anticorps primaire sans importance, à savoir., Dirigé contre un épitope qui présente une localisation cellulaire différente de l'épitope d'intérêt (vs nucléaire cytoplasmique). L'anticorps non pertinent doit être de la même espèce et isotype que l'anticorps primaire d'intérêt. Des contrôles supplémentaires pour les expériences IHC peuvent inclure l'utilisation d'échantillons de tissUES connues pour exprimer (animaux transgéniques) ou non (animaux knock-out) l'épitope d'intérêt. Cela peut constituer une référence utile et aider à optimiser la procédure IHC.
Une limitation principale de techniques IF est qu'ils ne peuvent être appliquées à fixe (morts) et / ou des cellules perméabilisées, à la fois la procédure potentiellement induire des artefacts. D'autres limitations de cette approche sont dues à l'utilisation d'un microscope pour l'observation des échantillons. Premièrement, comme la résolution optique de épifluorescence et confocale microscopes est limitée, l'emplacement ou co-localisation des protéines détectées ne devraient pas être sur-interprété. Deuxièmement, photoblanchiment, ie. la décoloration de l'intensité de fluorescence au cours du temps lorsqu'ils sont exposés à la lumière, est essentiellement due à la génération d'espèces réactives de l'oxygène dans l'échantillon lors de l'excitation de fluorescence qui, à son tour, conduit à la destruction photochimique du fluorophore. Photoblanchiment peut être réduit par: a) maintenir les échantillons à l'abri dela lumière pendant l'expérience SI et de stockage jusqu'à leur observation; b) en utilisant un agent de antifade (espèces réactives de l'oxygène des charognards) dans le milieu de montage; c) la réduction de l'intensité et / ou la durée de la lumière d'excitation; d) augmentation de la concentration des fluorophores ou d'utiliser une faible concentration d'un fluorochrome avec une efficacité quantique élevé; et e) en utilisant des fluorophores robustes qui sont moins sujettes au photoblanchiment (ie. Alexa Fluors, Seta Fluors, ou DyLightFluors). Troisièmement, autofluorescence est souvent due à la présence de co-enzymes flavines (DCP et FMN: absorption, 450 nm; émission, 515 nm) et les nucléotides de pyridine réduits (NADH: absorption, 340 nm; émission, 460 nm) dans les cellules de mammifères. En outre, l'utilisation d'aldéhydes, en particulier le glutaraldéhyde, pour fixer les échantillons, peut se traduire par des niveaux élevés d'auto-fluorescence. Ceci peut être minimisé par lavage des échantillons avec du borohydrure de sodium à 0,1% dans du PBS avant incubation avec l'anticorps et / ou en sélectionnant des sondes et des filtres optiques que maximize le signal de fluorescence par rapport à l'autofluorescence. Quatrièmement, la fluorescence chevauchement (également appelée purge à travers, croisé ou diaphonie) est principalement due à des propriétés spectrales d'émission des fluorophores car ils présentent souvent très larges bandes passantes, différents profils spectraux, asymétriques, ainsi que différentes longueurs d'onde de pic d'émission et le nombre de maxima. Fluorescence chevauchement se produit lorsque l'on travaille avec plusieurs fluorophores (étiquetage multicolore) et est caractérisé par l'émission d'un fluorophore dans le canal (filtre) d'un autre fluorophore. Artefacts purge travers doivent être minimisés car ils compliquent souvent l'interprétation des SI résultats, en particulier dans le cas de co-localisation ou les études quantitatives. Comme l'équilibrage de l'émission des fluorophores peut être que légèrement améliorée par la procédure si, transpercement peut principalement être réduite au moment de l'acquisition de l'image en utilisant une fluorescence jeux de filtres optimisés et / ou détecteur photomultiplicateur afin de bien separmangé les profils spectraux des fluorophores. À cet égard, la microscopie confocale est bien adapté à l'imagerie multicolore, parce qu'elle permet la différenciation des spectres d'émission de fluorescence de fluorophores individuelles en dirigeant chaque signal à un canal de détection particulier. En outre, la microscopie confocale permet d'ajuster le gain, la tension de photomultiplicateur, ou la puissance du laser pour les canaux individuels de détection pour l'acquisition séquentielle (un seul fluorophore à la fois) de l'étiquetage. Idéalement, les contrôles seule étiquette doivent être effectués pour quantifier la purge à travers et éventuellement enlever des calculs. Un contrôle sans anticorps secondaires (contrôle de fond) peut être préparé de fixer les limites de gain de signal et le décalage de chaque canal d'acquisition d'image optimale. Il peut également être utilisé pour le traitement post-acquisition d'image de fond correcte (autofluorescence).
