

Introduction
Cyanobacteriën zijn een evolutionair oud en divers stam van bacteriën gevonden in bijna elke natuurlijke omgeving op aarde. In de mariene ecosystemen zijn ze bijzonder overvloedig en spelen een belangrijke rol in vele nutriëntenkringlopen, goed voor ongeveer de helft van koolstof fixatie 1, de meerderheid van stikstofbinding 2 en honderden miljoenen tonnen koolwaterstof productie 3 in de oceanen jaarlijks. Chloroplasten het organel verantwoordelijk is voor de fotosynthese in eukaryote algen en planten, zijn waarschijnlijk zijn geëvolueerd van een cyanobacterie die werd opgeslokt door een gastheer organisme 4. Cyanobacteria nuttig modelorganismen gebleken voor de studie van de fotosynthese, elektronentransport 5 en biochemische wegen, waarvan vele zijn geconserveerd in planten. Bovendien cyanobacteriën worden steeds meer gebruikt voor de productie van levensmiddelen, biobrandstoffen 6, 7 elektriciteit en industriële verbindingen 8, door hun highly efficiënte omzetting van water en CO 2 op biomassa met behulp van zonne-energie 9. Veel soorten kunnen worden gekweekt op niet-landbouwgrond met een minimum aan voedingsstoffen en zeewater, wat suggereert dat cyanobacteriën zou kunnen worden geteeld op grote schaal zonder dat de landbouwproductie. Bepaalde soorten zijn ook bronnen van natuurlijke producten, waaronder antischimmel, antibacteriële en anti-kanker verbindingen 10,11.
Het vermogen om mutanten te genereren is essentieel voor het begrijpen van cyanobacteriën fotosynthese, biochemie en fysiologie en essentieel voor de ontwikkeling van stammen voor industriële doeleinden. De meeste gepubliceerde studies genereren genetisch gemodificeerde stammen door insertie van een antibioticum resistentie cassette in de plaats van belang. Dit beperkt het aantal mutaties die in een rek kan worden ingevoerd, aangezien slechts enkele antibioticaresistentie cassettes zijn beschikbaar voor gebruik in cyanobacteriën. Stammen die genen die antibioticum resistance kan niet worden gebruikt voor industriële productie ter openbare vijvers, die waarschijnlijk de enige kosten-effectieve manier tot biobrandstoffen en andere laagwaardige producten 12 produceren. Het genereren van ongemarkeerde mutanten overwint deze beperkingen. Ongemarkeerde mutanten bevatten geen vreemd DNA, tenzij opzettelijk inbegrepen en kan meerdere malen worden gemanipuleerd. Daarom is het mogelijk om zoveel veranderingen in een stam genereren gewenst. Bovendien kunnen polaire effecten van genen stroomafwaarts van de modificatieplaats worden geminimaliseerd, waardoor nauwkeuriger modificatie van het organisme 13.
Mutante stammen, zelfmoord plasmiden met twee DNA-fragmenten identiek aan gebieden in de cyanobacteriën chromosoom flankeren het gen verwijderd genereren (aangeduid als de 5 'en 3' flankerende gebieden) eerst geconstrueerd. Twee genen worden vervolgens tussen de flankerende gebieden. Eén daarvan codeert voor een antibioticumresistentie-eiwit; de tweede codeert SacB, die produces levansucrase, een verbinding verleent gevoeligheid voor sucrose. In de eerste stap van de werkwijze, gekenmerkt mutanten waarbij sommige stammen bevattende vreemd DNA, gegenereerd. Het plasmide construct gemengd met de cyanobacteriën cellen en het DNA is natuurlijk opgenomen door het organisme. Transformanten worden geselecteerd door groei op agarplaten die het geschikte antibioticum en het mutante genotype geverifieerd door PCR. Zelfmoord plasmiden kan niet repliceren binnen de stam van belang. Daarom zal antibiotica resistente kolonies ontstaan door een recombinatiegebeurtenis waarbij het gen van interesse in het chromosoom ingevoegd. Om ongemarkeerde mutanten te genereren, wordt de gemerkte mutant vervolgens gemengd met een tweede zelfmoord plasmide dat alleen de 5 'en 3' flankerende gebieden. Indien insertie van vreemd DNA is vereist, een plasmide dat bestaat uit de 5 'en 3' flankerende gebieden met een cassette die de genen van belang ingevoegd tussen deze DNA-fragmenten, kan worden gebruikt. Selectie is via groei op agar platen met sucrose. Aangezien sucrose is dodelijk voor cellen wanneer het sacB gen product tot expressie wordt gebracht, de enige cellen die overleven zijn die waarbij een tweede recombinatiegebeurtenis heeft plaatsgevonden, waarbij de sucrose gevoeligheid gen naast het antibioticum resistentiegen, werd uit de gerecombineerde chromosoom en op het plasmide. Als gevolg van de recombinatie uitwisseling worden de flankerende gebieden en alle DNA daartussen ingevoegd in het chromosoom.
We hebben met succes gebruik gemaakt van deze methoden om meerdere chromosomale mutaties in dezelfde stam van Synechocystis sp genereren. PCC6803 (hierna Synechocystis) 13,14, enkelvoudige puntmutaties te introduceren in een gen van interesse 13 en expressie van gencassettes. Terwijl de generatie van ongemarkeerde knockouts heeft voorafgaand aangetoond dat ons werk in Synechocystis 15,16, een gedetailleerde methode, geholpen dooreen visuele presentatie van de kritische stappen, is niet voor iedereen beschikbaar. We hebben ook gebruikt dezelfde methode voor het genereren van duidelijke knockouts in een ander model cyanobacterie, Synechococcus sp. PCC7002 (hierna Synechococcus). Dit protocol voorziet in een duidelijke, eenvoudige methode voor het genereren van mutanten en een snelle protocol voor het valideren en opslaan van deze stammen.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Voorbereiding van Cultuur Media
- Bereid BG11 Medium volgens Castenholz, 1988 17.
- Bereid voorraad oplossingen van 100x BG11, sporenelementen en ijzeren voorraad (tabel 1).
- Bereid afzonderlijke oplossingen van fosfaatvoorraadoplossing, Na 2 CO 3 beelden, N - [tris (hydroxymethyl) methyl] -2-aminoethaansulfonzuur (TES) buffer en NaHCO3 (Tabel 1).
