

Introduction
Cyanobakterier er en evolutionært gammel og forskelligartet phylum af bakterier, der findes i næsten alle naturlige miljø på Jorden. I marine økosystemer, de er særligt rigelige og spiller en central rolle i mange næringsstoffernes kredsløb, der tegner sig for omkring halvdelen af kulstof fiksering 1, at størstedelen af kvælstof fiksering 2 og hundreder af millioner af tons kulbrinter produktion 3 i oceanerne årligt. Chloroplaster, det organel ansvarlig for fotosyntesen i eukaryote alger og planter, vil sandsynligvis have udviklet sig fra en cyanobakterie, der blev opslugt af en værtsorganisme 4. Cyanobakterier har vist sig nyttige modelorganismer til studiet af fotosyntese, elektrontransport 5 og biokemiske veje, hvoraf mange er konserveret i planter. Desuden cyanobakterier i stigende grad brugt til produktion af fødevarer, biobrændstoffer 6, elektricitet 7 og industrielle forbindelser 8, på grund af deres highly effektiv omdannelse af vand og CO 2 til biomasse ved hjælp af solenergi 9. Mange arter kan dyrkes på ikke-landbrugsarealer med minimale næringsstoffer og havvand, tyder på, at cyanobakterier potentielt kunne dyrkes i stor skala uden at påvirke landbrugsproduktionen. Visse arter er også kilder til naturlige produkter, herunder svampemidler, antibakterielle og anti-cancer-forbindelser 10,11.
Evnen til at generere mutanter er nøglen til at forstå cyanobakteriel fotosyntese, biokemi og fysiologi, og afgørende for udviklingen af stammer til industrielle formål. Hovedparten af offentliggjorte undersøgelser generere genetisk modificerede stammer ved indsættelse af en antibiotikaresistens kassetten i stedet af interesse. Dette begrænser antallet af mutationer, der kan indføres i en stamme, som kun få antibiotikaresistens kassetter er tilgængelige til brug i cyanobakterier. Stammer, der indeholder gener, der giver antibiotisk resistance kan ikke anvendes til industriel produktion i åbne damme, som sandsynligvis vil være de eneste omkostningseffektive midler til at producere biobrændstoffer og andre lav værdi produkter 12. Frembringelsen af umærkede mutanter overvinder disse begrænsninger. Umarkerede mutanter indeholder ingen fremmede DNA, medmindre forsætligt inkluderet, og kan manipuleres flere gange. Derfor er det muligt at generere så mange ændringer i en stamme som ønsket. Derudover kan polære virkninger på gener nedstrøms for modifikationsstedet minimeres, hvilket tillader mere præcis modifikation af organismen 13.
For at generere mutantstammer, selvmord plasmider indeholdende to DNA-fragmenter identiske med regionerne i den cyanobakterielle kromosom flankerer genet, der skal slettes (såkaldte 5 'og 3' flankerende regioner) er først konstrueret. To gener indsættes derefter mellem disse flankerende regioner. En af disse koder for en antibiotikaresistens-protein; den anden koder SacB, der produces levansucrase, en forbindelse der giver følsomhed over for saccharose. I det første trin af processen, markerede mutanter, dvs. stammer indeholdende nogle fremmede DNA, genereres. Plasmidkonstruktionen blandes med de cyanobakterielle celler og DNA'et optages naturligt af organismen. Transformanter selekteres ved vækst på agarplader indeholdende det passende antibiotikum og mutant genotype verificeres ved PCR. Selvmord plasmider kan ikke replikere i stamme af interesse. antibiotiske resistente kolonier vil derfor resultere fra en rekombinationshændelse, hvorved genet af interesse i indsat i kromosomet. For at generere umarkerede mutanter, er den markante mutant derefter blandet med en anden selvmord plasmid indeholdende kun de 5 'og 3' flankerende regioner. Men hvis indsættelse af fremmed DNA er påkrævet, et plasmid bestående af 5'- og 3'-flankerende regioner med en kassette, der indeholder generne af interesse indsat mellem disse DNA-fragmenter, kan anvendes. Selektion er via vækst på agarplader indeholdende sucrose. Som saccharose er letal for celler, når sacB genproduktet udtrykkes, er de eneste celler, der overlever, er dem, hvor en anden rekombinationshændelse har fundet sted, hvorved sucrose følsomhed gen, udover antibiotikaresistensgenet, er blevet rekombineret ud af kromosom og på plasmidet. Som en konsekvens af den rekombinatoriske udveksling, er de flankerende regioner og enhver DNA mellem dem indsat i kromosomet.
Vi har med succes anvendt disse metoder til at generere flere kromosomale mutationer i samme stamme af Synechocystis sp. PCC6803 (i det følgende benævnt Synechocystis) 13,14, til at indføre enkelt punktmutationer i et gen af interesse 13 og til ekspression af gen-kassetter. Mens generation af umærkede knockouts er påvist forud for vores arbejde i Synechocystis 15,16, en detaljeret metode, hjulpet afen visuel præsentation af de kritiske trin, ikke er offentligt tilgængelige. Vi har også anvendt den samme metode til generering af markerede knockouts i en anden model cyanobakterien, Synechococcus sp. PCC7002 (i det følgende benævnt Synechococcus). Denne protokol giver en klar, enkel metode til at generere mutanter og en hurtig protokol for validering og opbevaring disse stammer.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Fremstilling af Kultur Medier
- Forbered BG11 medium ifølge Castenholz, 1988 17.
- Forbered stamopløsninger af 100x BG11, sporstoffer og jern lager (tabel 1).
- Udarbejde særskilte opløsninger af phosphatpufret lager, Na 2CO 3 lager, N - [Tris (hydroxymethyl) methyl] -2-aminoethansulfonsyre (TES) puffer og NaHCO3 (tabel 1).
- Autoklaver fosfat og Na 2 CO 3 bestande. Filter-sterilisere TES buffer og NaHCO3 med 0,2 um filtre.