En conclusion, la méthode décrite fournit un protocole standard simple pour realizati facilessur des immunocoloration sur des sections de la glande mammaire. Néanmoins, les principales étapes d'une expérience IHC doivent être optimisés pour chaque antigène / anticorps, couple, afin de visualiser la coloration spécifique et pour réduire au minimum les signaux de fond non spécifiques. Le procédé décrit comprend également plusieurs méthodes de base pour le post-traitement de la plupart des images obtenues. Immunodétection à base de fluorescence est une méthode puissante avec une large gamme d'applications de la localisation cellulaire d'un antigène à un diagnostic. De nouvelles avancées dans ces approches seront atteints avec le développement futur de nouveaux fluorophores, des dispositifs d'acquisition et des techniques de microscopie, à l'image de détails auparavant non observées des structures et des processus biologiques.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs déclarent qu'ils ont aucun intérêt financier concurrentes.
Acknowledgments
Les auteurs sont reconnaissants à l'installation de noyau d'imagerie INRA MIMA2 (INRA, UMR1198, Jouy-en-Josas) et au personnel de l'unité IERP (UE 0907, l'INRA, Jouy-en-Josas) pour les soins et les installations animal. Nous tenons également à remercier IH Mather, MC Neville et S. Tooze de nous fournir antibodie très utile.
Materials
| Name | Company | Catalog Number | Comments |
| Dissection | |||
| Pins | |||
| Ethanol | |||
| Scissors | |||
| Scalpel and adapted blades | |||
| Ice | |||
| Towel paper | |||
| Tissue sample preparation | Company | Catalog Number | Comments/Description |
| Phosphate Buffered Saline (pH7.4) | Sigma | P-3813 | |
| Paraformaldehyde (PFA, 32% EM grade, 100 ml) | Electron Microscopy Sciences | 15714-S | personnal protection equipment required WARNING: this product will expose you to Formaldehyde Gas, a chemical known to cause cancer |
| OCT compound/Tissue Tek | Sakura | 4583 | |
| Sucrose (D-saccharose) | VWR | 27480.294 | |
| Plastic molds | Dominique Dutscher | 39910 | |
| Liquid nitrogen | |||
| Cryostat/sample support | Leica | CM3050S | |
| Razor blades (SEC35) | Thermo Scientific | 152200 | |
| Slide box | |||
| Glass slides Superfrost/Superfrost Ultra Plus | Thermo Scientific | 10143560W90/1014356190 | |
| Brushes | |||
| IHC | Company | Catalog Number | Comments/Description |
| Super Pap Pen | Sigma | Z377821-1EA | |
| Permanent marker (black) | |||
| 50 mM NH4Cl in PBS | Sigma | A-0171 | |
| 0.1 M glycine in PBS | VWR | 24403.367 | |
| Antigen Retrieval solution: Tris 100 mM 5% urea pH9.6 | |||
| Heater (up to 100°C) | |||
| Bovine Serum Albumin (BSA) | Sigma | A7906-100G | |
| Vectashield (anti-fading mounting medium) without DAPI/with DAPI | Vector Laboratories | H-1000/H-1200 | |
| Glass coverslips 22x50mm (microscopy grade) | VWR | CORN2980-225 | |
| Nail polish | |||
| Primary antibodies | Company | Catalog Number | Comments/Description |
| Rabbit anti-mouse caseins (#7781; 1:50 dilution) | generously gifted by M.C. Neville (University of Colorado Health Sciences Center, USA) |
||
| Mouse anti-cytokeratin 8 (CK8, clone 1E8, 1:50 dilution) | Biolegend (Covance) | MMS-162P | |
