

Introduction
蓝藻是地球上几乎每一个自然环境中的细菌的进化古老而多样化的门。在海洋生态系统中,他们都特别丰富,许多营养循环中发挥关键作用,约占一半的固碳1,在海洋中的大多数固氮2和数百万吨的油气产量3每年。叶绿体,细胞器负责在真核藻类和植物的光合作用,很可能已经从由宿主生物体4吞噬蓝藻演变。蓝藻已经证明是有用的模式生物进行光合作用,电子传递5和生化途径,其中许多植物中是保守的研究。此外蓝藻越来越多地被用于生产食品,生物燃料6,电力7和工业化合物8,由于它们的喜的水和CO 2 ghly有效转化生物质使用太阳能9。许多物种可以在非耕地用最少的营养物和海水中培养,这表明蓝藻可能在大规模生长,而不会影响农业生产。某些物种也是天然产品,包括抗真菌剂,抗菌剂和抗癌化合物10,11的源。
产生突变的能力是关键了解蓝藻光合作用,生物化学和生理学,以及必不可少的菌株用于工业用途的发展。大多数已发表的研究中产生遗传上通过抗生素抗性盒的插入修饰的菌株到感兴趣的部位。这限制了可被引入到菌株,因为只有少数的抗生素抗性盒可用于在蓝藻使用突变的数量。含有基因的菌株抗生素赋予重sistance不能用于工业生产中的开放的池塘,这很可能是唯一的成本效益的方法,以产生生物燃料和其它低价值产品12。未标记突变体的产生克服了这些限制。未标记的突变体不包含外源DNA的,除非有意包括在内,并且可以操作多次。因此,可以根据需要在一个菌株,以产生尽可能多的改变。此外,在变形例的网站的下游基因极性影响可以最小化,从而使机体13的更精确的修饰。
为了产生突变体菌株,自杀质粒在蓝藻染色体含有两个DNA片段相同的区域侧翼的基因被删除(称为5'和3'侧翼区)的第一构造。然后两个基因这些侧翼区域之间插入。其中的一个编码抗生素抗性蛋白;第二编码将SacB,这督促集体企业果聚糖蔗糖酶的化合物,赋予蔗糖敏感性。在此过程中的第一阶段中,标记的突变体,含有一些外源DNA 即菌株产生。该质粒构建体与蓝藻细胞混合和该DNA是由生物体吸收自然。转化体是由生长在含有适当的抗生素和通过PCR验证了突变体基因型琼脂平板上选择。自杀质粒不能感兴趣的菌株中复制。因此,任何抗生素抗性菌落,将导致从由此感兴趣的基因中插入染色体中的重组事件。以产生未标记突变体中,明显的突变,然后用仅包含5'和3'侧翼区的第二自杀质粒混合。然而,如果需要的外源DNA的插入,由5'和3'侧翼用含有这些DNA片段之间插入目的基因的盒的区域的质粒,可以使用。塞莱ction是,通过含有蔗糖琼脂平板上生长。作为当sacB基因产物被表达蔗糖是致死的细胞,该存在的唯一的细胞是其中已经发生了第二次重组事件,由此,蔗糖敏感性基因,除了在抗生素抗性基因,已被重组出的染色体上的质粒。作为重组交换的结果,侧翼区和它们之间的任何DNA被插入到染色体。
我们已经成功地使用这些方法来产生在集胞藻的同一菌株的多个染色体突变。 6803(以下简称为集胞藻属 )13,14,引入单点突变到感兴趣13的基因和用于基因盒的表达。而代无标记击倒之前已经提供了工作在集胞藻 15,16,详细的方法证实,由辅助的关键步骤的视觉呈现,是不公开的。我们还以另一种模式蓝藻, 聚球藻申请代明显击倒同样的方法。 PCC7002(以下简称为聚球藻 )。该协议提供了产生突变体和用于验证和存储这些菌株快速协议的清晰,简单的方法。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1.培养基的制备
- 根据Castenholz,1988年17准备BG11培养基。
- 准备100X BG11的储备液,微量元素铁的股票( 表1)。
- 制备磷酸盐股票的单独的溶液, 的 Na 2 CO 3的库存,N - [三(羟甲基)甲基] -2-氨基乙磺酸(TES)缓冲液和碳酸氢钠 ( 表1)。
- 高压灭菌的磷酸盐和Na 2 CO 3的股票。过滤消毒TES缓冲液和碳酸氢钠 0.2微米的过滤器。