- Forbered BG11 ved at kombinere 976 ml vand, 10 ml 100x BG11, 1 ml sporstoffer og 1 ml jern lager og autoklaver opløsningen. Efter denne løsning er afkølet til stuetemperatur, tilsæt 1 ml fosfat bestand, 1 ml Na 2 CO 3 lager og 10 ml NaHCO3.
- For BG11 fast medium, tilsættes 15 g agar og 700 ml vand til en flask. Til den anden kolbe, tilsættes 3 g Na 2 S 2 O 3, 226 ml vand, 10 ml 100x BG11, 1 ml af sporstoffer og 1 ml jern lager. Autoklavér begge opløsninger. Efter disse løsninger er afkølet til stuetemperatur, kombinere dem og der tilsættes 1 ml fosfat bestand, 1 ml Na 2 CO 3 bestand, 10 ml TES-puffer, og 10 ml NaHCO3.
Anmærkning: Opløsninger fremstilles separat for at undgå udfældning af visse salte.
- Til selektion på saccharose fremstilles en 50% (vægt / volumen) sucroseopløsning. Filtersteriliser opløsningen med 0,2 um filtre og tilføje til BG11 (100 ml 50% sucrose til 900 ml BG11) til frembringelse BG11 / 5% sucrose plader.
Bemærk: Du må ikke tilføje NaHCO3 til BG11 / 5% sucrose agarplader. Tilføj Na 2CO 3 som normalt. - Til dyrkning af Synechococcus tilsættes 10 ml 1 M 4- (2-hydroxyethyl) piperazin-1-ethansulfonsyre, N - (2-hydroxyethyl) piperazin NR42 - (2-ethansulfonsyre) (HEPES) og 1 ml af vitamin B12 (tabel 1) til 1 I BG11-medium.
Bemærk: Transformation af stammer dyrket i kommercielt tilgængelig BG11 medier er betydeligt mindre effektiv end i BG11 medier opskrifter beskrevet her, og kan derfor ikke anbefales.
2. Vækst af cyanobakteriel stammer
- Kultur stammer i 100 ml koniske kolber med en maksimal volumen på 50 ml og ryst ved 120 rpm. Seal BG11 plader med Parafilm og punktering tre små huller i siden af pladen for at tillade gas udveksling. Inkuber alle stammer ved 30 ° C under fluorescerende pærer i en fotobioreaktor ved en lysintensitet mellem 20-40 pmol fotoner m-2 sek -1.
- Anvendelse af de bedste sterile teknikker. Håndter alle cyanobakterielle stammer i en laminar flow hætte.
Bemærk: Dette er især vigtigt, når stammerne dyrkes med medier indeholdende saccharose, som let kan contaminated.
3. Frembringelse af plasmidkonstrukter
- Design sæt primere, herunder de nødvendige restriktionsenzymsteder, der anvender primer design software såsom Primer3 (http://frodo.wi.mit.edu/primer3/), til amplifikation to ~ 1 kb regioner 5 'og 3' for gen af interesse. Rådfør genom sekvens af de cyanobakterielle arter via Cyanobase (http://genome.kazusa.or.jp/cyanobase). Se tabel 2 for alle anvendte primere her. Ved udformningen primere overveje følgende faktorer:
- Sikre, at amplificerede regioner omfatter 5 'og 3' regioner af genet, der vil blive muteret, fx figur 1.
- Må ikke mutere intergeniske regioner for at undgå utilsigtet mutation af antisense og ikke-kodende RNA. Til generering af mutanter i Synechocystis henvises til listen over transkriptionelle startsteder dokumenteret i Mitschke et al. 2011 18, for at undgå mutation af antisenseeller ikke-kodende RNA.
- Ved valg flankerende regioner ikke omfatter hele den åbne læseramme af tilstødende gener som ekspression af disse gener i Escherichia coli kan forstyrre kloning.
- Amplificere produkter ved PCR under anvendelse high fidelity DNA polymerase ifølge producentens anvisninger.
Bemærk: Det er vores erfaring dette enzym producerer få fejl.- Nedsat 50 pi PCR-reaktioner indeholdende HF buffer og enten 0, 1,5 eller 3 pi DMSO. Bruger 100 ng genomisk DNA pr reaktion. Bruge et program bestående af et initialt denatureringstrin på 98 ° C i 30 sek, 35 runder af 98 ° C i 10 sek, 67 ° C i 30 sek, 72 ° C i 30 sek, efterfulgt af en endelig forlængelse trin på 72 ° C i 5 min. Dette giver typisk ensartede produkter.
- Kontrollere PCR-produkter og prøver fordøjet med-enzymer for den korrekte størrelse via gelelektroforese. Kør 1% (w / v) agarosegeler indeholdende 0,02%(Vol / vol) ethidiumbromid i 45 minutter ved 100 V.
ADVARSEL: Ethidiumbromid er en potentiel mutagen og bør håndteres med passende beskyttelse. - Oprense PCR-produkter under anvendelse af en DNA-oprensningskit ifølge producentens anvisninger. Brug også dette kit til oprensning af plasmid fragmenter, herunder stykker skåret fra agarose geler. Eluere oprensede DNA i 14 pi vand.
- For kloningstrin, inkuber restriktionsendonuklease reaktionsblandingerne ved 37 ° C i> 1 time i et samlet volumen på 30 pi i overensstemmelse med producentens anvisninger.
- Til ligering trin, afsnøre DNA-fragmenter ved stuetemperatur i> 1 time i et samlet volumen på 20 pi indeholdende 5 pi oprenset fordøjet plasmid, 12 pi oprenset fordøjet insert, 2 pi buffer og 1 pi ligase.
- Forbered Escherichia coli DH5a transformerede celler ifølge den følgende fremgangsmåde.