| Mouse anti-cytokeratin 14 (CK14, cloneLL002, 1:50 dilution) | Thermo Scientific | MS-115-P0/P1 | |
| Rabbit anti-butyrophilin (1:300 dilution) | generously gifted by I.H. Mather (Department of Animal and Avian Sciences University of Maryland College Park, USA) | ||
| Rabbit anti-Stx6 (1:50 dilution) | generously gifted S. Tooze (Cancer Research UK, London Research Institute, London, UK) |
||
| Rabbit anti-VAMP4 (1:50 dilution) | Abcam | ab3348 | |
| Secondary antibodies | Company | Catalog Number | Comments/Description |
| Rhodamine-conjugated goat anti-rabbit IgG (H + L) (1:300 dilution) | Jackson ImmunoResearch Laboratories | 111-025-003 | |
| Counterstains | Company | Catalog Number | Comments/Description |
| Bodipy 493/503 | Life Technologies (Molecular Probes) | D-3922 | |
| DAPI (4-6-diamidino-2-phenylindole) | Life Technologies (Molecular Probes) | D-1306 | |
| Observation/Image capture | Company | Catalog Number | Comments/Description |
| conventional fluorescence microscope | Leica Leitz DMRB microscope |
Standard filters for FITC, Rhodamine and DAPI emissions, ×63 oil-immersion objective (NA 1.3), DP50 imaging camera (Olympus), CellˆF software (Olympus) |
|
| Laser Scanning Microscope (confocal microscopy) | Zeiss LSM 510 microscope |
Plan-Apochromat ×63 oil-immersion objective (NA 1.4), CLSM 510 software, Confocal facilities, MIMA2 Platform, INRA Jouy-en-Josas, France, http://mima2.jouy.inra.fr/mima2) | |
| Image treatment | Company | Catalog Number | Comments/Description |
| ImageJ 1.49k software | Free software |
References
- Watson, C. J., Khaled, W. T. Mammary development in the embryo and adult: a journey of morphogenesis and commitment. Development. , 135-995 (2008).
- Smith, G. H. Experimental mammary epithelial morphogenesis in an in vivo model: evidence for distinct cellular progenitors of the ductal and lobular phenotype. Breast Cancer Res Treat. 39, 21-31 (1996).
- Van Keymeulen, A., et al. Distinct stem cells contribute to mammary gland development and maintenance. Nature. 479, 189-193 (2011).
- Oakes, S. R., Gallego-Ortega, D., Ormandy, C. J. The mammary cellular hierarchy and breast cancer. Cell Mol Life Sci. 71, 4301-4324 (2014).
- Visvader, J. E., Stingl, J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes & development. 28, 1143-1158 (2014).
- Robinson, G. W. Cooperation of signalling pathways in embryonic mammary gland development. Nat Rev Genet. 8, 963-972 (2007).
- Cowin, P., Wysolmerski, J. Molecular mechanisms guiding embryonic mammary gland development. Cold Spring Harb Perspect Biol. 2, a003251 (2010).
- Brisken, C., O'Malley, B. Hormone action in the mammary gland. Cold Spring Harb Perspect Biol. 2, a003178 (2010).
- Gjorevski, N., Nelson, C. M. Integrated morphodynamic signalling of the mammary gland. Nat Rev Mol Cell Biol. 12, 581-593 (2011).
- Daniel, C. W., Smith, G. H. The mammary gland: a model for development. J Mammary Gland Biol Neoplasia. 4, 3-8 (1999).
- Howlett, A. R., Bissell, M. J. The influence of tissue microenvironment (stroma and extracellular matrix) on the development and function of mammary epithelium. Epithelial Cell Biol. 2, 79-89 (1993).
- Edwards, G., Streuli, C. Signalling in extracellular-matrix-mediated control of epithelial cell phenotype. Biochem Soc Trans. 23, 464-468 (1995).