- Autoclaaf het fosfaat en Na 2 CO 3 voorraden. Filter-steriliseren TES buffer en NaHCO3 met 0,2 um filters.
- Bereid BG11 door het combineren van 976 ml water, 10 ml van 100x BG11, 1 ml van sporenelementen en 1 ml van de ijzeren voorraad en autoclaaf de oplossing. Nadat deze oplossing afkoelen tot kamertemperatuur, voeg 1 ml fosfaatvoorraadoplossing, 1 ml Na 2 CO 3 voorraad en 10 ml NaHCO3.
- Voor BG11 vast medium, voeg 15 g agar en 700 ml water tot een flask. Om de tweede kolf, voeg 3 g Na 2 S 2 O 3, 226 ml water, 10 ml van 100x BG11, 1 ml van sporenelementen en 1 ml ijzeren voorraad. Autoclaaf beide oplossingen. Nadat deze oplossing tot kamertemperatuur afgekoeld, combineren en voeg 1 ml fosfaatvoorraadoplossing, 1 ml Na 2 CO 3 beelden, 10 ml TES-buffer en 10 ml NaHCO3.
Noot: oplossingen worden afzonderlijk bereid precipitatie van bepaalde zouten voorkomen.
- Voor selectie op sucrose Bereid een 50% (w / v) sucrose-oplossing. Filter steriliseren van de oplossing met 0,2 urn filters en toevoegen BG11 (100 ml van 50% sucrose tot 900 ml BG11) tot BG11 / 5% sucrose platen produceren.
Opmerking: Weet NaHCO3 niet BG11 / 5% sucrose agar platen toe te voegen. Voeg Na 2 CO 3 normaal. - Voor het kweken van Synechococcus voeg 10 ml van 1 M 4- (2-hydroxyethyl) piperazine-1-ethaansulfonzuur, N - (2-hydroxyethyl) piperazine NR42, - (2-ethaansulfonzuur) (HEPES) en 1 ml van vitamine B 12 (tabel 1) tot 1 liter BG11 medium.
Opmerking: Transformatie van stammen gekweekt in BG11 handel verkrijgbare media aanzienlijk minder efficiënt dan in BG11 media recepten beschreven en wordt daarom niet aanbevolen.
2. De groei van cyanobacteriën stammen
- Cultuur stammen in 100 ml erlenmeyers met een maximaal volume van 50 ml en schud bij 120 rpm. Seal BG11 platen met Parafilm en punctie drie kleine gaten in de zijkant van de plaat om gasuitwisseling mogelijk te maken. Incubeer alle stammen bij 30 ° C onder TL-lampen in een fotobioreactor bij een lichtintensiteit tussen 20-40 umol fotonen m -2 sec -1.
- Gebruik best steriele technieken. Behandel alle cyanobacteriën stammen in een laminaire stroming kap.
Opmerking: Dit is vooral belangrijk als stammen zijn gekweekt met medium dat sucrose, die gemakkelijk kan worden Contaminated.
3. Productie van plasmide constructen
- Ontwerp primersets, inclusief de vereiste restrictie-enzymplaatsen, middels primer design software zoals Primer3 (http://frodo.wi.mit.edu/primer3/), twee ~ 1 kb regio 5 'en 3' van de te amplificeren gen van belang. Raadpleeg de genoomsequentie van de cyanobacteriën soorten via Cyanobase (http://genome.kazusa.or.jp/cyanobase). Zie Tabel 2 voor alle primers hier gebruikt. Bij het ontwerpen van primers rekening met de volgende factoren:
- Controleer of geamplificeerde gebieden omvatten 5 'en 3' gebieden van het gen die worden gemuteerd, bijvoorbeeld figuur 1.
- Niet muteren intergene gebieden onbedoelde mutatie van antisense en niet-coderende RNA's te vermijden. Voor het genereren van mutanten in Synechocystis, verwijzen naar de lijst van transcriptie startplaatsen gedocumenteerd in Mitschke et al., 2011 18, om de mutatie van antisense voorkomenof niet-coderende RNA's.
- Bij het kiezen flankerende gebieden omvatten niet het volledige open leesraam van aangrenzende genen expressie van deze genen in Escherichia coli kunnen verstoren klonen.
- Amplificeren door PCR producten met hoogwaardige DNA polymerase volgens de aanwijzingen van de fabrikant.
Opmerking: In onze ervaring dit enzym produceert weinig fouten.- Opgericht 50 pi PCR-reacties met HF buffer en ofwel 0, 1,5 of 3 pl DMSO. Gebruik 100 ng van genomisch DNA per reactie. Gebruik een programma dat bestaat uit een eerste denaturatiestap van 98 ° C gedurende 30 sec, 35 rondes van 98 ° C gedurende 10 sec, 67 ° C gedurende 30 sec, 72 ° C gedurende 30 seconden, gevolgd door een laatste verlenging stap van 72 ° C gedurende 5 minuten. Dit geeft typisch consistente producten.
- Verifiëren PCR producten en monsters gedigesteerd met endonuclease enzymen voor de juiste maat via gelelectroforese. Run 1% (w / v) agarose gel met daarin 0,02%(V / v) ethidium bromide gedurende 45 minuten bij 100 V.
LET OP: Ethidiumbromide is een potentiële mutagene stof en moeten met de nodige bescherming worden behandeld. - Zuiver PCR producten met behulp van een DNA-zuivering kit volgens instructies van de fabrikant. Ook gebruik maken van deze kit voor de zuivering van plasmide-fragmenten, waaronder stukken gesneden uit agarosegels. Elueren gezuiverde DNA in 14 pi water.
- Voor kloneringsstappen, incubeer restrictie endonuclease reactiemengsels bij 37 ° C gedurende> 1 uur in een totaal volume van 30 pi volgens de instructies van de fabrikant.
- Voor ligatie stappen ligeren DNA-fragmenten bij kamertemperatuur gedurende> 1 uur in een totaal volume van 20 ul, bevattende 5 ui gezuiverde gedigereerde plasmide, 12 ui gezuiverd gedigereerd insert, 2 ui buffer en 1 ui ligase.
- Bereid Escherichia coli DH5a getransformeerde cellen volgens de volgende werkwijze.