- Grow en overnatning E. coli
- Pode 400 ml LB i en 1 liters konisk kolbe indeholdende 6 ml 1 M MgCI2 (tabel 1) med 1 ml overnatskultur.
- Grow kulturen ved 37 ° C ved 220 rpm i ca. 4 timer, eller indtil OD 600 nm når 0,4-0,6.
- Placer celler på is i 1 time.
- Centrifuger ved 2.800 xg i 10 minutter for at pelletere cellerne ved 4 ° C.
- Fjern supernatanten og resuspender i 160 ml opløsning A (tabel 1) og inkuberes på is i 20 minutter.
- Centrifuger ved 2.800 xg i 10 minutter for at pelletere cellerne ved 4 ° C.
- Fjern supernatanten og resuspender i 4 ml opløsning A + glycerol (tabel 1).
- Forbered 50 pi prøver, fryse i flydende N2, opbevares ved -80 ° C.
- Bland 5 ul ligeringsblanding med 50 pi kompetente celler og inkuberes i 1 time på is.
- Varmechok cellerne ved 42 ° C i 90 sek, følgenwed ved inkubation på is i 2 min.
- Tilføj 950 pi LB-medium (tabel 1) og inkuberes ved 37 ° C i 1 time.
- Portion 50 og 200 pi på plader med passende antibiotika, enten ampicillin (100 ug / ml) og / eller kanamycin (30 ug / ml).
ADVARSEL: Både kanamycin og ampicillin er giftige og bør håndteres med passende beskyttelse. - Pick og inkuberes enkelte kolonier i 2 ml LB-medium podet med den passende antibiotikum.
- Oprens alle plasmider under anvendelse af en miniprep plasmid oprensningskit ifølge producentens anvisninger.
- Generere plasmider, i dette specifikke eksempel for udskære cpcC1C2 gener, ifølge de følgende trin.
- Opformere 1.012 bp 5 'flankerende region (venstre fragment) under anvendelse af primerne cpcC1C2leftfor og cpcC1C2leftrev (Se trin 3.2, tabel 2). Fjerne en lille mængde af PCR-reaktionen og bekræfte, omkorrekte størrelse produkt er blevet forstærket via gelelektroforese (trin 3.3). Fordøje dette fragment og pUC19 med Xbal og BamHI (trin 3.5).
- Rens begge præparater (trin 3.4), ligere (trin 3.6), transformere (trin 3.7) og oprette fire 2 ml LB flydende kulturer med ampicillin (100 mg / ml) fra separate kolonier for plasmid rensning via minipreps (trin 3.8).
- Check for insertion af fragmentet i pUC19 via Xbal / BamHI-fordøjelse og gelelektroforese (trin 3.3). Bånd på 2.660 bp og 1.012 bp indikerer korrekt indføring af indsatsen i plasmidet.
- Amplificere 1.016 bp 3'-flankerende region (højre fragment) under anvendelse af primerne cpcC1C2rightfor og cpcC1C2rightrev (Se trin 3.2, tabel 2). Fjerne en lille mængde af PCR-reaktionen og bekræfte, om den korrekte størrelse produkt er blevet amplificeret via gelelektroforese (trin 3.3). Fordøje dette fragment og pUC19 med Sac I og Eco RI (step 3.5).
- Rens begge præparater (trin 3.4), ligere (trin 3.6), transformere (trin 3.7) og oprette fire 2 ml LB flydende kulturer med ampicillin (100 mg / ml) fra separate kolonier for plasmid rensning via minipreps (trin 3.8).
- Check for insertion af fragmentet i pUC19 via SacI / EcoRI-fordøjelse (trin 3.5) og gelelektroforese (trin 3.3). Bånd på 2.660 bp og 1.016 bp indikerer korrekt indføring af indsatsen i plasmidet.
Bemærk: Xba I / Bam HI sites for kloning af 5'-regionen og Sac I / Eco RI til kloning af 3'-regionen i pUC19 anvendes så vidt muligt. Hvis det er muligt, altid omfatte et BamHI-sted på den reverse primer for 5'-området eller den fremadrettede primer for 3'-regionen for at sikre, at senere kloningstrin er lettere at udføre. - Sekvens begge indsatse at bestemme, om sekvensen er korrekt anvendelse af primere spænder over indføringsstedet, fx M13 fremad og M13 revers (tabel 2). Rækkefølgen skal være korrekt for at sikre ingen fejl indføres i flankerende regioner.
- Udskære venstre fragmentet fra pUC19 via Xbal / BamHI-fordøjelse. Fordøje pUC19 + højre fragment med Xbal / BamHI (trin 3.5).
- Oprens 1.012 bp venstre fragment og 3676 bp pUC19 + højre fragment fra en agarosegel (trin 3.3) via excision af DNA'et under anvendelse af en skalpel.
- Rens begge præparater (trin 3.4), ligere (trin 3.6), transformere (trin 3.7) og oprette fire 2 ml LB flydende kulturer med ampicillin (100 mg / ml) fra separate kolonier for plasmid rensning via minipreps (trin 3.8).
- Check for insertion af fragmentet i pUC19 + højre fragment via XbaI / BamHI-fordøjelse (trin 3.5) og gelelektroforese (trin 3.3). Bånd på 3676 bp og 1.012 bp indikerer korrekt indsættelse af indsatsen ind i plasmidet (se dette som plasmid B).
Bemærk: npt1 / sacB kassette behøver ikke at være oprenset fra agarosegeler siden pUM24cm koder for et protein, der giver chloramphenicolresistens. Derfor hvis kolonier dyrkes på LB / ampicillin / kanamycin-agarplader den eneste mulige kombination, der vil føre til resistente kolonier er inkorporering af npt1 / sacB kassetten i plasmid B. - Renses begge præparater (trin 3.4), ligeres (trin 3.6), transformation (trin 3.7) og oprette fire 2 ml LB flydende kulturer med ampicillin (100 ug / ml) og kanamycin (30 ug / ml) fra separate kolonier til plasmidoprensning via minipreps (trin 3.8).