- Hennighausen, L., Robinson, G. W. Think globally, act locally: the making of a mouse mammary gland. Genes & development. 12, 449-455 (1998).
- Topper, Y. J., Freeman, C. S. Multiple hormone interactions in the developmental biology of the mammary gland. Physiol Rev. 60, 1049-1106 (1980).
- Brisken, C., et al. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc Natl Acad Sci U S A. 95, 5076-5081 (1998).
- Ormandy, C. J., Binart, N., Kelly, P. A. Mammary gland development in prolactin receptor knockout mice. J Mammary Gland Biol Neoplasia. 2, 355-364 (1997).
- Oakes, S. R., Rogers, R. L., Naylor, M. J., Ormandy, C. J. Prolactin regulation of mammary gland development. J Mammary Gland Biol Neoplasia. 13, 13-28 (2008).
- Hennighausen, L., Robinson, G. W. Information networks in the mammary gland. Nat Rev Mol Cell Biol. 6, 715-725 (2005).
- Kouros-Mehr, H., Werb, Z. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev Dyn. 235, 3404-3412 (2006).
- Khaled, W. T., et al. The IL-4/IL-13/Stat6 signalling pathway promotes luminal mammary epithelial cell development. Development. 134, 2739-2750 (2007).
- Asselin-Labat, M. L., et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol. 9, 201-209 (2007).
- Barcellos-Hoff, M. H., Aggeler, J., Ram, T. G., Bissell, M. J. Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development. 105, 223-235 (1989).
- Robinson, G. W., McKnight, R. A., Smith, G. H., Hennighausen, L. Mammary epithelial cells undergo secretory differentiation in cycling virgins but require pregnancy for the establishment of terminal differentiation. Development. 121, 2079-2090 (1995).
- Streuli, C. H., Bissell, M. J. Mammary epithelial cells, extracellular matrix, and gene expression. Cancer Treat Res. 53, 365-381 (1991).
- Streuli, C. H., et al. Laminin mediates tissue-specific gene expression in mammary epithelia. J Cell Biol. 129, 591-603 (1995).
- Boudreau, N., Sympson, C. J., Werb, Z., Bissell, M. J. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science. 267, 891-893 (1995).
- Pullan, S., et al. Requirement of basement membrane for the suppression of programmed cell death in mammary epithelium. J Cell Sci. 109 (Pt 3), 631-642 (1996).
- Schmidhauser, C., Bissell, M. J., Myers, C. A., Casperson, G. F. Extracellular matrix and hormones transcriptionally regulate bovine beta-casein 5' sequences in stably transfected mouse mammary cells. Proc Natl Acad Sci U S A. 87, 9118-9122 (1990).
- Streuli, C. H., et al. Stat5 as a target for regulation by extracellular matrix. J Biol Chem. 270, 21639-21644 (1995).
- Sollner, T., et al. SNAP receptors implicated in vesicle targeting and fusion. Nature. 362, 318-324 (1993).
- Jahn, R., Scheller, R. H. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 7, 631-643 (2006).
- Weber, T., et al. SNAREpins: minimal machinery for membrane fusion. Cell. 92, 759-772 (1998).
- Sollner, T., Bennett, M. K., Whiteheart, S. W., Scheller, R. H., Rothman, J. E. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 75, 409-418 (1993).
- McNew, J. A. Regulation of SNARE-mediated membrane fusion during exocytosis. Chem Rev. 108, 1669-1686 (2008).
- Wang, C. C., et al. VAMP8/endobrevin as a general vesicular SNARE for regulated exocytosis of the exocrine system. Mol Biol Cell. 18, 1056-1063 (2007).
- Chat, S., et al. Characterisation of the potential SNARE proteins relevant to milk product release by mouse mammary epithelial cells. Eur J Cell Biol. 90, 401-413 (2011).
- Reinhardt, T. A., Lippolis, J. D. Bovine milk fat globule membrane proteome. J Dairy Res. 73, 406-416 (2006).