- Grow een overnachting E. coli
- Inoculeren van 400 ml LB in een 1 L erlenmeyer met 6 ml 1 M MgCl2 (tabel 1) met 1 ml overnacht cultuur.
- Groeien de cultuur bij 37 ° C bij 220 rpm gedurende ongeveer 4 uur of tot OD 600 nm 0,4-0,6 bereikt.
- Plaats cellen op ijs gedurende 1 uur.
- Centrifugeer bij 2800 xg gedurende 10 minuten om de cellen te pelleteren bij 4 ° C.
- Verwijder supernatant en resuspendeer in 160 ml oplossing A (Tabel 1) en incubeer op ijs gedurende 20 min.
- Centrifugeer bij 2800 xg gedurende 10 minuten om de cellen te pelleteren bij 4 ° C.
- Verwijder supernatant en resuspendeer in 4 ml Oplossing A + glycerol (tabel 1).
- Bereid 50 pi aliquots invriezen in vloeibare N2, bewaar bij -80 ° C.
- Meng 5 ui ligatiemengsel met 50 pi competente cellen en incubeer gedurende 1 uur op ijs.
- Heat shock de cellen bij 42 ° C gedurende 90 sec, followed door incubatie op ijs gedurende 2 minuten.
- Voeg 950 pl LB-medium (Tabel 1) en incubeer bij 37 ° C gedurende 1 uur.
- Aliquot 50 en 200 ui op platen met het geschikte antibioticum, ofwel ampicilline (100 ug / ml) en / of kanamycine (30 ug / ml).
LET OP: Zowel kanamycine en ampicilline zijn giftig en moeten met de nodige bescherming worden behandeld. - Pick en incubeer enkele kolonies in 2 ml LB media geënt met de juiste antibiotica.
- Gij alle plasmiden onder toepassing van een miniprep plasmide zuivering kit volgens instructies van de fabrikant.
- Genereren plasmiden, in dit specifieke voorbeeld kloppen de cpcC1C2 genen, volgens de volgende stappen.
- Versterken de 1012 bp 5 'flankerende gebied (linker fragment) met behulp van primers cpcC1C2leftfor en cpcC1C2leftrev (Zie stap 3.2, tabel 2). Verwijderen van een kleine hoeveelheid van de PCR reactie en bevestigen of dejuiste grootte product is versterkt via gelelektroforese (stap 3.3). Verwerk deze fragment en pUC19 met XbaI en BamHI (stap 3,5).
- Zuiver beide preparaten (stap 3,4), ligeren (stap 3,6), transformatie (stap 3,7) en stel vier 2 ml vloeibaar LB-kweken met ampicilline (100 ug / ml) van afzonderlijke kolonies voor plasmidezuivering via minipreps (stap 3,8).
- Controleer op insertie van het fragment in pUC19 via XbaI / BamHI digestie en gel elektroforese (stap 3,3). Banden van 2660 bp en 1012 bp geven correcte invoering van de insert in het plasmide.
- Versterken de 1016 bp 3 'flankerende gebied (rechts fragment) met behulp van primers cpcC1C2rightfor en cpcC1C2rightrev (Zie stap 3.2, tabel 2). Verwijderen van een kleine hoeveelheid van de PCR reactie en bevestigen dat de juiste grootte product werd geamplificeerd via gelelektroforese (stap 3,3). Verteren dit fragment en pUC19 met Sac I en Eco RI (step 3.5).
- Zuiver beide preparaten (stap 3,4), ligeren (stap 3,6), transformatie (stap 3,7) en stel vier 2 ml vloeibaar LB-kweken met ampicilline (100 ug / ml) van afzonderlijke kolonies voor plasmidezuivering via minipreps (stap 3,8).
- Controleer op insertie van het fragment in pUC19 via SacI / EcoRI digestie (stap 3,5) en gelelektroforese (stap 3,3). Banden van 2660 bp en 1016 bp geven correcte invoering van de insert in het plasmide.
Opmerking: XbaI / BamHI sites voor klonering van de 5 'regio en SacI / EcoRI voor het kloneren van het 3'-gebied in pUC19 gebruikt waar mogelijk. Indien mogelijk, altijd een BamHI-plaats op het omgekeerde primer voor het 5'-gebied en de voorwaartse primer voor het 3'-gebied zodat latere klonering stappen makkelijker uit te voeren. - Sequentie beide inserts te bepalen of de sequentie juist is met behulp van primers overspannen de insertieplaats, bijv M13 voorwaartse en M13 reverse (tabel 2). De volgorde moet correct zijn om te controleren of er geen fouten worden geïntroduceerd in flankerende gebieden.
- Uitsnijden links fragment uit pUC19 via XbaI / BamHI digestie. Digest het pUC19 + juiste fragment met XbaI / BamHI (stap 3,5).
- Zuiver het 1012 bp fragment linker en 3676 bp pUC19 + juiste fragment uit een agarosegel (stap 3,3) via excisie van het DNA met behulp van een scalpel.
- Zuiver beide preparaten (stap 3,4), ligeren (stap 3,6), transformatie (stap 3,7) en stel vier 2 ml vloeibaar LB-kweken met ampicilline (100 ug / ml) van afzonderlijke kolonies voor plasmidezuivering via minipreps (stap 3,8).
- Controleer op insertie van het fragment in pUC19 + juiste fragment via XbaI / BamHI digestie (stap 3,5) en gelelektroforese (stap 3,3). Banden van 3676 bp en 1012 bp duiden juiste plaatsing van het inzetstuk in het plasmide (zie dit als plasmide B).
Opmerking: De NPT1 / sacB-cassette niet te worden gezuiverd uit agarosegels sinds pUM24cm codeert voor een eiwit verleent tegen chlooramfenicol. Dus als kolonies gekweekt op LB / ampicilline / kanamycine agarplaten de enige combinatie die leidt tot resistente kolonies opname van de NPT1 / sacB-cassette in plasmide B. - Zuiver beide preparaten (stap 3,4), ligeren (stap 3,6), transformatie (stap 3,7) en stel vier 2 ml vloeibaar LB-kweken met ampicilline (100 ug / ml) en kanamycine (30 ug / ml) van afzonderlijke kolonies voor plasmidezuivering via minipreps (stap 3.8).