- Check for indsættelse af npt1 / sacB kassetten i plasmid B via Bam HI fordøjelse (trin 3.5) og gelelektroforese (trin 3.3). Bands af 4688 bp og 3894 bp indikerer korrekt isætning af the indsætte i plasmidet (se dette som plasmid A).
- Alternativt stumpe ende af npt1 / sacB kassette og klon til en anden restriktionsendonukleasested mellem venstre og højre fragmenter i plasmid B. npt1 / sacB kassette skal klones mellem venstre og højre fragmenter.
Bemærk: Hvis ekspression af et fremmed kassette er påkrævet, bør dette indsættes mellem venstre og højre fragmenter af plasmid B. Dette plasmid derefter anvendes i de umarkerede knockout trin.
4. generation af Afmærkede Synechocystis og Synechococcus mutanter
- Oprette en ny kultur ved at pode en løkke fuld af celler i 30-50 ml BG11 medium. Grow kulturen i 2-3 dage til OD 750 nm = 0,2 til 0,6.
Bemærk: Typisk individuelle kolonier er for små til at bruge til inokulering og eksponering af de enkelte celler til selv lave niveauer af lys vil resultere i photoinhibition og udvælgelsefor lette resistente mutanter. - Centrifuge 1-2 ml af kulturen ved 2.300 xg i 5 min og kassér supernatanten. Må ikke centrifugeres nogen cyanobakterielle kulturer ved> 2300 xg, da dette kan beskadige cellerne. Vask pelleten gang med BG11-medium.
Bemærk: Du må ikke resuspender celler ved vortex, da dette kan resultere i tab af pili, som er afgørende for DNA-optagelse. Resuspender celler ved forsigtig pipettering. - Tilføj BG11-medium til et slutvolumen på 100 pi. Overfør celler til en 14 ml rundbundet rør.
- Tilsæt 1 ug plasmid A til cellerne og blandes ved en let banken. Tilføj <10 pi plasmid.
Bemærk: Fortrinsvis bør plasmidet være i en koncentration på> 100 ng / pl men koncentrationer lavere end dette er tilstrækkelige til vellykket transformation. - Lay rør ned vandret i inkubatoren. Inkuber kulturer i 4-6 timer.
Bemærk: Celler kan kort blandes ved at trykke hver 1-2 timer, men dette er ikke væsentligt. Prøver kan anbringes i enrysteinkubator selv om dette ikke i væsentlig grad forbedre effektiviteten. - Spred alikvoter af cellekulturen / plasmid-DNA-blanding på BG11 agarplader uden antibiotika. Typisk 20 pi og 80 pi alikvoter spredes på separate plader.
- ~ 24 timer senere tilsættes 2,5-3 ml 0,6% agar opløsning i vand indeholdende kanamycin (per 20 ml: 0,12 g agar, 100 pi 100 mg / ml kanamycin) til agarpladen. Afkøl denne løsning til ~ 42 ° C, og tilsæt til kanten af agarpladen. Vip pladen så løsningen udgør en endnu »topagar» lag på overfladen.
- Inkubér agarplader for en yderligere periode. Kolonier skal være synlig efter ca. 7 dage.
Bemærk: Agar plader kan stables 3 højt i en inkubator. Typisk opnås hundredvis af kolonier pr transformation. - Streak individuelle kolonier på BG11 + kanamycin (30 ug / ml) agarplader. Opdel agarplade i 6 sektorer og bruge en stump ende tandstik til streak udkolonierne end hver enkelt sektor. Opnåelse enkelte kolonier er ikke vigtigt, blot vækst af transformanterne.
- Bekræft markant knockout ved PCR ved anvendelse af Taq-DNA-polymerase ifølge producentens anvisninger. Tilsæt 2 pi MgCl2 (25 mM) pr reaktion.
- Fjerne en lille del af cellerne og overføres til et rør indeholdende 50 pi vand og ~ 20 425-600 um glasperler. Ryst i en vibrator i 5 min ved ~ 2.000 rpm. Centrifuger ved 15.700 xg i 5 min, og 5 pi supernatant per 50 pi PCR-reaktion.
Bemærk: Du må ikke resuspender løsning. Celleresterne skal forblive på bunden af røret.
- Fjerne en lille del af cellerne og overføres til et rør indeholdende 50 pi vand og ~ 20 425-600 um glasperler. Ryst i en vibrator i 5 min ved ~ 2.000 rpm. Centrifuger ved 15.700 xg i 5 min, og 5 pi supernatant per 50 pi PCR-reaktion.
- Godkend mutanter
- Design primere, som spænder over knockout region anvendelse af primer design software (såsom Primer3). Design primere der starter ved ~ 200 bp på hver side af knockout-regionen.
Bemærk: Primere til kontrol cpcC1C2 mutant er skitseret i tabel 2og benævnes cpcC1C2for og cpcC1C2rev. - Amplificere produkter ved hjælp af et program, der består af et indledende denatureringstrin på 95 ° C i 2 minutter, 35 runder af 95 ° C i 1 min, 60 ° C i 1 min, 72 ° C i 1 min pr kb af sekvensen, efterfulgt af en final extension trin på 72 ° C i 5 min. Omfatte en vildtype kontrol. Dette giver typisk ensartede produkter.
- Kontroller genotypen via gelelektroforese. Markeret knockout transformanter vil vise et bånd på ~ 4 kb (0,2 kb fra både venstre og højre fragmenter plus npt1 / sacB kassette) og fraværet af vildtype-båndet (figur 2).
Bemærk: I visse tilfælde observeres ikke en ~ 4 kb bånd i den markerede mutanten grund af den store størrelse af denne PCR-produkt. Men hvis en bånd svarende til den forventede størrelse af vildtype ikke overholdes derefter typisk denne stamme er en markant knockout.
