Note: This video protocol is based on an associated publication9. Specific reagents, material and equipment necessary to perform all the steps described in this protocol are listed in the materials section. The recipes and information for preparation of all required solutions are itemized in Table 1.
1. Pouring of Gradient Tube Gels
- Assembling the Casting System
- Using a flame-heated razor blade or scalpel, cut a 20 ml serological pipette along an orthogonal section 10 cm from the top of the dispensing tip. Ensure that the cut surface is smooth. Keep the bottom piece (with the tip) and discard the top piece. Clamp the bottom pipette piece vertically (conical tip facing down) to a support stand.
CAUTION: Do not touch the heated blade metal, use proper handles or a plier to avoid burns. - Cut 3 cm of flexible tubing and connect it to the cut pipet by stretching it over the dispensing tip. Connect the flow control valve at the other end of the tube.
- Link the bottom of the valve to the dispensing tip of the gradient mixer using a piece of tubing. Keep the valve open.
- Set the gradient mixer on top of a magnetic stirrer. Place magnetic bars inside the mixing chambers.
- Position the dispensing tip of the gradient mixer 5-10 cm higher than the top of the pipette piece to allow for the gel to be poured by gravity.
- Test the system for leaks with deionized water. If no leaks are found, dry the system by blowing air inside it before continuing.
- Cover the top of the pipette piece with two or more layers of tape. Puncture a hole through the tape layers in the center of the circular opening. Ensure that the puncture is large enough for the glass tube to pass tightly through.
- Slide the glass tube, where the gel will be cast, through the punctured hole, stretching the tape so the glass fits tightly. Insert the glass tube so that the bottom of the tube is 2 mm above the pipette bottom.
- Check to see if the glass tube is held and centralized and is not touching the pipette wall once the system is prepared. The system is now ready for casting.
- Using a flame-heated razor blade or scalpel, cut a 20 ml serological pipette along an orthogonal section 10 cm from the top of the dispensing tip. Ensure that the cut surface is smooth. Keep the bottom piece (with the tip) and discard the top piece. Clamp the bottom pipette piece vertically (conical tip facing down) to a support stand.
- Casting Gel
- Prepare gel buffer, APS and Coomassie solutions (solutions 1-3, Table 1).
- Prepare 12% T (solution 4, Table 1) and 1% T (solution 5, Table 1) separating gel solutions. Add 10 µl of Coomassie solution in the 12% T separating gel solution. Add APS and TEMED in both solutions immediately before pouring in gradient mixer.
CAUTION: polyacrylamide is highly neurotoxic and gloves must be worn. - Make sure the dispensing valve of the gradient mixer is closed. Add the 12% T separating gel solution to the mixing chamber farthest from the dispensing tip. Open the valve between both chambers and let the solution flow into the next chamber.
- Close the valve and pipette the dislocated 12% T separating gel solution back to the previous chamber. This is done to avoid air bubbles between chambers.
- Add the 1% T separating gel solution to the mixing chamber closest to the dispensing tip. Turn on the magnetic stirrer.
- Open both gradient mixer valves at the same time to allow the separating gel solutions to be mixed and to flow into the pipette and glass tube.
- Once the separating gel solutions in the gradient mixer are depleted, uncouple the tubing from the mixer and couple it to a 10 ml syringe. Make sure the end of the tube remains above the liquid level in the pipette until the syringe is firmly attached.
- Use the syringe to gently push the remaining separating gel solution from the tubing into the pipette and glass tube. Close the pipette valve before allowing air bubbles into it.
- Gently add a layer of about 0.25 ml of water-saturated butanol (solution 6, Table 1) on top of the gel solution inside the glass tube to seal the polymerization reaction from oxygen in air and to flatten the top meniscus.
- Clean the pouring apparatus immediately with deionized water (do not use detergent).
- Wait overnight for complete gel polymerization at room temperature.
- Detaching Gel Tube from Device and Storing
- On the following day, carefully peel the tape off the top of the device.
- Disconnect the valve from the tubing under the pipette. Release the pipette from the clamp and perform steps 1.3.3. and 1.3.4. over a sink.