- 通过组合976毫升的水,将10毫升100倍BG11的1毫升痕量元素和1ml的铁库存准备BG11和高压灭菌的溶液中。后该溶液冷却到室温,加入1毫升磷酸盐库存,将1ml 的 Na 2 CO 3的库存和10毫升碳酸氢钠的。
- 为BG11固体培养基,加入琼脂15克和700毫升水于一个FLASK。向第二个烧瓶中,加入的 Na 2 S 3克2- O 3 226毫升水,将10毫升100倍BG11的1毫升痕量元素和1ml的铁库存。高压灭菌两种解决方案。之后这些解决方案已经冷却至室温,将它们合并并加入1毫升磷酸盐库存,将1ml 的 Na 2 CO 3的库存,将10毫升TES缓冲液,和10毫升碳酸氢钠的。
注意:解决方案是单独制备,以避免某些盐的沉淀。
- 对蔗糖选择,制备50%(重量/体积)的蔗糖溶液。过滤消毒用0.2微米的过滤该溶液,并加入到BG11(100毫升50%的蔗糖到900毫升BG11的),以产生BG11 / 10%蔗糖平板上。
注意:不要碳酸氢钠添加到BG11 / 5%蔗糖琼脂平板上。加的 Na 2 CO 3为正常。 - 对于聚球藻的培养加入10毫升的1摩尔的4-(2-羟乙基)哌嗪-1-乙磺酸,N-的- (2-羟乙基)哌嗪NR42 - (2-乙磺酸)(HEPES)和1ml维生素B 12( 表1)的1升BG11培养基中。
注意:在市售的BG11培养基中培养菌株的转化是显著少效率比在这里描述的BG11培养基配方,因此不建议。
2.蓝藻菌生长
- 在100毫升锥形瓶中培养菌株为50毫升最大体积并在120rpm摇动。密封用Parafilm和穿刺三个小的孔BG11板在板的一侧,以允许气体交换。孵育在30℃下,在光生物反应器的荧光灯所有菌株在光强度20-40微摩尔光子微米之间-2秒-1。
- 最好使用无菌技术。处理层流罩所有蓝藻菌。
注意:这是特别重要的,当菌株是用含有蔗糖的培养基中培养,可以很容易地沾污ated。
3.质粒构建的生成
- 设计组引物,包括所需的限制酶位点,使用引物设计软件,例如引物3(http://frodo.wi.mit.edu/primer3/),以扩增的两〜1 kb的区域的5'和3'感兴趣的基因。通过Cyanobase咨询蓝藻物种的基因组序列(http://genome.kazusa.or.jp/cyanobase)。 见表2此处使用的所有引物。设计引物时考虑以下因素:
- 确保扩增区域包括将要突变的基因, 例如 , 图1的5'和3'区域。
- 不发生变异间隔区,以避免反义和非编码RNA的无意的突变。用于产生在集胞藻突变体,是指在Mitschke 等记载的转录起始位点的列表。,2011 18,以避免反义的突变或非编码RNA。
- 当选择侧翼区不包括相邻基因作为这些基因在大肠杆菌中的表达的整个开放阅读框可以与克隆干扰。
- 扩增按照制造商的说明使用高保真DNA聚合酶产品通过PCR。
注:在我们的经验,这种酶会产生一些错误。- 建立含有HF缓冲液和DMSO的任0,1.5或3微升50微升PCR反应。使用100纳克每反应的基因组DNA。使用由98℃初始变性步骤的程序,30秒,35轮的98℃10秒,67℃,30秒,72℃30秒,随后的72℃最后延伸步骤下进行5分钟。这通常给予一致的产品。
- 验证与核酸内切酶消化通过凝胶电泳正确大小的PCR产品和样品。运行1%(重量/体积)含0.02%琼脂糖凝胶上(体积/体积)溴化乙锭45分钟在100伏
注意:溴化乙锭是一种潜在的诱变剂,应使用适当的保护处理。 - 纯化使用根据制造商的说明的DNA纯化试剂盒的PCR产物。还可以使用此试剂盒质粒片段,包括从琼脂糖凝胶裁片的净化。在水14微升洗脱纯化的DNA。
- 用于克隆步骤,根据制造商的说明孵育限制性内切酶反应的混合物在37℃下为> 1小时在30微升的总体积。
- 用于连接的步骤,在室温下在20μl的总体积> 1小时后,含有5微升纯化消化的质粒,12微升纯化消化的插入物,缓冲液2微升和1微升连接酶连接DNA片段。
- 根据以下方法制备大肠杆菌 DH5α转化体细胞。