- Robenek, H., et al. Butyrophilin controls milk fat globule secretion. Proc Natl Acad Sci U S A. 103, 10385-10390 (2006).
- Fujimoto, T., Ohsaki, Y., Cheng, J., Suzuki, M., Shinohara, Y. Lipid droplets: a classic organelle with new outfits. Histochem Cell Biol. 130, 263-279 (2008).
- Mather, I. H., Keenan, T. W. Origin and secretion of milk lipids. J Mammary Gland Biol Neoplasia. 3, 259-273 (1998).
- Heid, H. W., Keenan, T. W. Intracellular origin and secretion of milk fat globules. Eur J Cell Biol. 84, 245-258 (2005).
- McManaman, J. L., Russell, T. D., Schaack, J., Orlicky, D. J., Robenek, H. Molecular determinants of milk lipid secretion. J Mammary Gland Biol Neoplasia. 12, 259-268 (2007).
- de Assis, S., Warri, A., Cruz, M. I., Hilakivi-Clarke, L. Changes in mammary gland morphology and breast cancer risk in rats. Journal of visualized experiments : JoVE. , (2010).
- Plante, I., Stewart, M. K., Laird, D. W. Evaluation of mammary gland development and function in mouse models. Journal of visualized experiments : JoVE. , (2011).
- Galio, L., et al. MicroRNA in the ovine mammary gland during early pregnancy: spatial and temporal expression of miR-21, miR-205, and miR-200. Physiol Genomics. 45, 151-161 (2013).
- Linzell, J. L., Peaker, M. The effects of oxytocin and milk removal on milk secretion in the goat. J Physiol. 216, 717-734 (1971).
- Knight, C. H., Peaker, M., Wilde, C. J. Local control of mammary development and function. Rev Reprod. 3, 104-112 (1998).
- Walid, M. S., Osborne, T. J., Robinson, J. S. Primary brain sarcoma or metastatic carcinoma? Indian J Cancer. 46, 174-175 (2009).
- Hue-Beauvais, C., et al. Localisation of caveolin in mammary tissue depends on cell type. Cell Tissue Res. 328, 521-536 (2007).
- McManaman, J. L., Neville, M. C. Mammary physiology and milk secretion. Adv Drug Deliv Rev. 55, 629-641 (2003).
- Mather, I. H., Jack, L. J. A review of the molecular and cellular biology of butyrophilin, the major protein of bovine milk fat globule membrane. J Dairy Sci. 76, 3832-3850 (1993).
- Heid, H. W., Winter, S., Bruder, G., Keenan, T. W., Jarasch, E. D. Butyrophilin, an apical plasma membrane-associated glycoprotein characteristic of lactating mammary glands of diverse species. Biochim Biophys Acta. 728, 228-238 (1983).
- Bock, J. B., Klumperman, J., Davanger, S., Scheller, R. H. Syntaxin 6 functions in trans-Golgi network vesicle trafficking. Mol Biol Cell. 8, 1261-1271 (1997).
- Tran, T. H., Zeng, Q., Hong, W. VAMP4 cycles from the cell surface to the trans-Golgi network via sorting and recycling endosomes. J Cell Sci. 120, 1028-1041 (2007).
- Wendler, F., Page, L., Urbe, S., Tooze, S. A. Homotypic fusion of immature secretory granules during maturation requires syntaxin 6. Mol Biol Cell. 12, 1699-1709 (2001).
- Wendler, F., Tooze, S. Syntaxin 6: the promiscuous behaviour of a SNARE protein. Traffic. 2, 606-611 (2001).
- Shitara, A., et al. VAMP4 is required to maintain the ribbon structure of the Golgi apparatus. Mol Cell Biochem. 380, 11-21 (2013).
- Thibault, C., Levasseur, M. C. La reproduction chez les mammifères et l'homme. , INRA Editions. 928 (2001).
- Truchet, S., Wietzerbin, J., Debey, P. Mouse oocytes and preimplantation embryos bear the two sub-units of interferon-gamma receptor. Mol Reprod Dev. 60, 319-330 (2001).