- Design primere, som spænder over knockout region anvendelse af primer design software (såsom Primer3). Design primere der starter ved ~ 200 bp på hver side af knockout-regionen.
- Hvis en vildtype-båndet er stadig til stede derefter igen streak stammen på enfrisk BG11 + kanamycin (30 ug / ml) agarplade og gentage PCR. Gentag re-udstrygning proces, indtil mutanten er adskilt, således at der ikke observeres nogen vildtype-båndet i PCR-reaktionen.
Bemærk: Forøgelse af mængden af kanamycin til en koncentration på 50 ug / ml, derefter 100 ug / ml er undertiden vigtig for at adskille en markant mutant fuldt. - Hvis stammen viser en markant mutant profil via PCR, så re-streak på en frisk BG11 + kanamycin (30 ug / ml) agar plade. Brug denne stamme til at generere den umarkerede knockout.
Bemærk: protokol kan bruges til at generere afmærkede mutanter med blot en antibiotisk resistens-kassette dvs. ved at erstatte npt1 / sacB kassette med netop den npt1 kassetten fra pUC18K 20 mellem venstre og højre fragmenter..
5. generation af markerede Synechocystis mutanter
- Opsæt en frisk kultur præget knockout ved at pode en løkke fuld af calen i 30-50 ml BG11 medium. Grow kulturen i 2-3 dage til OD 750 nm = 0,2 til 0,6.
- Centrifuge 10 ml af kulturen ved 2300 xg i 5 minutter og kassere supernatanten. Der vaskes én gang med BG11-medium.
Bemærk: Du må ikke resuspender celler ved vortex, da dette kan resultere i tab af pili, som er afgørende for DNA-optagelse. Resuspender celler ved forsigtig pipettering. - Tilføj BG11 til et endeligt volumen på 200 pi. Overfør celler til en 14 ml rundbundet rør.
- Tilsæt 1 ug plasmid B DNA til cellerne og blandes ved en let banken.
- Inkuber prøverne for 4-6 timer. Lay rør ned vandret.
Bemærk: Celler kan kort blandes ved at trykke hver 1-2 timer, men dette er ikke væsentligt. Prøver kan anbringes i en rysteinkubator selvom dette ikke forbedrer effektiviteten. - Tilsættes 1,8 ml BG11-medium og inkuberes prøver i i alt 4 dage under omrystning. Dette er tilstrækkelig tid til at tillade rekombination at forekomme i de mange kromosomale kopier.
- Plate portioner af transformationsblandingen på BG11 / 5% saccharose agarplader. Plade 50 pi, 10 pi og 1 pi pr agarplade. Hvis en koloni græsplæne vises på alle disse agarplader fortynde opløsningen yderligere og alikvot af friske plader. Kolonier skal være synlig efter ca. 7 dage.
- Patch 30-50 individuelle kolonier på BG11 + kanamycin (30 ug / ml) agarplader først og BG11 / 5% saccharose agarplader andet ved hjælp af en stump ende tandstikker. Eventuelle bakterier, der vokser på BG11 / 5% sucrose plader, men ikke BG11 + kanamycinplader er potentielle umarkerede knockouts. Bakterier vokser på begge plader er sandsynligvis saccharose resistente på grund af en mutation i sacB genet.
- Kontroller umarkerede knockouts med de samme primere og metode, som blev anvendt til at kontrollere de markerede knockouts. F.eks cpcC1C2for og cpcC1C2rev (tabel 2) til at kontrollere cpcC1C2 umarkerede knockout. En umærket knockout viser et bånd på en agarosegel svarende to vildtype-størrelse minus den deleterede region (figur 2).
- Hvis stammen viser en umærket mutant profil via PCR (trin 4.11.2) og gelelektroforese (figur 2), så re-streak på en frisk BG11 agarplade uden antibiotika.
6. Langsigtet Opbevaring af stammer
- Opsætning af en frisk kultur af stamme ved podning en løkke fuld af celler i 30-50 ml BG11-medium. Grow kulturen i 3-4 dage til OD 750 nm = 0,4 til 0,7.
- Vask cellerne en gang med BG11 og resuspender i ~ 2 ml BG11.
- Tilsættes 0,8 ml koncentreret celler til ét rør. Derpå tilsættes 0,2 ml 80% filtersteriliseret glycerol.
- Valgfrit: Tilføj 0,93 ml koncentreret celler til et andet rør. Tilsættes 0,07 ml DMSO til dette rør.
ADVARSEL: DMSO er giftigt og bør håndteres med passende beskyttelse. - Opbevar begge rør ved -80 ° C. For at genoplive stammer fjerne røret og skrabe nogle celler med en stump tandbryde på en agarplade uden antibiotika. Streak ud som normale anvendelse af en steril løkke.

Figur 1: Plasmidkonstruktion til generering af mærket og umærket knockouts, fx cpcC1 og cpcC2 i Synechocystis (A) Region Synechocystis genomet, hvor (B) cpcC1 og cpcC2 og tilstødende gener er placeret.. Fremhævet i sort er den region af genomet, der skal slettes i mutanten. (C) områder af genomet, som er amplificeret ved PCR. Den 5'-flankerende region (vist med blåt) og 3 'flankerende region (angivet med rødt) forstærkes med restriktionsendonukleasesites til kloning ind i pUC19. Den 5 '(eller 3') flankerende region udskæres ud af pUC19 og indsat i pUC19 + 3 '(eller 59) flankerende region plasmid for at generere plasmid B. (D) Den npt1 / sacB kassette fra pUM24 udskæres via Bam HI fordøjelse og indsættes mellem 5 'og 3' flankerende regioner til at generere Plasmid A. Klik her for at se et større version af denne figur.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Plasmid design er afgørende for en vellykket generation af både markerede og umarkerede mutanter. Figur 1 giver et eksempel af plasmid A og B bruges til at generere en sletning mutant i Synechocystis generne cpcC1 og cpcC2 13. I hvert tilfælde de 5'- og 3'-flankerende regioner er ca. 900-1.000 bp. Reduceret flankerende regioner kan bruges, selv den mindste vi har med succes trialed har været ca. 500 bp. Plasmid B kan også indeholde en genkassette mellem 5 'og 3' ~ 1 kb flankerende regioner eller en modificeret version af den native gensekvens.