- 成长隔夜E.大肠杆菌
- 接种400毫升的LB中含有6毫升的1摩尔的MgCl 2( 表1)用1毫升过夜培养物的1升锥形瓶中。
- 生长的培养在37℃下以220rpm进行约4小时或直至OD 600nm处达到0.4-0.6。
- 细胞置于冰上1小时。
- 离心机在2,800×g离心10分钟以沉淀细胞在4℃。
- 160 ml溶液A( 表1)取出上清,重悬并在冰上孵育20分钟。
- 离心机在2,800×g离心10分钟以沉淀细胞在4℃。
- 在4毫升溶液A +甘油( 表1)取出上清,重悬。
- 准备50微升等分,冻结液态氮,储存在-80℃。
- 混合5微升连接混合物用50μl感受态细胞,并孵育在冰上1小时。
- 热休克细胞在42℃下90秒,弗洛通过在冰上温育结婚2分钟。
- 添加950微升LB培养基( 表1)中,并在37℃孵育1小时。
- 等分试样50和200微升在平板上用适当的抗生素,或者氨苄青霉素(100微克/ ml)和/或卡那霉素(30微克/毫升)。
注意:卡那霉素和氨苄青霉素是有毒的,应适当的保护处理。 - 挑并与合适的抗生素接种2毫升LB培养基孵育单个菌落。
- 纯化使用根据制造商的说明一小量制备质粒纯化试剂盒的所有质粒。
- 产生的质粒,在用于敲除cpcC1C2基因这个具体的例子,根据下面的步骤。
- 扩增使用引物cpcC1C2leftfor和cpcC1C2leftrev(参见步骤3.2, 表2)的1012 bp的5'侧翼区(左片段)。除去PCR反应的少量,并确认是否正确尺寸产品已通过凝胶电泳(步骤3.3)被放大。消化该片段和pUC19用Xba I和Bam HI(步骤3.5)。
- 既净化制剂(步3.4),结扎(步骤3.6),转换(步骤3.7),并通过小量(步骤3.8)设置了4个2毫升LB与质粒净化独立的殖民地氨苄青霉素(100微克/毫升)的液体培养。
- 检查通过的XbaⅠ/ 巴姆 HI消化和凝胶电泳(步骤3.3)片段到pUC19的插入。 2,660 bp和1012 bp的条带指示正确引入插入到质粒。
- 扩增使用引物cpcC1C2rightfor和cpcC1C2rightrev(参见步骤3.2, 表2)的1016 bp的3'侧翼区(右片段)。除去PCR反应的少量,并确认是否正确尺寸产品已通过凝胶电泳(步骤3.3)被放大。消化该片段和pUC19用Sac I和EcoRⅠ(STEP 3.5)。
- 既净化制剂(步3.4),结扎(步骤3.6),转换(步骤3.7),并通过小量(步骤3.8)设置了4个2毫升LB与质粒净化独立的殖民地氨苄青霉素(100微克/毫升)的液体培养。
- 检查通过萨克 I / RI 生态消化(步骤3.5)和凝胶电泳(步骤3.3)片段到pUC19的插入。 2,660基点人和1 016 bp的条带指示正确引入插入到质粒。
注:XbaⅠ位 / 巴姆 HI位点'为3的克隆区域和萨克 I / RI 生态 '区域5克隆到pUC19尽可能使用。如果可行的话,始终包含在对于5'区域或3正向引物'区的反向引物一个BamHⅠ位点,以确保以后的克隆步骤是更容易执行。 - 序列的两个插入件,以确定该序列使用引物跨越插入部位, 例如 ,M1为正确3前进和M13反 向( 见表2)。顺序必须是正确的,以确保没有错误被引入侧翼区。
- 消费的来自pUC19通过的XbaⅠ/ 巴姆 HI消化左侧的片段。消化的pUC19 +正确的片段用XbaI / 巴姆 HI(步骤3.5)。
- 从净化通过使用手术刀刀片DNA切除的琼脂糖凝胶(步骤3.3)的1012 bp的左片段和3,676基点的pUC19 +右片段。
- 既净化制剂(步3.4),结扎(步骤3.6),转换(步骤3.7),并通过小量(步骤3.8)设置了4个2毫升LB与质粒净化独立的殖民地氨苄青霉素(100微克/毫升)的液体培养。
- 检查片段插入通过的XbaⅠ/ 巴姆 HI消化(步骤3.5)和凝胶电泳(步骤3.3)的pUC19 +右片段的插入。 3,676 bp和1012 bp的条带指示插入正确插入质粒(请参阅本质粒B)。