- Controleer voor het inbrengen van de NPT1 / sacB cassette in plasmide B via BamHI spijsvertering (stap 3.5) en gelelektroforese (stap 3.3). Banden van 4688 bp en 3894 bp duiden juiste plaatsing van the invoegen in het plasmide (noemen dit plasmide A).
- Alternatief stompe einde de NPT1 / sacB-cassette en klonen in een andere restrictie-endonuclease plaats tussen de linker en rechter fragmenten in plasmide B. De NPT1 / sacB-cassette worden gekloneerd tussen de linker en rechter fragmenten.
Opmerking: Als expressie van een vreemd cassette dan dient dit te worden ingevoegd tussen de linker en rechter fragmenten van plasmide B. Dit plasmide wordt vervolgens gebruikt in de vrijstaande knockout stappen.
4. Generatie van Marked Synechocystis en Synechococcus Mutanten
- Het opzetten van een nieuwe cultuur door het enten van een lus vol cellen in 30-50 ml BG11 medium. Groeien de kweek gedurende 2-3 dagen bij OD 750 nm = 0,2 tot 0,6.
Opmerking: Typisch individuele kolonies te klein om te gebruiken voor vaccinatie en blootstelling van individuele cellen zelfs lage lichtniveaus leidt tot fotoinhibitie en selectievoor lichte resistente mutanten. - Centrifuge 1-2 ml van de kweek bij 2300 xg gedurende 5 minuten en gooi de supernatant. Gebruik geen cyanobacteriën culturen niet centrifugeren bij> 2300 xg, omdat dit de cellen kunnen beschadigen. Was de pellet één keer met BG11 medium.
Opmerking: Weet cellen niet mengen door vortexen, omdat dit kan leiden tot verlies van pili die essentieel zijn voor DNA-opname zijn. Resuspendeer cellen door zachte pipetteren. - Voeg BG11 medium tot een eindvolume van 100 pl. Overdracht cellen om een 14 ml ronde bodem buis.
- Voeg 1 ug plasmide A aan de cellen en meng door zachtjes te tikken. Toevoegen <10 ul plasmide.
Opmerking: Bij voorkeur is het plasmide moet op een concentratie van> 100 ng / gl, maar lagere concentraties dan deze geschikt zijn voor succesvolle transformatie. - Leg buizen naar beneden horizontaal in de incubator. Incubeer kweken gedurende 4-6 uur.
Opmerking: Cellen kunnen kort gemengd door tikken om de 1-2 uur, maar dit is niet essentieel. Monsters kunnen in worden geplaatstschudincubator hoewel dit niet significant te verbeteren. - Spreid monsters van de celkweek / plasmide-DNA mengsel op BG11 agarplaten zonder antibiotica. Typisch 20 ul en 80 ul aliquots verdeeld op afzonderlijke platen.
- ~ 24 uur later, voeg 2,5-3 ml 0,6% agar in water bevattende kanamycine (20 per ml: 0,12 g agar, 100 ul 100 mg / ml kanamycine) de agarplaat. Koel de oplossing tot ~ 42 ° C, en aan de rand van de agarplaat. Kantel het bord, zodat de oplossing vormt een nog 'top agar' laag op het oppervlak.
- Incubeer agar platen voor een nieuwe periode van tijd. Kolonies moet zichtbaar zijn na ongeveer 7 dagen.
Opmerking: Agar platen kunnen worden gestapeld 3 hoog in een incubator. Typisch honderden kolonies verkregen per transformatie. - Streak individuele kolonies op BG11 + kanamycine (30 ug / ml) agarplaten. Verdeel de agar plaat in 6 sectoren en gebruik maken van een stomp uiteinde tandenstoker streak uitde kolonies over elke individuele sector. Verkrijgen van afzonderlijke kolonies niet belangrijk, alleen de groei van de transformanten.
- Bevestig duidelijke knockout met PCR met gebruik van Taq DNA polymerase volgens de aanwijzingen van de fabrikant. Voeg 2 pl MgCl2 (25 mM) per reactie.
- Verwijderen van een klein deel van de cellen en overbrengen naar een buis die 50 gl water en ~ 20 425-600 urn glasparels. Schud in een vibrator voor 5 min bij 2000 rpm ~. Centrifugeer bij 15.700 xg gedurende 5 minuten en gebruik 5 pl supernatant per 50 ul PCR reactie.
Let op: De oplossing niet mengen. Celafval moet blijven op de bodem van de buis.
- Verwijderen van een klein deel van de cellen en overbrengen naar een buis die 50 gl water en ~ 20 425-600 urn glasparels. Schud in een vibrator voor 5 min bij 2000 rpm ~. Centrifugeer bij 15.700 xg gedurende 5 minuten en gebruik 5 pl supernatant per 50 ul PCR reactie.
- valideren mutanten
- Ontwerp van primers die de knock-out gebied bestrijken middels primer design software (zoals Primer3). Ontwerp primers vanaf ~ 200 bp weerszijden van de knockout regio.
Opmerking: Primers voor het verifiëren van de cpcC1C2 mutant zijn aangegeven in tabel 2en worden aangeduid als cpcC1C2for en cpcC1C2rev. - Amplify producten met een programma dat bestaat uit een eerste denaturatiestap van 95 ° C gedurende 2 minuten, 35 rondes van 95 ° C gedurende 1 min, 60 ° C gedurende 1 minuut, 72 ° C gedurende 1 minuut per kb sequentie, gevolgd door een laatste verlenging stap van 72 ° C gedurende 5 minuten. Onder meer een wild-type controle. Dit geeft typisch consistente producten.
- Controleer het genotype via gelelektroforese. Gemarkeerde knockout transformanten zal een band van ~ 4 kb (0,2 kb uit de linker- en rechter fragmenten plus de NPT1 / sacB-cassette) en de afwezigheid van de wild-type band (Figuur 2) tonen.
Opmerking: In sommige gevallen is een ~ 4 kb band niet waargenomen in het aangegeven mutante vanwege de grote omvang van dit PCR product. Indien een band die overeenkomt met de verwachte grootte van het wild-type niet wordt waargenomen, meestal deze soort is een duidelijke knockout.
- Ontwerp van primers die de knock-out gebied bestrijken middels primer design software (zoals Primer3). Ontwerp primers vanaf ~ 200 bp weerszijden van de knockout regio.