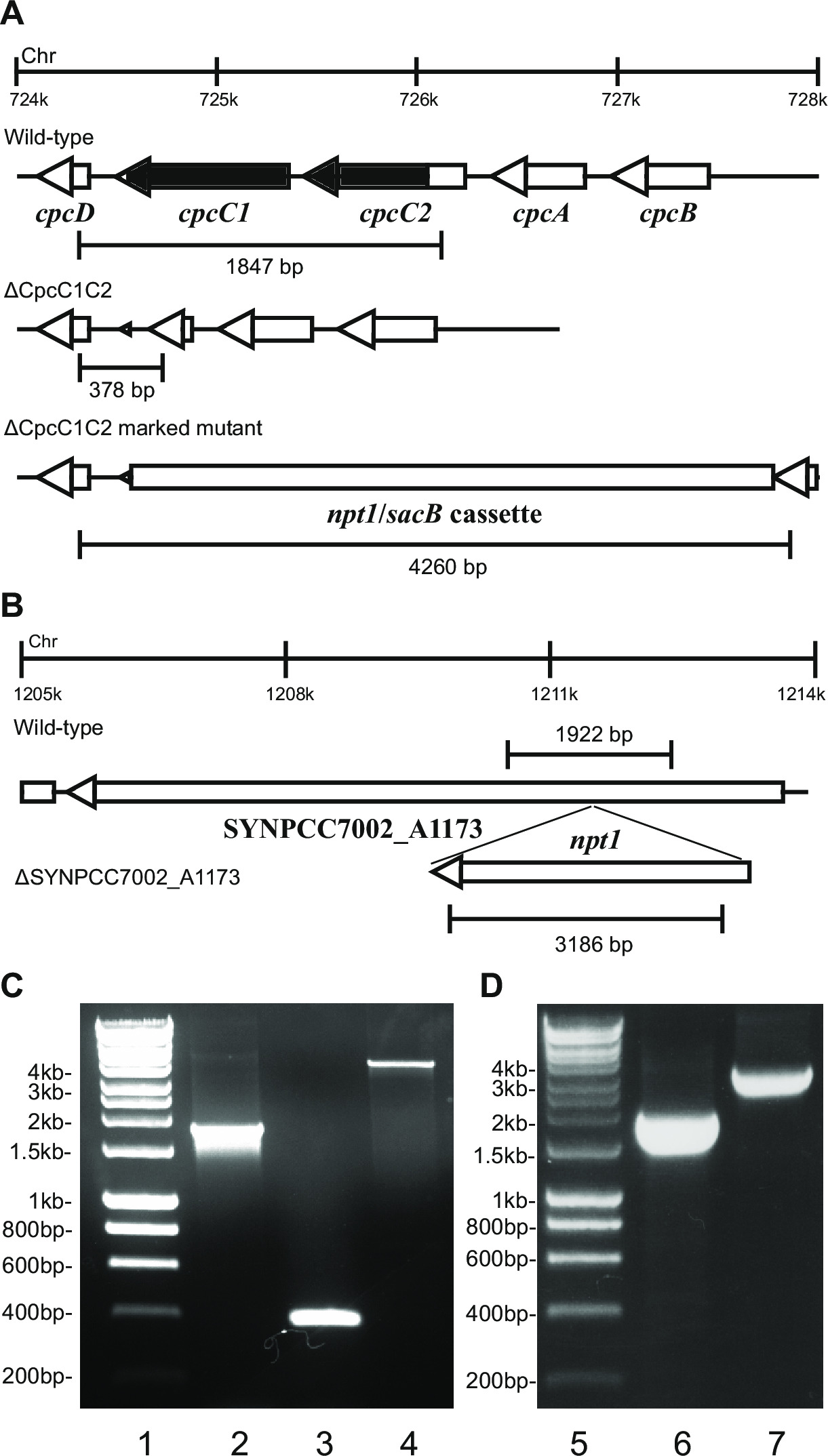
Figur 2: Kontrol af mærket og umærket mutanter, fx cpcC1 / cpcC2
Efter transformation af plasmid A i cellerne, vil typisk flere hundrede kolonier vises på en plade efter ca. 7-10 dage. Kolonier er <1 mm i diameter og vil ikke stige i størrelse for de næste par uger. Derfor er det vigtigt at anvende en stump ende tandstikker at fjerne kolonien og stribefri det på en frisk BG11 + kanamycin-agarplade. Cirka halvdelen af de re-stribede kolonier vil vokse efter 4-6 dage. Hvis generne er ikke-essentielle og mutanter demonstrere vækst svarende til den vildtypestammen under vedvarende lys på 20-40 pmol fotoner m-2 sek-1 (f.eks terminale oxidase mutanter i Lea-Smith et al. 2013 14) (figur 3), så alle kromosomerne bør indeholde en kopi af t han npt1 / sacB kassette indsat sekvens, som bestemt via PCR. Hvis generne er ikke-essentielle og mutanter udviser en langsom vækst fænotype under vedvarende lys på 20-40 pmol fotoner m-2 sek-1 (f.eks fykobilisom deficiente mutanter i Lea-Smith et al. 2014 13) (figur 3), derefter flere runder af re-udstrygning på BG11 agarplader med gradvist øgede mængder kanamycin er afgørende for at opnå et segregeret markeret mutant. Når et segregeret mutant opnås dette bør re-stribet på en frisk BG11 plus kanamycin agarplade at sikre, at adskillelse er færdig. Hvis gentagne runder af striber ikke resulterer i et segregeret markeret mutant så genet sandsynligvis afgørende for overlevelse. Figur 4 giver en oversigt over de eksperimentelle trin involveret i umærket mutant generation.