注意:NPT1 /的sacB盒不必从琼脂糖凝胶中纯化,因为pUM24cm编码赋予氯霉素抗性的蛋白质。因此,如果菌落在LB生长/氨苄青霉素/卡那霉素的琼脂平板上的唯一可能的组合,这将导致抗性菌落是掺入NPT1 /的sacB盒的入质粒B. - 净化两种制剂(步3.4),结扎(步骤3.6),转换(步骤3.7),并设置4个2毫升LB与质粒净化独立的殖民地氨苄青霉素(100微克/毫升)和卡那霉素(30微克/毫升)的液体培养通过小量(步骤3.8)。
- 检查通过巴姆 HI消化(步骤3.5)和凝胶电泳(步骤3.3)的NPT1 /卡带的sacB插入到质粒B点。 4,688 bp和3894 bp的条带表明日的正确插入E将到质粒(请参阅本质粒A)。
- 可替代地,钝端NPT1 /的sacB盒和克隆到质粒B的左侧和右侧片段之间的不同限制性内切酶位点的NPT1 /的sacB盒必须在左边和右边的片段之间进行克隆。
注:如果需要的外盒的表达那么这应该质粒B的左,右片段此质粒然后在未标记的基因敲除步骤中使用之间插入。
4.生成标记的蓝藻和聚球藻的突变体
- 通过接种环满格为30-50毫升BG11培养基中建立了一个新的文化。生长2-3天的文化OD 750nm的 = 0.2〜0.6。
注意:通常单个菌落太小用于接种和单个细胞暴露于甚至低光水平将导致光抑制和选择光耐药突变。 - 离心机1-2毫升培养2,300×g离心5分钟并弃上清。在> 2300 XG不要离心任何蓝藻文化,因为这可能会损坏电池。以BG11培养基洗涤沉淀一次。
注意:不要涡旋,因为这可能会导致菌毛这对DNA的摄取必需的损失悬浮细胞。轻柔吹打悬浮细胞。 - BG11培养基添加到100微升的最终体积。转移细胞到14毫升的圆底管中。
- 添加1微克质粒A对细胞和通过轻轻敲击混合。添加<10微升质粒。
注:优选质粒应是> 100毫微克/微升的浓度,但浓度低于此是足够的成功转型。 - 在孵化器水平放置管子下来。孵育培养4-6小时。
注:细胞可以通过点击每1-2小时进行简单混合,但是这不是必不可少的。样品可以被放置在一个振荡培养箱虽然这并不显著提高效率。 - 散布在BG11琼脂平板上的细胞培养/质粒DNA混合物的等分无抗生素。通常20微升和80微升等分被分散在不同的板块。
- 〜24小时后,加水含卡那霉素2.5-3毫升0.6%琼脂溶液(每次20毫升0.12克琼脂,100微升的100毫克/毫升卡那霉素)琼脂板上。冷却此溶液至〜42℃,并加入到琼脂平板的边缘。倾斜板,使该溶液形成表面上的偶数'顶层琼脂'层。
- 孵育琼脂平板上的时间再延长一段时间。经过大约7天后殖民地应该是可见的。
注:琼脂平板可以堆叠3高的孵化器。通常情况下200个菌落都是每变换得到。 - 上BG11 +卡那霉素(30微克/毫升)的琼脂平板上划线单个菌落。划分琼脂平板成6个扇区,用钝端牙签出来连胜殖民地放在每个单独的部门。获得单菌落不是转化的重要,只是增长。
- 确认根据制造商的说明使用Taq DNA聚合酶通过PCR标记基因敲除。加入2微升氯化镁 (25毫米),每反应。
- 除去细胞的一小部分,并转移到含有50微升水和〜20 425-600微米的玻璃珠的管中。撼动振动器在〜2000转5分钟。离心机以15,700xg xg离心5分钟,并用5微升上清液每50μlPCR反应。
注意:不要悬浮液。细胞碎片需要留在管的底部。
- 除去细胞的一小部分,并转移到含有50微升水和〜20 425-600微米的玻璃珠的管中。撼动振动器在〜2000转5分钟。离心机以15,700xg xg离心5分钟,并用5微升上清液每50μlPCR反应。
- 验证突变
- 利用引物设计软件(如:引物),它涵盖了淘汰赛区域设计引物。设计引物起始于淘汰赛地区〜200 bp的两侧。