- Indien een wildtype band aanhoudt dan opnieuw strook de druk op eenverse BG11 + kanamycine (30 ug / ml) agar plaat en herhaal de PCR. Herhaal de re strepen proces tot de mutant is gescheiden zodat er geen wild-type band waargenomen in de PCR reactie.
Noot: Het verhogen van kanamycine tot een concentratie van 50 ug / ml, vervolgens 100 ug / ml soms essentieel om een sterke mutant volledig scheiden. - Als de stam een mutant gekenmerkt profiel via PCR toont vervolgens opnieuw strook op een verse BG11 + kanamycine (30 ug / ml) agarplaat. Gebruik deze stam naar de vrijstaande knock-out te genereren.
Opmerking: Het protocol kan worden gebruikt om mutanten met gemerkt onder antibioticaresistentie cassette genereren dwz door vervanging van de NPT1 / sacB-cassette met z'n NPT1 cassette uit pUC18K 20 tussen de linker en rechter fragmenten..
5. Het genereren van gemarkeerde Synechocystis Mutants
- Het opzetten van een nieuwe cultuur van de gemarkeerde knock-out door het enten een lus vol cells in 30-50 ml BG11 medium. Groeien de kweek gedurende 2-3 dagen bij OD 750 nm = 0,2 tot 0,6.
- Centrifuge 10 ml van de kweek bij 2300 xg gedurende 5 minuten en gooi de supernatant. Was eenmaal met BG11 medium.
Opmerking: Weet cellen niet mengen door vortexen, omdat dit kan leiden tot verlies van pili die essentieel zijn voor DNA-opname zijn. Resuspendeer cellen door zachte pipetteren. - BG11 aan een eindvolume van 200 pl. Overdracht cellen om een 14 ml ronde bodem buis.
- Voeg 1 ug plasmide B DNA aan de cellen en meng door zachtjes te tikken.
- Incubeer de monsters gedurende 4-6 uur. Leg buizen naar beneden horizontaal.
Opmerking: Cellen kunnen kort gemengd door tikken om de 1-2 uur, maar dit is niet essentieel. Monsters kunnen in een schudincubator geplaatst hoewel dit niet te verbeteren. - Voeg 1,8 ml BG11 medium en incubeer de monsters gedurende in totaal 4 dagen onder schudden. Dit is voldoende tijd om recombinatie optreden in de verschillende chromosomale kopieën.
- Plaat aliquots van de transformatie mengsel op BG11 / 5% sucrose agar platen. Plaat 50 ul, 10 ul en 1 pi per agarplaat. Als een kolonie gazon weergegeven op deze agarplaten verdun de oplossing verder en aliquot op verse platen. Kolonies moet zichtbaar zijn na ongeveer 7 dagen.
- Patch 30-50 individuele kolonies op BG11 + kanamycine (30 ug / ml) agarplaten eerste en BG11 / 5% sucrose agar platen seconde, met een stomp uiteinde tandenstoker. Alle bacteriën die groeien op BG11 / 5% sucrose borden maar niet BG11 + kanamycine platen zijn potentiële ongemarkeerde knockouts. Bacteriën groeien op beide platen waarschijnlijk sucrose resistent te wijten aan een mutatie in het sacB gen.
- Controleer ongemarkeerde knockouts met behulp van dezelfde primers en methode werd gebruikt om de gemarkeerde knockouts controleren. Bv cpcC1C2for en cpcC1C2rev (tabel 2) voor het verifiëren van de cpcC1C2 ongemarkeerde knock-out. Een ongemarkeerd knock-out zal een band te laten zien op een agarosegel overeenkomstige to het wildtype minus het gedeleteerde gebied (Figuur 2).
- Wanneer de stam een mutant gemarkeerde profiel via PCR (stap 4.11.2) en gelelektroforese (figuur 2) toont vervolgens opnieuw strook op een verse BG11 agarplaat zonder antibiotica.
6. Lange termijn Opslag van de stammen
- Het opzetten van een nieuwe cultuur van de stam door het enten een lus vol cellen in 30-50 ml BG11 medium. Groeien de cultuur voor 3-4 dagen om OD 750nm = 0,4-0,7.
- Was de cellen eenmaal met BG11 en resuspendeer in ~ 2 ml BG11.
- Voeg 0,8 ml geconcentreerde cellen aan één buis. Voeg vervolgens 0,2 ml van 80% glycerol filter gesteriliseerd.
- Optioneel: Voeg 0,93 ml geconcentreerde cellen aan een andere buis. Voeg 0,07 ml DMSO deze buis.
LET OP: DMSO is giftig en moeten met de nodige bescherming worden behandeld. - Bewaar beide buizen bij -80 ° C. Te doen herleven stammen te verwijderen de buis en schraap sommige cellen met een stomp tandpick op een agar plaat zonder antibiotica. Streak als normaal met een steriele lus.

Figuur 1: plasmide constructie voor het genereren van gemarkeerde en ongemarkeerde knockouts, bijvoorbeeld cpcC1 en cpcC2 in Synechocystis (A) Regio van de Synechocystis genoom waarbij (B) cpcC1 en cpcC2 en aangrenzende genen zich bevinden.. Zwart weergegeven is het gebied van het genoom te verwijderen in de mutant. (C) gebieden van het genoom die worden geamplificeerd met PCR. De 5 'flankerende gebied (aangegeven in blauw) en 3' flankerende gebied (aangeduid in het rood) worden geamplificeerd met restrictie-endonucleaseplaatsen voor kloneren in pUC19. De 5 '(of 3') flankerend gebied wordt uitgesneden uit pUC19 en ingevoegd in het pUC19 + 3 '(of 59;) flankerend gebied plasmide plasmide B. (D) De NPT1 / sacB-cassette uit pUM24 uitgesneden via BamHI digestie en ingevoegd tussen de 5 'en 3' flankerende regio A. Plasmide genereren Klik hier om een vergroting versie van deze figuur.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Plasmide ontwerp is essentieel voor succesvolle productie van zowel gemarkeerde en ongemarkeerde mutanten. Figuur 1 geeft een voorbeeld van plasmide A en B gebruikt om een deletie mutant in het Synechocystis genen cpcC1 en cpcC2 13 genereren. In elk geval de 5 'en 3' flankerende gebieden ongeveer 900-1,000 bp. Verminderde flankerende gebieden kunnen worden gebruikt, hoewel de kleinste hebben we met succes uitgetest ongeveer 500 bp is. Plasmide B kan eveneens een gen cassette tussen de 5 'en 3' ~ 1 kb flankerende gebieden of een gewijzigde versie van de natieve gensequentie.