- Couple a 10 ml syringe filled with deionized water to the flexible tubing. Push water through the tubing into the pipette, slowly sliding the polymerized gel out of the pipette.
- Hold the pipette horizontally and carefully secure the gel sliding out of the open end.
- Cut off excess polyacrylamide beneath and around the glass tube using a razor blade. Make sure to create a smooth end on the gel within the tube, by making it flush with the tip of the glass tube.
- Dispense any remaining butanol from the top side of the gel and set the gel tube in an uncapped 15 ml centrifuge tube. Mix 0.5 ml gel buffer (solution 1, Table 1) with 4.5 ml ultra-pure water and add about 2 ml of the solution to both the top side (within glass tube) and the bottom side (in centrifuge tube) of gel tube.
- Cover the full top side of the centrifuge tube, including the glass tube opening, with parafilm to prevent buffer evaporation. Gels can be stored at 4 °C for about 2 weeks.
2. Sample Preparation
- Weigh two mouse hearts and mince them in a Petri dish using a razor blade.
- Add the processed tissue to a mortar and add sufficient liquid nitrogen to cover the sample. Pulverize the tissue with a pestle, adding more liquid nitrogen when it evaporates.
- Once the tissue is thoroughly ground, let the nitrogen evaporate and add 1 ml of lysis buffer (solution 7, Table 1) at 4 °C for every 100 mg of tissue. Move solution into a centrifuge tube.
- Centrifuge at 2,000 x g for 5 min at 4 °C. Carefully collect the supernatant in a new tube and discard the pellet. Quantify the protein concentration in the supernatant using the bicinchoninic acid method12, or as preferred. Note: Native Cell lysates or subcellular fractions may be prepared as preferred. CN-GELFrEE is compatible with various different native sample preparations.
- Prepare the solubilization buffer solution and loading solution (solutions 8 and 9, Table 1).
- If the sample is a pellet, resuspend it in 50-150 µl of solubilization buffer. Keep the sample on ice for 15 min.
- Buffer exchange the resuspended sample or the cell lysate with solubilization buffer using a 30 kDa-MWCO ultra centrifugal filter.
- Centrifuge the protein sample at 4 °C and 10,000 x g for 5-10 min or until the retained fraction volume is down to approximately 20 µl. Discard the flow-through.
- Add 400 µl of solubilization buffer to the retained fraction and mix by pipetting. Centrifuge at 4 °C and 10,000 x g for 5-10 min or until the retained fraction is down to approximately 20 µl. Discard the flow-through.
- Repeat step 2.9 two additional times.
- Dilute the filtered sample (approximately 20 µl) in 100 µl of solubilization buffer.
- Add 20 µl of loading solution to sample.
3. CN-GELFrEE Device
- To perform CN-GELFrEE fractionation, manufacture the device parts by referring to a machine shop. Manufacture the device parts in Teflon. Please refer to Figure 1 for all information necessary to manufacture the parts and to Table 2 for the number of necessary parts for one device.
Safety Considerations: Wear appropriate personal protective equipment. Care should be taken around the high-voltage power supplies, and all junctions should be correctly mounted. Do not touch the device or any adjacent wet area while voltage is on and maintain the surrounding bench area dry during the electrophoresis. Furthermore, the power supply should be completely turned off when handling the device and/or collecting fractions. - Prepare the anode buffer and cathode buffer (solutions 10-11, Table 1) and keep them at 4 °C or in ice.
- Fit the top end of the gel tube (end without gel) through a spacer, an O-ring and a second spacer, in a "sandwich" fashion.
- Add the buffer chamber (cathode) after the second spacer, pass the screws fully through the side holes, add and tighten nuts to both sides. Ensure that the top end of the glass tube is fixed before the top aperture of the buffer chamber, not directly under it or past it.
- Fit the bottom end of the gel tube through a spacer, an O-ring, the collection chamber, and a second spacer.
- Add the buffer chamber (anode) after the second spacer, pass the screws fully through the side holes, add and tighten nuts to both sides. Ensure that the bottom end of the gel tube is fixed past the top aperture of the buffer chamber, close but not touching the back wall.