注意:引物用于验证cpcC1C2突变体在表2中概述和被称为cpcC1C2for和cpcC1C2rev。 - 扩增使用程序由95℃的初始变性步骤中2分钟,35轮95℃1分钟,60℃1分钟,72℃每序列kb的1分钟,随后一个产品5分钟的72℃最后延伸步骤。包括野生型对照。这通常给予一致的产品。
- 验证通过凝胶电泳基因型。标记基因敲除的转化体将呈现〜4kb的(从左边两个和右侧片段加上NPT1 /的sacB盒0.2 kb的)和不存在的野生型带( 图2)的频带。
注意:在某些情况下,一个〜4kb的频带中没有明显的突变体观察到的由于大尺寸该PCR产物。但是,如果没有则观察到对应于野生型的预期大小的带通常该菌株是一个显着的敲除。
- 利用引物设计软件(如:引物),它涵盖了淘汰赛区域设计引物。设计引物起始于淘汰赛地区〜200 bp的两侧。
- 如果野生型带仍然存在,然后重新条纹上的应变新鲜BG11 +卡那霉素(30微克/毫升)琼脂制版及重复PCR。重复重新划线过程,直到使得没有野生型带在PCR反应中观察到的突变体是分开。
注意:卡那霉素的量增加至50微克/ ml的浓度,然后100微克/毫升,有时为了充分偏析显着的突变是必不可少的。 - 如果应变显示了通过PCR有明显的突变配置文件,然后重新上连胜新鲜BG11 +卡那霉素(30微克/毫升)琼脂平板上。使用该菌株生成无人盯防的淘汰赛。
注意:该协议可以被用于产生与只抗生素抗性盒标记的突变体,即通过从pUC18K 20只与NPT1盒更换的左,右片段之间的NPT1 /的sacB盒。
5.代无标记蓝藻突变体
- 通过接种环全C的设置标记基因敲除的新鲜培养厄尔成30-50毫升BG11培养基。生长2-3天的文化OD 750nm的 = 0.2〜0.6。
- 离心机将10ml培养物2300×g离心5分钟并弃上清。以BG11培养基洗一次。
注意:不要涡旋,因为这可能会导致菌毛这对DNA的摄取必需的损失悬浮细胞。轻柔吹打悬浮细胞。 - 添加BG11到200微升的最终体积。转移细胞到14毫升的圆底管中。
- 添加1微克质粒B核酸向细胞通过轻轻敲击混合。
- 孵育样品4-6小时。水平放置管子下来。
注:细胞可以通过点击每1-2小时进行简单混合,但是这不是必不可少的。样品可以被放置在振荡培养箱尽管这并不提高效率。 - 添加1.8毫升BG11培养基中孵育样品,共4天振荡培养。这是足够的时间,以允许在多个染色体副本发生重组。
- 上BG11 / 5%蔗糖琼脂平板转化混合物的板等分试样。板50微升,10微升和每琼脂板1微升。如果一个殖民地的草坪出现在所有这些琼脂平板上稀释新鲜平板解决方案进一步和分装。经过大约7天后殖民地应该是可见的。
- 补丁琼脂平板上首次+ BG11卡那霉素(30微克/毫升)30-50单个菌落和BG11 / 5%蔗糖琼脂平板上第二,使用钝端牙签。生长在BG11 / 5%蔗糖板但不BG11 +卡那霉素板块都是潜在的无标记击倒任何细菌。细菌在两个平板上生长很可能是蔗糖抗性由于sacB基因的突变。
- 验证使用相同的引物和方法,被用于检查标记击倒未标记击倒。 例如 cpcC1C2for和cpcC1C2rev( 表2)用于检验cpcC1C2无标记基因敲除。一个没有标记的淘汰赛将显示在琼脂糖凝胶相应吨乐队Ø野生型大小减去删除区( 图2)。
- 如果应变示出通过PCR未标记的突变体轮廓(步骤4.11.2)和凝胶电泳( 图2),然后重新条纹上的新鲜BG11琼脂平板上无抗生素。
6株长期储存
- 通过接种环满格为30-50毫升BG11培养基一起成立了应变的一个新的文化。生长3-4天的文化OD 750nm的 = 0.4〜0.7。
- 在约2ml BG11的BG11和悬浮细胞洗涤一次。
- 添加0.8毫升浓缩细胞的一个管。然后加入0.2毫升80%过滤消毒的甘油。
- 可选0.93毫升浓细胞添加到另一个管。添加0.07毫升DMSO中至该管。
注意:DMSO是有毒的,应使用适当的保护处理。 - 储存在-80℃的两个管。为了重振株取出管刮去部分细胞用钝齿挑到琼脂平板无抗生素。条纹出正常使用无菌循环。