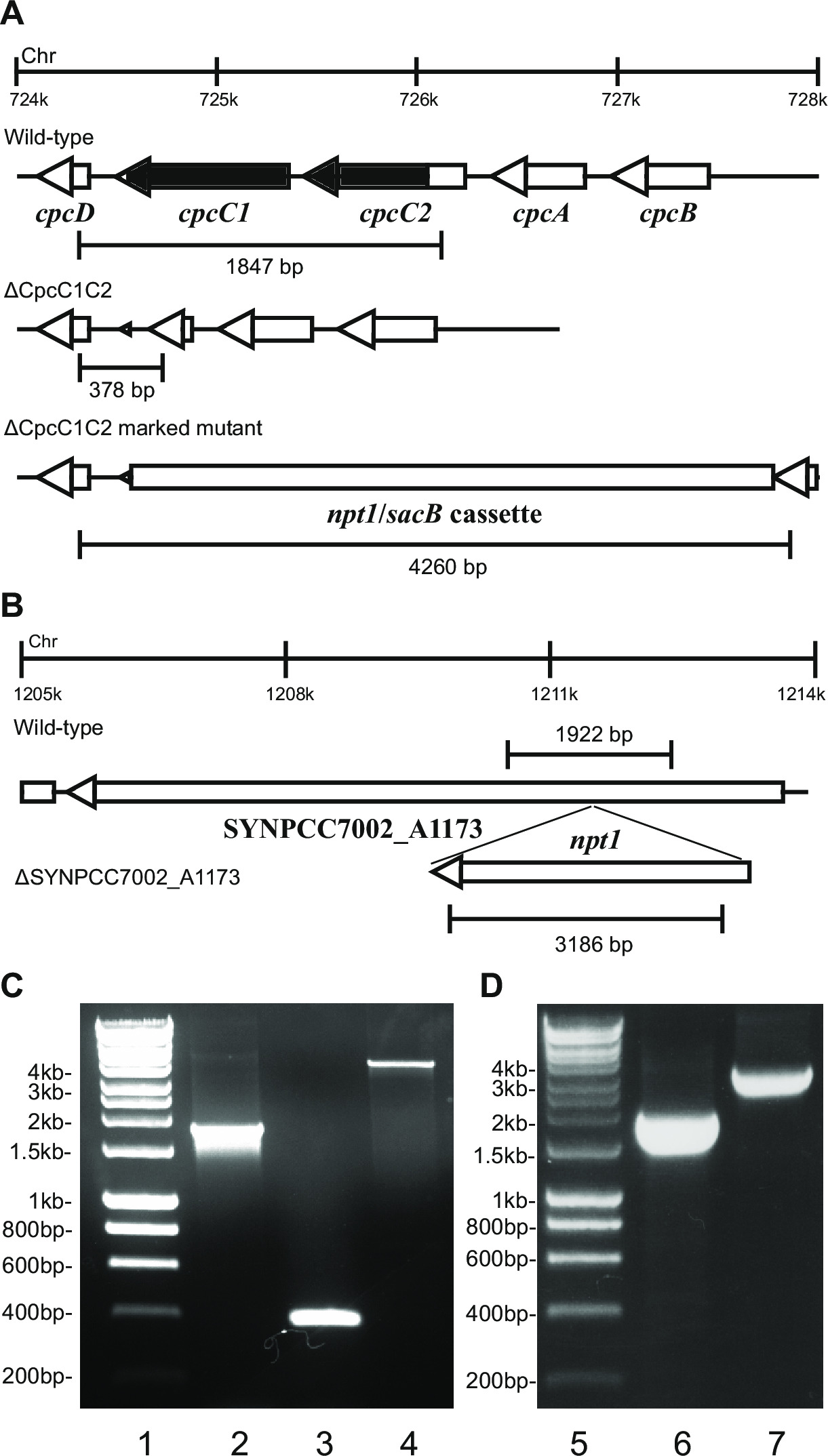
Figuur 2: Verificatie van gemarkeerde en ongemarkeerde mutanten, bijvoorbeeld cpcC1 / cpcC2
Na transformatie van plasmide A in de cellen wordt meestal verscheidene kolonies op een plaat na ongeveer 7-10 dagen. Kolonies <1 mm in diameter en niet groter worden voor de volgende weken. Daarom is het van cruciaal belang voor een stomp uiteinde tandenstoker gebruiken om de kolonie en streak het op een frisse BG11 + kanamycine agar plaat te verwijderen. Ongeveer de helft van de re-gestreepte kolonies zal groeien na 4-6 dagen. Als genen niet essentieel en mutanten tonen groei vergelijkbaar met de wild-type stam onder voortdurend licht van 20-40 umol fotonen m-2 s -1 (bijv terminal oxidase mutanten in Lea-Smith et al., 2013 14) (Fig 3), dan zijn alle chromosomen zou een kopie van t bevatten hij NPT1 / sacB-cassette sequentie bepaald via PCR. Als genen niet essentieel en mutanten vertonen een trage groei fenotype onder continu licht van 20-40 umol fotonen m-2 s -1 (bijv phycobilisome mutanten in Lea-Smith et al., 2014 13) (figuur 3), dan verschillende rondes van opnieuw uitstrijken op BG11 agarplaten met geleidelijk grotere hoeveelheden kanamycine zijn essentieel om een afzonderlijk gemerkt mutant te verkrijgen. Zodra een afzonderlijk mutant wordt verkregen moet dit opnieuw uitgestreken op een verse BG11 plus kanamycine agarplaat zodat segregatie voltooid. Indien herhaalde ronden van streaking niet tot een afzonderlijk gemerkt mutant dan het gen waarschijnlijk essentieel voor overleving. Figuur 4 geeft een overzicht van de experimentele stappen die alleen mutant generatie.