/54001/54001fig3highres.jpg "Width =" 700 "/>
Figur 3:. Vækst i Synechocystis mutanter Eksempler på mutanter, der demonstrerer (A) tilsvarende vækst til vildtype og (B) langsommere vækst end vildtype. Den ΔCOX mutant mangler cytochrom oxidase skyldes sletning af CtaC1D1E1 gener. Den ΔCyd mutant mangler quinol oxidase skyldes sletning af CydAB gener. Oliven mutant mangler en del af fykobilisom grund sletning af CpcABC1C2D gener. Prøver i (B) blev boblet med luft for at lette vækst. Gengivet fra data offentliggjort i Lea-Smith et al, 2013 14 og 2014 13 (www.plantphysiol.org; Copyright American Society of Plant Biologer).. Klik her for at se en større version af dette tal.
f.eks Figur 2. Hvis der indsættes en genkassette i kromosomet derefter typisk en højere andel af kanamycinresistente og saccharose-resistente kolonier observeret. Disse mutanter kan vokse på saccharose grund af en mutation i sacB genet. Hvis der ikke kanamycin sensitive og saccharose-resistente kolonier genereres derefter genkassetten er skadelig for cellen.

Figur 4: Generering af mærkede og umærkede mutanter i Synechocystis Skematisk detaljering (A) rekombination og (B) eksperimentelle trin involveret i mutant generation.. Plasmid A bliver først blandet med celler. Efter inkubation på agarplader indeholdende kanamycin, kolonier, hvor en rekombinationshændelse forekommer mellem 5'end 3'-flankerende regioner (vist med blåt og rødt, henholdsvis) og den homologe sekvens i kromosomet, isoleres. Desuden er det npt1 / sacB kassette mellem 5 'og 3' flankerende regioner indsat i kromosomet. Efter adskillelse genereres en markant mutant. Markeret mutantceller blandes derefter med plasmid B, som kan indeholde enten (C) 1: 5 'og 3' flankerende regioner; 2: 5'- og 3'-flankerende regioner med en ekspressionskassette indeholdende gener af interesse indsat mellem disse sekvenser; 3: 5'- og 3'-flankerende regioner med vildtypesekvensen med de ønskede nukleotidændringer indsat mellem disse sekvenser. En anden homolog rekombinationsbegivenhed forekommer mellem 5 'og 3' flankerende regioner og de homologe regioner i kromosomet, hvilket resulterer i fjernelse af npt1 / sacB kassette og enten umærket knockout eller en mutant med en insertion eller ændret vild-type region indført i kromosomet. Klik her for at se en større version af dette tal.
| Stock løsning opskrifter | |
| Kemisk | Mængde (g) |
| 100x BG11 (per L) | |
| NaNO 3 | 149,6 |
| MgSO4 .7H 2 O | 7,49 |
| CaCl2 .2H 2 O | 3.6 |
| Citronsyre | 0,6 |
| Tilføj 1,12 ml 0,25 M Na2EDTA, pH 8,0 | |
| 0,25 M Na2EDTA, pH 8,0 (100 ml) | |
| na 2 | 9.3 |
| Sporstoffer (per 100 ml) | |
| H 3 BO 3 | 0,286 |
| MnCI2 .4H 2 O | 0,181 |
| ZnSO4 .7H 2 O | 0.022 |
| Na 2 MoO 4 .2H 2 O | 0,039 |
| CuSO4 .5H 2 O | 0,008 |
| Co (NO3) 2 .6H 2 O | 0,005 |
| Iron stock (per 100 ml) | |
| Ferriammoniumcitrat | 1.11 |
| Phosphat lager (per 100 ml) | |
| K 2 HPO 4 | 3.05 |
| Na 2 CO 3 lager (100 ml) | |
| Na 2 CO 3 | 2 |
| TES-puffer, pH 8,2 (100 ml) | |
| TES | 22.9 |
| NaHCO3 lager (per 100 ml) | |
| NaHCO3 | 8.4 |
| HEPES, pH 8,2 (per 500 ml) | |
| HEPES | 119,15 |
| Vitamin B 12 (per 50 ml) | |
| cyanocobalamin | 0.02 |
| Luria Bertani media (Per 500 ml) | |
| Luria Bertani bouillon | 12.5 |
| 1 M MgCI2 (100 ml) | |
| MgCl2 .6H 2 O | 20,33 |
| MnCI2 .4H 2 O | 0,395 |
| CaCl2 .2H 2 O | 1,47 |
| 2- (N -Morpholino) ethansulfonsyre hydrat, 4-Morpholineethanesulfonic acid (MES) | 0,4265 |
| Opløsning A + glycerol | |
| 10 ml opløsning A | |
| 1,5 ml glycerol |
Tabel 1: Løsninger anvendt i denne undersøgelse.