图1:质粒构建新一代标记和未标记击倒, 如 cpcC1和cpcC2在集胞藻 (A) 集胞基因组,其中(B)cpcC1和cpcC2和邻近的基因位于的区域。以黑色突出显示是在突变要被删除的基因组的区域。这是通过PCR扩增的基因组中的(C)的网站。 5'侧翼区(蓝色表示)和3'侧翼区(以红色表示)被放大用限制性内切酶位点克隆到pUC19上。 5'(或3')的侧翼区域被切除了的pUC19的并插入pUC19中+ 3'(或59)侧翼区质粒产生质粒B.(D)从pUM24的NPT1 /卡带的sacB通过巴姆 HI消化切和插入的5'和3'的侧翼区产生质粒A. 请点击这里查看大图版本这个数字。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
质粒设计是成功产生两个标记和未标记的突变体是至关重要的。 图1给出了质粒A的一个例子和B用于产生在集胞藻属基因cpcC1和cpcC2 13的缺失突变体。在每种情况下,5'和3'侧翼区是约900-1,000碱基对。可用于降低侧翼区虽然我们已经试验成功最小的已经约500基点。质粒B也可以可以包含5'和3'〜1kb的侧翼区或天然基因序列的修改版本之间的基因盒。
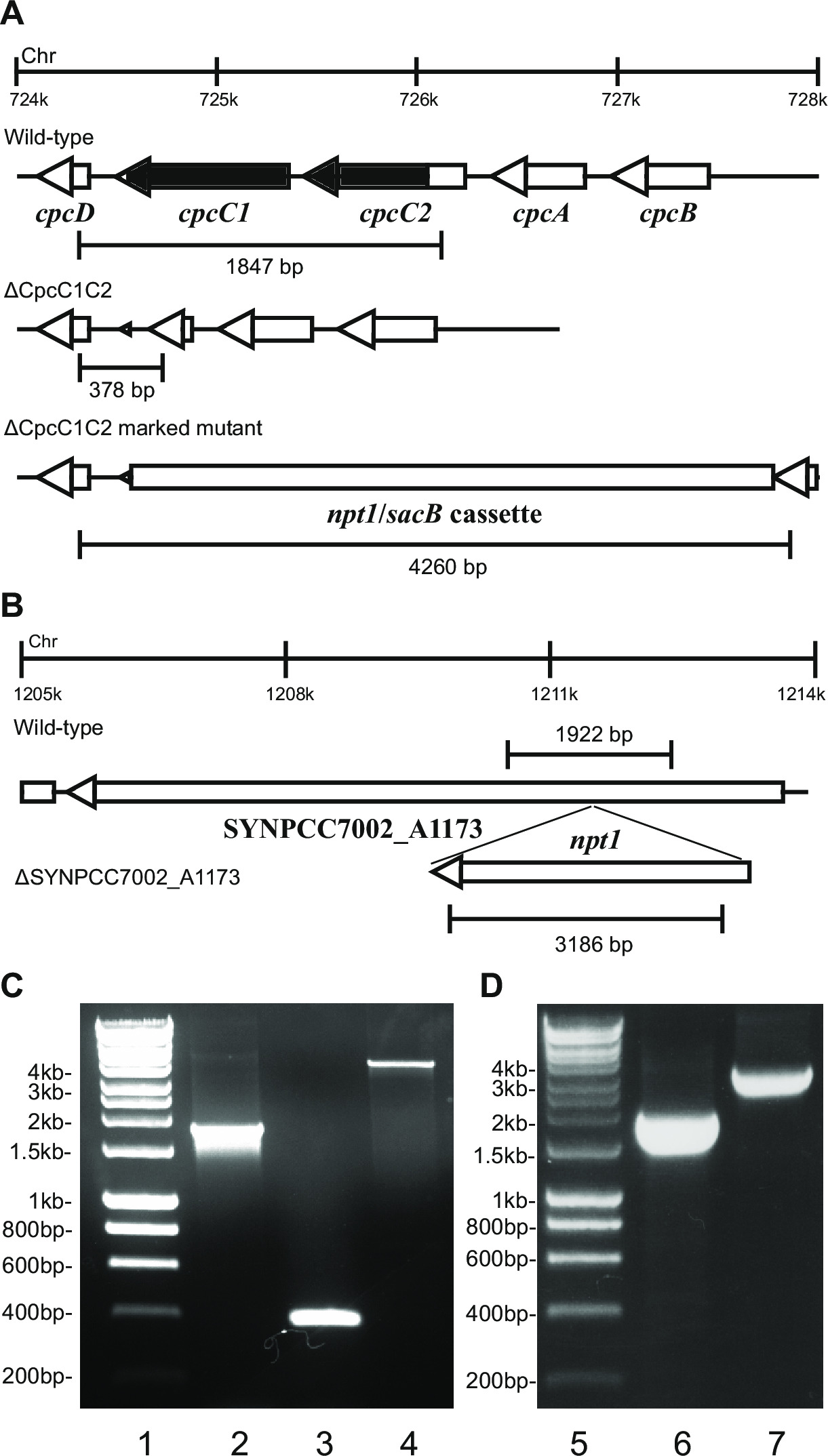
图2:标记和未标记的突变体的验证, 如 cpcC1 / cpcC2
当质粒的转化进入细胞内,通常有几百殖民地将出现在一盘后约7-10天。菌落<1毫米直径大小在未来几周内将不会增加。因此,它是用钝端牙签去除新鲜BG11 +卡那霉素琼脂平板上菌落和条纹是至关重要的。大约再挑染殖民地半会后4-6天生长。如果基因的非必需和突变体在连续光的20-40微摩尔光子米-2秒-1表明类似于野生型菌株的生长( 例如 ,在LEA-Smith等人 ,2013 14终端氧化酶突变体)( 图3),则所有的染色体应包含t的拷贝他NPT1 /卡带的sacB插入序列,通过PCR检测。如果基因的非必需和突变体证明在连续光的缓慢生长表型的20-40微摩尔光子米-2秒-1( 如藻胆体在LEA-Smith等缺陷突变体2014年13)( 图3),然后几轮对卡那霉素的量的逐渐增加BG11琼脂平板上重新划线是为了获得一个隔离标记突变体是必不可少的。一旦获得一个隔离突变这应在新鲜BG11加的卡那霉素琼脂平板上再划线,以确保偏析已完成。如果条纹的重复几轮不允许一个隔离标记突变体的结果,那么该基因是生存可能是必不可少的。 图4给出了涉及在未标记突变体产生的实验步骤的概要。
/54001/54001fig3highres.jpg“WIDTH =”700“/>
图 3: 蓝藻 突变体 生长突变体的例子证明这(A)相似的生长野生型和(B)比野生型增长放缓。所述ΔCOX突变体缺乏细胞色素氧化酶,由于CtaC1D1E1基因的缺失。所述ΔCyd突变体缺少苯二酚氧化酶由于CydAB基因的缺失。橄榄突变体缺乏藻胆体的一部分,由于CpcABC1C2D基因的缺失。在(B)的样品用空气进行鼓泡,以促进生长。 从公布的LEA-Smith 等数据中再现2013年14和2014年13(www.plantphysiol.org;版权的美国植物生物学家协会)。 请点击此处查看该图的放大版本。
例如 图2,如果一个基因盒被插入到染色体中,然后通常抗性和蔗糖抗性菌落观察卡那霉素的比例较高。这些突变体可以在蔗糖上生长由于sacB基因的突变。如果未生成卡那霉素敏感和蔗糖抗性菌落则基因盒是有害的细胞。

图4:在 集胞 标记和未标记的突变体的产生原理图,详细说明(A)的重组和(B)涉及的突变体的产生实验步骤。质粒首先用细胞混合。在含卡那霉素的琼脂平板上,菌落,其中的5'的之间发生重组事件孵育之后d 3处'侧翼区(分别为蓝色和红色表示,),并在染色体的同源序列,是分离的。另外,5'和3'侧翼区域之间的NPT1 /的sacB盒插入染色体中。以下偏析产生的显着的突变体。标记突变体细胞,然后用质粒B混其可以包含(C)的1:5'和3'侧翼区; 2:5'和3'侧翼区与含有这些序列之间插入目的基因的表达盒; 3:5'和3'侧翼区与这些序列之间插入了所希望的核苷酸改变的野生型序列。第二同源重组事件的5'和3'侧翼区和在染色体的同源区之间发生,从而导致去除NPT1 /的sacB盒的,要么无标记的敲除或插入的突变体或改变的野生典型导入染色体ê地区。 请点击此处查看该图的放大版本。