/54001/54001fig3highres.jpg "Width =" 700 "/>
Figuur 3:. Groei van Synechocystis mutanten Voorbeelden van mutanten die aantonen (A) vergelijkbare groei wild-type en (B) tragere groei dan wildtype. De ΔCOX mutant ontbreekt cytochroom oxidase te wijten aan schrapping van de CtaC1D1E1 genen. De ΔCyd mutant ontbreekt quinol oxidase als gevolg van schrapping van de CydAB genen. De olijf mutant ontbreekt een deel van de phycobilisome als gevolg van schrapping van de CpcABC1C2D genen. Monsters (B) werden met lucht geborreld bevorderen van groei. Overgenomen uit gegevens die zijn gepubliceerd in Lea-Smith et al, 2013 14 en 2014 13 (www.plantphysiol.org; Copyright American Society of Plant Biologen).. Klik hier om een grotere versie van deze figuur te bekijken.
zoals figuur 2. Als een gen cassette in het chromosoom is doorgaans een groter aantal kanamycine resistente en sucrose resistente kolonies worden waargenomen wordt ingebracht. Deze mutanten kunnen groeien op sucrose als gevolg van een mutatie in het sacB gen. Indien geen kanamycine gevoelige sucrose resistente kolonies worden vervolgens gegenereerd de gencassette is schadelijk voor de cel.

Figuur 4: Genereren van gemarkeerde en ongemarkeerde mutaties in Synechocystis Schematische detaillering (A) recombinatie en (B) experimentele stappen van mutant generatie.. Een Plasmide wordt eerst gemengd met cellen. Na incubatie op agarplaten die kanamycine, kolonies waarbij een recombinatiegebeurtenis plaats tussen de 5 'eend 3 'flankerende gebieden (aangegeven in blauw en rood, respectievelijk) en de homologe sequentie in het chromosoom, geïsoleerd. Bovendien wordt de NPT1 / sacB-cassette tussen de 5 'en 3' flankerende gebieden ingebracht in het chromosoom. Na scheiding een duidelijke mutant gegenereerd. Gemarkeerd mutante cellen worden vervolgens gemengd met plasmide B die ofwel (C) 1 kan bevatten: de 5 'en 3' flankerende gebieden; 2: de 5 'en 3' flankerende gebieden met een expressiecassette die genen van belang ingevoegd tussen deze sequenties; 3: de 5 'en 3' flankerende gebieden de wild-type sequentie met de gewenste nucleotide veranderingen ingevoegd tussen deze sequenties. Een tweede homologe recombinatie optreedt tussen de 5 'en 3' flankerende gebieden en de homologe gebieden in het chromosoom, waardoor verwijdering van de NPT1 / sacB-cassette en hetzij de vrijstaande knockout mutant of een insertie of gewijzigde wild-type regio geïntroduceerd in het chromosoom. Klik hier om een grotere versie van deze figuur te bekijken.
| Stock oplossing recepten | |
| Chemisch | Bedrag (g) |
| 100x BG11 (per L) | |
| NaNO3 | 149,6 |
| 4 MgSO .7H O 2 | 7.49 |
| CaCl 2 .2H O 2 | 3.6 |
| Citroenzuur | 0.6 |
| Voeg 1,12 ml 0,25 M Na2 EDTA, pH 8,0 | |
| 0,25 M Na2 EDTA, pH 8,0 (per 100 ml) | |
| na 2 | 9.3 |
| Sporenelementen (per 100 ml) | |
| H 3 BO 3 | 0,286 |
| MnCl2 .4H O 2 | 0,181 |
| ZnSO 4 .7H O 2 | 0,022 |
| Na 2 MoO 4 .2H O 2 | 0,039 |
| CuSO4 .5H 2 O | 0,008 |
| Co (NO 3) 2 2 O .6H | 0,005 |
| Ijzeren voorraad (per 100 ml) | |
| Ferric ammoniumcitraat | 1.11 |
| Fosfaat voorraad (per 100 ml) | |
| K 2 HPO 4 | 3.05 |
| Na 2 CO 3 stock (per 100 ml) | |
| Na 2 CO 3 | 2 |
| TES-buffer, pH 8,2 (per 100 ml) | |
| TES | 22.9 |
| NaHCO3 voorraad (per 100 ml) | |
| NaHCO3 | 8.4 |
| HEPES, pH 8,2 (per 500 ml) | |
| HEPES | 119,15 |
| Vitamine B12 (Per 50 ml) | |
| cyanocobalamine | 0.02 |
| Luria Bertani medium (per 500 ml) | |
| Luria Bertani bouillon | 12.5 |
| 1 M MgCl2 (per 100 ml) | |
| MgCl2 .6H O 2 | 20.33 |
| MnCl2 .4H O 2 | 0,395 |
| CaCl 2 .2H O 2 | 1.47 |
| 2- (N -Morpholino) ethaansulfonzuur hydraat, 4-Morpholineethanesulfonic zuur (MES) | 0,4265 |
| Oplossing A + glycerol | |
| 10 ml oplossing A | |
| 1,5 ml glycerol |
Tabel 1: Oplossingen die in deze studie.
| grondverf | Volgorde |
| cpcC1C2leftfor | GTAC TCTAGA GCGGCTAAATGCTACGAC |
| CPCC1C2leftrev | GATC GGATCC GCGGTAATTGTTCCCTTTGA |
| cpcC1C2rightfor | GATC GAGCTC TGCACTGGTCAGTCGTTC |
| cpcC1C2rightrev | GACT GAATTC ATCGTTGCTTGAACGGTCTC |
| M13 forward | TGTAAAACGACGGCCAGT |
| M13 reverse | CAGGAAACAGCTATGAC |
| cpcC1C2for | GTTTTCATTGGCATCGGTCT |
| cpcC1C2rev | ATGTCCCAGGAACGACTGAC |
| A1173for | AGCAAACCGTTTTTGTGACC |
| A1173rev | TGCAAGGTGGCGAACTGTAT |
Tabel 2:. Primers gebruikt in deze studie Restrictie-endonucleaseplaatsen zijn onderstreept.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
De meest kritische stappen in het genereren van ongemarkeerde mutanten zijn: 1) voorzichtig plasmide ontwerp om ervoor te zorgen alleen de beoogde regio is veranderd; 2) ervoor te zorgen dat de monsters blijven axenic, vooral wanneer gekweekt op sucrose; 3) plateren getransformeerde cellen gemerkt mutant generatie aanvankelijk op BG11 agarplaten ontbreekt antibiotica, gevolgd door toevoeging van agar plus antibiotica 24 uur later; 4) het kweken gemarkeerd mutanten voor 4 volle dagen voorafgaand aan de beplating op BG11 plus sucrose agar platen: 5) ervoor te zorgen dat de gemarkeerde mutanten zijn volledig gescheiden en 6) grondig bevestiging van de genotype van mutante stammen. Voor deze laatste stap, aanvullende primers ontworpen om een deel van het verwijderde gebied te amplificeren, kan worden gebruikt om te garanderen dat deze verwijderd is. Southern blotting, terwijl omslachtig, kunnen ook worden gebruikt. Echter, onze ervaring is dat de procedure die in dit artikel is voldoende voor een goede controle van de mutanten. Deze procedure is ook gebruikt om duidelijke mutanten in Synechococ genererencus elongatus PCC7942. Echter herhaalde omzetting van dit cyanobacterie uitdaging gebleken.