- Set the CN-GELFrEE device vertically with the cathode buffer chamber upwards using a support stand with clamps.
- Add anode buffer to the anode buffer chamber (bottom) and cathode buffer to the cathode buffer chamber (top). Keep all buffers at 4 °C.
- Remove air bubbles from inside the glass tube by gently tapping the glass tube and check the system for leakage.
- Make sure the platinum electrodes are in contact with the buffers in the chambers and that both gel tube ends are immersed in the buffer within each chamber.
- Connect power cords from the power supply to the respective banana plugs: negative on the cathode buffer chamber (top) and positive on the anode buffer chamber (bottom).
- Load the sample using a gel load tip to top surface of the gel, inside the gel tube.
- Run the gel at 1 W constant, to avoid overheating, until the red dye front is at the bottom of the gel tube.
CAUTION: Turn off power supply before continuing. - Discard anode and cathode buffers.
- Untighten the nuts by the anode buffer chamber and disassemble the bottom spacer and the chamber, maintaining a spacer and the O-ring. Place the bottom end of the gel tube inside the collection chamber, before the top aperture, not past or directly under it. Push the spacer and O-ring close to the collection chamber.
- Cut a piece of cellulose dialysis membrane and rehydrate it by immerging in anode buffer. Cover the aperture between the collection chamber and the bottom spacer using the dialysis membrane.
- Assemble the anode buffer chamber after the second spacer, add and tighten the screws and bolts.
- Lay the device horizontally over an ice bucket. Add fresh anode and cathode buffers to the buffer chambers and check the system for leakage.
- Add 150 µl of anode buffer into the collection chamber. Reconnect the power cords to the respective banana plugs.
- Turn on the power supply at 3 mA constant current. The dye front should then start to elute into the buffer in the collection chamber.
- When the entire dye front is eluted, turn off the power supply and collect the first fraction (0 min), transferring it into a low-protein binding microcentrifuge tube.
- Refill the collection chamber with 150 µl of anode buffer. Keep all the collected fractions on ice.
- Collect fractions after the following time intervals from collection of the first fraction: 2 min, 4 min, 6 min, 8 min, 10 min, 15 min, 20 min, 25 min, 30 min, 45 min and 60 min. Do not account for time while the power supply is off. Do not forget to turn off the power supply before collecting the fractions and to refill the collection chamber with 150 µl of anode buffer between samples. Turn power supply on again after collection to start eluting the next fraction.
- Dispense the anode and cathode buffers and add fresh solutions to the buffer chambers after 60 min. Collect fractions at 90 min and 120 min.
- Change anode and cathode buffers every hour.
- Run a clear native or a blue native PAGE with the fractions and stain it as preferred. Follow the manufacturer's instructions or preferred protocol.
- Run a reducing SDS-PAGE and stain it as preferred. Follow the manufacturer's instructions or preferred protocol.
- Clean up the GELFrEE fractions and submit to native mass spectrometry analyses as described previously9.

Figure 1: CN-GELFrEE device. (A) GELFrEE device parts drawings and (B) photograph of manufactured parts; (C) CN-GELFrEE device assembly scheme: (1) buffer chamber, (2) spacer, (3) O-ring, (4) gel tube, (5) collection chamber, and (6) dialysis membrane.