| 原液配方 | |
| 化学 | 量(g) |
| 100X BG11(每升) | |
| 硝酸钠 | 149.6 |
| 硫酸镁4·7H 2 O | 7.49 |
| 氯化钙2·2H 2 O | 3.6 |
| 柠檬酸 | 0.6 |
| 加入1.12毫升0.25摩尔的 Na 2 EDTA,pH值8.0 | |
| 的0.25M 的 Na 2 EDTA,pH为8.0(每100毫升) | |
| 娜2 | 9.3 |
| 微量元素(每100毫升) | |
| H 3 BO 3 | 0.286 |
| 氯化锰2·4H 2 O | 0.181 |
| 硫酸锌4·7H 2 O | 0.022 |
| 的 Na 2的MoO 4·2H 2 O | 0.039 |
| 硫酸铜 ·5H 2 O | 0.008 |
| 有限公司(NO 3)2·6H 2 O | 0.005 |
| 铁件(每100毫升) | |
| 柠檬酸铁铵 | 1.11 |
| 磷酸盐股票(每100毫升) | |
| K 2 HPO 4 | 3.05 |
| 的 Na 2 CO 3股(每100毫升) | |
| 的 Na 2 CO 3 | 2 |
| TES缓冲液,pH值8.2(每100毫升) | |
| TES | 22.9 |
| 碳酸氢钠股票(每100毫升) | |
| 碳酸氢钠 | 8.4 |
| HEPES,pH值8.2(每500毫升) | |
| HEPES | 119.15 |
| 维生素B 12(每50毫升) | |
| 氰钴胺素 | 0.02 |
| 卢里亚BERTANI媒体(每500毫升) | |
| 卢里亚肉汤BERTANI | 12.5 |
| 1M的氯化镁 (每100毫升) | |
| 氯化镁2·6H 2 O | 20.33 |
| 氯化锰2·4H 2 O | 0.395 |
| 氯化钙2·2H 2 O | 1.47 |
| 2-(N-吗啉代)乙磺酸水合物,4-吗啉乙磺酸(MES) | 0.4265 |
| 解决方案A +甘油 | |
| 滴加10ml一个 | |
| 1.5毫升甘油 |
表1:本研究中使用的解决方案。
| 底漆 | 序列 |
| cpcC1C2leftfor | GTAC TCTAGA GCGGCTAAATGCTACGAC |
| CPCC1C2leftrev | GATC GGATCC GCGGTAATTGTTCCCTTTGA |
| cpcC1C2rightfor | GATC GAGCTC TGCACTGGTCAGTCGTTC |
| cpcC1C2rightrev | GACT GAATTC ATCGTTGCTTGAACGGTCTC |
| M13正向 | TGTAAAACGACGGCCAGT |
| M13反向 | CAGGAAACAGCTATGAC |
| cpcC1C2for | GTTTTCATTGGCATCGGTCT |
| cpcC1C2rev | ATGTCCCAGGAACGACTGAC |
| A1173for | AGCAAACCGTTTTTGTGACC |
| A1173rev | TGCAAGGTGGCGAACTGTAT |
表2:本研究中限制性内切酶位点使用的引物有下划线。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