Bij een zeer mutanten niet kan worden gescheiden dan verschillende omgevingsomstandigheden hoge CO 2, weinig licht (<20 umol fotonen m-2 s-1) of extra nutriënten (bijvoorbeeld glucose) kunnen worden getest. Bijvoorbeeld, de toevoeging van glucose is noodzakelijk om fotosysteem II mutanten te 21 genereren. Als gemarkeerd mutanten nooit volledig daarna scheiden van het gen is waarschijnlijk essentieel voor de levensvatbaarheid. Er zijn echter voorbeelden uit de literatuur waar een aantal onderzoeksgroepen niet een gen knockout zijn (bijvoorbeeld Vipp in Synechocystis) 22, maar andere groepen later blijkt dat het gen niet essentieel 23. Dit kan te wijten aan verschillen in de wild-type stammen of onjuist plasmide ontwerp, waardoor polaire effecten op aangrenzende essentiële genen. Als een mutant niet volledigscheiden adviseren wij het plasmide dat het NPT1 cassette uit pUC18K 20 tussen de linker en rechter fragmenten worden gebruikt voor transformatie. Het is gemakkelijker om de aanwezigheid van banden die overeenkomen met het wild-type en mutant door PCR verifiëren, omdat dit fragment is ongeveer 1,2 kb, in vergelijking met de 3,8 kb NPT1 / sacB-cassette. Dit resultaat is belangrijk bewijs aantoont dat het gen essentieel.
Genereren van ongemarkeerde mutanten met ingevoegde expressiecassettes is over het algemeen een grotere uitdaging dan de ontwikkeling van de knock-out stammen. We drukken gewoonlijk genen onder controle van de sterke promoter cpcBAC1C2D 13. In sommige gevallen kan dit de kans op succesvolle insertie van het gen cassette verlagen wanneer overexpressie van een eiwit is schadelijk voor de cel. Zwakkere promotors dient dan te worden getest. In het algemeen hebben we opgemerkt dat hoe groter het gen-cassette is, hoe moeilijker het is om invastpakken het in het genoom. We zijn niet in staat om gen cassettes groter dan 5 kb plaatst geweest. Er moet ook rekening worden gehouden bij het kiezen van sites om expressiecassettes te voegen in het genoom. Neutrale sites die geen invloed hebben op levensvatbaarheid van de cellen of groei moet worden gebruikt. Voorbeelden omvatten Synechocystis phaAB en PHACE, die de eiwitten codeert de polyhydroxybutyraat 24,25 biosyntheseroute coderen. Meer recent een uitgebreide lijst van neutrale sites in Synechocystis is geïdentificeerd 26.
Genereren van ongemarkeerde mutanten in cyanobacteriën is een langzaam proces, waarbij ongeveer 5-7 weken of alle stappen goed geleidend. Dit is lager dan de standaard werkwijze voor het genereren gemarkeerde knockouts gebruikt door de meeste onderzoeksgroepen onderzoeken cyanobacteriën. De flexibiliteit van de mogelijkheid om verdere mutaties te introduceren in alleen mutanten gedeeltelijk compenseert dit, aangezien additionele plasmiden containing verschillende cassettes resistentie tegen verschillende antibiotica, hoeven niet te worden geconstrueerd. Voor onderzoeksdoeleinden kan meerdere genen muteren soms nodig om volledig karakteriseren van de functionele rol van eiwitten. Bijvoorbeeld identificeerden we een schadelijk fenotype alleen op deletie van de twee eindstandige oxidase elektron putten gelokaliseerd in het thylakoidmembraan, aangezien verlies van slechts één van deze complexen kunnen worden gecompenseerd door de activiteit van de andere 14. Ontwikkeling van een stam voor industriële toepassingen zal ook vereisen meerdere wijzigingen aan een stam, niet alleen voor de introductie van vreemde genen, maar ook om fotosynthetische efficiëntie, licht oogsten optimalisatie en verwijdering van concurrerende trajecten te verhogen voor het gewenste substraat.
De belangrijkste factor die de snelheid van ongemarkeerde mutant generatie is de langzame divisie tijd van model cyanobacteriën soorten, tussen 8-20 uur afhankelijk van de lichtomstandigheden. Under hogere lichtintensiteiten en de CO 2-concentraties, groei is sneller. Er bestaat echter een risico dat mutante stammen die noch hoogtepunt of CO 2 kan verdragen worden geselecteerd tegen, of dat mutante stammen zal ongewenste veranderingen vóór fenotypische karakterisatie ondergaan. Dus dit is niet aan te raden. Toch zou het zeer voordelig zijn indien een snellere protocol alleen mutanten te genereren ontwikkeld. Kortom, dit zou de ontwikkeling van de stammen voor zowel fundamenteel onderzoek en toegepaste applicaties te vergemakkelijken. Dergelijke stammen kunnen worden gebruikt voor biobrandstof biomassa of chemische productie of begrijpen vele aspecten van cyanobacteriën biochemie, genetica en fysiologie.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
We zijn dankbaar voor de Environmental Services Association Education Trust, de synthetische biologie in Cambridge Synbio fonds en het ministerie van Sociale Rechtvaardigheid en Empowerment, de regering van India, voor financiële steun.
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. 3rd Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