| Primer | sekvens |
| cpcC1C2leftfor | GTAC TCTAGA GCGGCTAAATGCTACGAC |
| CPCC1C2leftrev | GATC GGATCC GCGGTAATTGTTCCCTTTGA |
| cpcC1C2rightfor | GATC GAGCTC TGCACTGGTCAGTCGTTC |
| cpcC1C2rightrev | GACT GAATTC ATCGTTGCTTGAACGGTCTC |
| M13 frem | TGTAAAACGACGGCCAGT |
| M13 revers | CAGGAAACAGCTATGAC |
| cpcC1C2for | GTTTTCATTGGCATCGGTCT |
| cpcC1C2rev | ATGTCCCAGGAACGACTGAC |
| A1173for | AGCAAACCGTTTTTGTGACC |
| A1173rev | TGCAAGGTGGCGAACTGTAT |
Tabel 2:. Primere anvendt i denne undersøgelse restriktionsendonukleasesteder er understreget.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
De mest kritiske trin i frembringelsen af umærkede mutanter er: 1) omhyggelig plasmid design for at sikre, at kun den målrettede region ændres; 2) at sikre, at prøverne er fortsat axenisk, især når dyrket på saccharose; 3) plating transformerede celler til mærket mutant generation oprindeligt på BG11 agarplader mangler antibiotika, efterfulgt af tilsætning af agar plus antibiotika 24 timer senere; 4) dyrkning markeret mutanter til 4 fulde dage før plating på BG11 plus saccharose agarplader: 5) sikre, at mærket mutanter er fuldt adskilte og 6) grundigt bekræfter genotype mutante stammer. Til dette sidste trin, yderligere primere designet til at amplificere en del af den deleterede region, kan anvendes til at sikre, at den er blevet fjernet. Southern blotting, mens besværlig, kan også anvendes. vores erfaring er dog, at proceduren i dette papir er tilstrækkeligt for korrekt kontrol af mutanter. Denne procedure er også blevet anvendt til at generere markante mutanter i Synechococcus elongatus PCC7942. Imidlertid har gentagen transformation af denne cyanobakterien vist sig udfordrende.
Hvis markeret mutanter ikke kan adskilles så forskellige miljøforhold høj CO 2, svagt lys (<20 pmol fotoner m-2 sek -1) eller yderligere næringsstoffer (dvs. glukose) kan testes. For eksempel tilsætning af glucose er afgørende for at generere fotosystem II mutanter 21. Hvis markeret mutanter aldrig helt adskille så genet er sandsynligvis afgørende for levedygtighed. Der er imidlertid eksempler fra litteraturen, hvor nogle forskningsgrupper har kunnet knockout et gen (for eksempel Vipp i Synechocystis) 22, kun for andre grupper til senere vise, at genet ikke er essentielt 23. Dette kunne skyldes forskelle i vildtypestammer eller forkert plasmid design, hvilket resulterer i polære virkninger på tilgrænsende, essentielle gener. Hvis en mutant ikke fuldt udadskille vi vil anbefale, at plasmid, der indeholder npt1 kassette fra pUC18K 20 mellem venstre og højre fragmenter bruges til transformation. Det er lettere at kontrollere tilstedeværelsen af bånd svarende til vildtype og mutant ved PCR, idet dette fragment er ca. 1,2 kb sammenlignet med 3,8 kb npt1 / sacB kassette. Dette resultat er et vigtigt stykke bevis for, at genet er afgørende.
Generering af umærkede mutanter med indsatte ekspressionskassetter er generelt mere udfordrende end udvikling af knockout-stammer. Vi udtrykker generelt gener under kontrol af den stærke cpcBAC1C2D promotor 13. I nogle tilfælde kan dette mindske chancerne for vellykket indsætning af genet kassetten, hvis over-ekspression af et protein er skadeligt for cellen. Svagere promotorer bør derefter testes. Generelt har vi observeret, at jo større genkassette er, jo vanskeligere er det at isert det i genomet. Vi har ikke været i stand til at indsætte genkassetter større end 5 kb. Man skal også tages i at vælge steder at indsætte ekspressionskassetter i genomet. Neutrale websteder, der ikke påvirker cellernes levedygtighed eller vækst bør anvendes. Eksempler i Synechocystis omfatter phaAB og phaCE, som koder for de proteiner, der koder for polyhydroxybutyrat biosyntesevejen 24,25. For nylig en omfattende liste af neutrale steder i Synechocystis er blevet identificeret 26.
Generation af umarkerede mutanter i cyanobakterier er en langsom proces, der tager cirka 5-7 uger, hvis alle trin gennemfører ordentligt. Dette er langsommere end den standard metode til generering afmærkede udslagsblanketter anvendes af flertallet af forskergrupper undersøger cyanobakterier. Men fleksibiliteten i at kunne indføre yderligere mutationer i umarkerede mutanter delvist kompenserer for dette, da yderligere plasmider containing en række kassetter giver resistens over for forskellige antibiotika, behøver ikke at være konstrueret. Til forskningsformål kan mutere multiple gener er undertiden nødvendigt for fuldt ud at karakterisere den funktionelle rolle af proteiner. For eksempel, vi identificeret en skadelig fænotype kun efter sletning af de to terminal oxidase elektron dræn lokaliseret til den thylakoid membran, da tab af kun én af disse komplekser kan der kompenseres for ved aktivitet af de øvrige 14. Udvikling af en stamme i industrielle applikationer vil også kræve flere ændringer af en stamme, ikke kun for introduktion af fremmede gener, men også at øge fotosyntetiske effektivitet, lys høst optimering og sletning af konkurrerende veje for den ønskede substrat.
Den største faktor, der begrænser hastigheden af umærket mutant generation er den langsomme division tidspunktet for model cyanobakterielle arter, mellem 8-20 timer afhængig af lysforholdene. Unwho højere lysintensiteter og CO 2 koncentrationer, væksten er hurtigere. Der er imidlertid en risiko for, at mutantstammer, der ikke kan tåle enten høj lys eller CO 2, udvælges imod, eller at mutantstammerne vil undergå uønskede ændringer før fænotypisk karakterisering. Dette er derfor ikke anbefales. Det ville imidlertid være yderst fordelagtigt, hvis en hurtigere protokol til at generere umarkerede mutanter blev udviklet. Samlet set vil det lette udviklingen af stammer for både grundforskning og anvendt applikationer. Sådanne stammer kunne bruges til biobrændstof, biomasse eller kemisk produktion eller i forståelsen mange aspekter af cyanobakteriel biokemi, genetik og fysiologi.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Vi er taknemmelige for Environmental Services Association Education Trust, den Syntetisk Biologi i Cambridge SynBio fond og Ministeriet for social retfærdighed og empowerment, Indiens regering, om økonomisk støtte.
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. 3rd Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