在无人盯防的产生突变体的最重要的步骤是:1)精心设计载体,以确保只有目标区域被改变; 2)确保样品保持无菌,尤其是当上培养的蔗糖; 3)电镀转化最初在缺乏抗生素,随后加入琼脂加抗生素24小时后的BG11琼脂平板上标注的突变体产生的细胞; 4)培养标志着前4整天突变体上镀BG11加蔗糖琼脂平板:5)确保标记突变体是完全隔离和6)确认彻底突变株的基因型。对于这最后的步骤中,设计用于扩增缺失区的一部分额外的引物,可以使用,以确保它已被删除。 Southern印迹,而费力,也可以使用。然而,我们的经验是,在本文中所述的程序是足够的突变体的适当的验证。这个过程也被用于产生Synechococ标记的突变体CUS细长7942。然而,该蓝藻的反复变换已证明具有挑战性。
如果标记的突变体不能分离然后不同环境条件下高的CO 2,低光(<20微摩尔光子米-2秒-1)或额外的营养( 即葡萄糖)可以进行测试。例如,在加入葡萄糖的是为了产生光系统II的突变体21是必不可少的。如果标记的突变体没有完全分离,然后该基因可能是对存活所必需的。但是,也有从那里一些研究小组已经无法敲除的基因的文献的例子(例如,VIPP在集胞藻属 )22,仅对于其它基团,以后来表明基因不是必需的23。这可能是由于在野生型菌株或不正确的质粒设计的差异,从而对相邻的,必需的基因的极性影响。如果一个突变的不完全偏析,我们将建议用于转化含有pUC18K 20的左,右片段之间的NPT1盒的质粒。这是比较容易验证对应于通过PCR野生型和突变型带的存在,因为该片段大约为1.2 kb的,相比于3.8kb的NPT1 /的sacB盒。这个结果是证据表明该基因是必不可少的一个重要部分。
插入表达盒未标记的突变体的产生通常比敲除菌株的开发更具挑战性。我们一般表现强劲的下cpcBAC1C2D子13的控制基因。在某些情况下,这可能会降低基因盒的成功插入的机会,如果过表达的蛋白质的是有害的细胞。那么弱促销员进行测试。一般来说,我们已经观察到较大的基因盒是,越难是在SERT入基因组中。我们一直无法插入基因盒超过500 KB的。护理还必须采取在选择位点以插入表达盒到基因组中。应使用不影响细胞生存力或生长中性点。在集胞藻属的实例包括phaAB和PHACE,其编码的编码聚羟基丁酸酯合成途径24,25的蛋白质。最近一个广泛的蓝藻中性的站点列表中已被确定26。
在蓝藻未标记突变体的产生是一个缓慢的过程,以约5-7周,如果所有的步骤都正确导通。这比产生由多数研究小组研究蓝藻利用标记击倒的标准方法慢。然而,能够采取进一步的突变引入未标记突变体部分的弹性补偿这一点,因为附加质粒续癌宁的范围内盒赋予对不同抗生素的抗性的,不必构成。用于研究目的变异的多个基因的能力有时是必要的,以便充分表征蛋白质的功能作用。例如,我们仅在局部的类囊体膜的两个末端氧化酶电子汇缺失鉴定有害表型,因为只有这些复合物中的一个丢失可能由其他14的活动进行补偿。用于工业应用的菌株的发展,也将需要多个修改的应变,而不是仅仅用于引入外源基因,但也增加光合效率,捕光优化和竞争性途径的缺失对所需基材。
根据光条件的限制未标记突变体产生的速度的主要因素是模型蓝藻物种的缓慢分裂时间,8-20小时之间。联合国明镜较高的光照强度和CO 2浓度,生长速度更快。然而,存在不能耐受或高光或CO 2突变株将针对被选择,或者说,突变菌株将经历前表型特征不希望的变化的危险。因此不建议这样做。然而,如果一个更迅速的协议来产生未标记突变体的开发将是非常有利的。总的来说,这将有利于菌株为基础研究和应用的应用程序的开发。这样的菌株可用于生物燃料,生物质或化学生产或在理解蓝藻生物化学,遗传学和生理学的许多方面。
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
我们感谢环境服务协会教育信托基金,在剑桥SynBio基金合成生物学和社会正义和赋权,印度政府部,给予资金支持。
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. 3rd Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