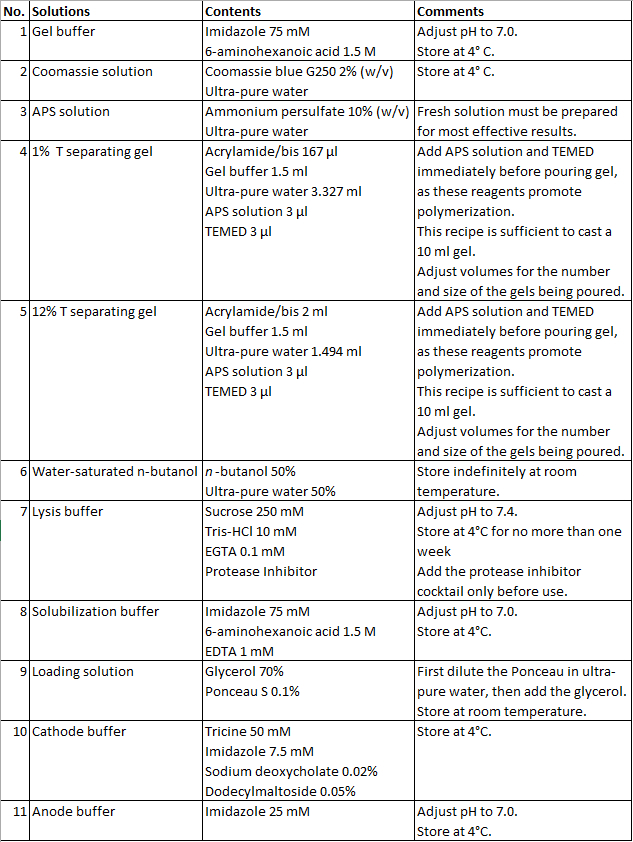
Table 1: Table of solutions.
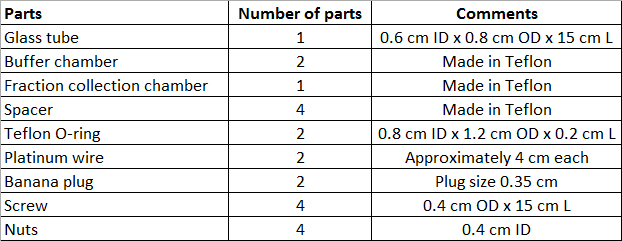
Table 2: Table of GELFrEE device parts.
200 µg of proteins extracted from cryogenically ground mouse hearts were fractionated natively into 14 fractions of 150 µl each, using the CN-GELFrEE method in a 1-12% T gel tube. An aliquot of 10 µl of each fraction was run on a CN-PAGE slab gel and silver stained. A clear depiction of the fractionation and resolution obtained with CN-GELFrEE fractionation is shown in Figure 2A. Fractions were collected over two hours and the gel assay shows a consistent pattern of increasing molecular weights ranging from ~30 to 500 kDa. Each fraction contains species ranging in mass over ~100 kDa. Fractions display a low overlap of molecular-weight-ranges that is increasingly reduced as more collections happen between fractions. The CN-PAGE slab gel shown here represents only 6% of the protein recovered from each 150 µl fraction.
Each of the fractions eluted natively from CN-GELFrEE were subsequently reduced and denatured prior to running them on an SDS-PAGE slab gel. In the reducing SDS-PAGE slab gel (Figure 2B), it is possible to see mass shifts in all fractions when compared to the respective native fractions (Figure 2A), indicating that intact protein complexes were disassembled and disulfide bridges were reduced.

Figure 2: CN-GELFrEE separation of mouse heart cryogenic grinding extract. (A) Clear Native PAGE of CN-GELFrEE fractions collected at time intervals 0 to 120 min. Left: native molecular-weight ladder. (B) Reducing SDS-PAGE of same fractions as A. Left: reduced and denatured molecular-weight ladder.
Fractions can then be cleaned and submitted to native mass spectrometry analyses9. Native MS spectrum of the homodimeric malate dehydrogenase, identified in some of the GELFrEE fractions, shows a charge distribution between 16+ and 20+ and a molecular mass of 72,843 Da (Figure 3A). The 18+ charge state was isolated and collisionally-activated, resulting in the ejection of monomeric malate dehydrogenase with a molecular mass of 36,421.7 (Figure 3B). Source activation was applied to the intact complex in order to induce monomer ejection, allowing isolation and further fragmentation of the monomers. The fragmentation map of the 11+ monomer can be observed in Figure 3C. CN-GELFrEE was proven to be compatible with mass spectrometry analyses, showing no disruption of the noncovalent protein−protein interactions of macromolecular assemblies.

Figure 3: Mass spectra and fragmentation map of the homodimer malate dehydrogenase. (A) MS1 spectrum of the intact complex, (B) MS2 spectrum exhibiting monomer ejected from the complex, and (C) a fragmentation map obtained from the fragmentation of the monomer. The red N indicates that the N-terminal is acetylated.
