

Introduction
Les cyanobactéries sont un phylum ancienne et diverse évolutionnaire des bactéries présentes dans presque tous les milieux naturels sur la Terre. Dans les écosystèmes marins , ils sont particulièrement abondants et jouent un rôle clé dans de nombreux cycles de nutriments, ce qui représente environ la moitié de la fixation du carbone 1, la majorité de l' azote fixation 2 et des centaines de millions de tonnes de la production d'hydrocarbures 3 dans les océans chaque année. Chloroplastes, l'organite responsable de la photosynthèse dans les algues et les plantes eucaryotes, sont susceptibles d'avoir évolué à partir d' une cyanobactérie qui a été englouti par un organisme hôte 4. Les cyanobactéries se sont révélés utiles des organismes modèles pour l'étude de la photosynthèse, le transport d'électrons 5 et les voies biochimiques, dont la plupart sont conservés dans les plantes. En plus des cyanobactéries sont de plus en plus utilisé pour la production de denrées alimentaires, les biocarburants 6, 7 et électricité industriels composés 8, en raison de leur salutconversion ghly efficace de l' eau et du CO 2 à la biomasse en utilisant l' énergie solaire 9. De nombreuses espèces peuvent être cultivées sur des terres non arables avec des nutriments minimaux et l'eau de mer, ce qui suggère que les cyanobactéries pourraient être cultivées à grande échelle sans affecter la production agricole. Certaines espèces sont également des sources de produits naturels, y compris les antifongiques, antibactériennes et anti-cancer composés 10,11.
La capacité de générer des mutants est essentielle pour comprendre la photosynthèse des cyanobactéries, la biochimie et la physiologie, et essentielle pour le développement de souches à des fins industrielles. La majorité des études publiées générer des souches génétiquement modifiées par insertion d'une cassette de résistance aux antibiotiques au site d'intérêt. Cela limite le nombre de mutations qui peuvent être introduites dans une souche, car seulement quelques cassettes de résistance aux antibiotiques sont disponibles pour une utilisation dans les cyanobactéries. Les souches contenant les gènes conférant aux antibiotiques resistance ne peut pas être utilisé pour la production industrielle dans les étangs ouverts, ce qui est susceptible d'être le seul moyen rentable pour produire des biocarburants et autres produits à faible valeur 12. La génération de mutants non marqués surmonte ces limitations. les mutants non marqués ne contiennent pas d'ADN étranger, à moins d'inclure intentionnellement, et peuvent être manipulées à plusieurs reprises. Par conséquent, il est possible de générer autant de modifications dans une souche telle que souhaitée. En outre, des effets polaires sur les gènes en aval du site de modification peuvent être minimisés, ce qui permet une modification plus précise de l'organisme 13.
Pour générer des souches de mutants, des plasmides suicides contenant au moins deux fragments d'ADN identiques à des régions dans le chromosome de cyanobactéries flanquant le gène à supprimer (appelée 5 'et 3' des régions adjacentes) sont tout d'abord construit. Deux gènes sont ensuite insérés entre ces régions adjacentes. L'un d'entre eux codant pour une protéine de résistance à un antibiotique; le second code SacB, qui produces levansucrase, un composé conférant une sensibilité au sucrose. Dans la première étape du procédé, les mutants marqués, les souches contenant l' ADN étranger certains sont générés. Le produit de construction de plasmide est mélangé avec les cellules de cyanobactéries, et l'ADN est repris naturellement par l'organisme. Les transformants sont sélectionnés par croissance sur des plaques d'agar contenant l'antibiotique approprié et le génotype mutant vérifié par PCR. plasmides de suicide ne peuvent pas se répliquer dans la souche d'intérêt. Par conséquent, toutes les colonies résistantes aux antibiotiques vont résulter d'un événement de recombinaison dans lequel le gène d'intérêt inséré dans le chromosome. Pour générer des mutants non marqués, le mutant marqué est ensuite mélangé avec un second plasmide suicide ne contenant que les régions flanquantes 5 'et 3'. Cependant, si l'insertion de l'ADN étranger est nécessaire, un plasmide consistant en 5 'et 3' des régions avec une cassette contenant les gènes d'intérêt inséré entre ces fragments d'ADN flanquant, peut être utilisé. Selection est par croissance sur des plaques de gélose contenant du saccharose. Sous forme de saccharose est létal pour les cellules lorsque le produit du gène sacB est exprimé, les seules cellules qui survivent sont ceux dans lesquels un deuxième événement de recombinaison a eu lieu, de sorte que le gène de la sensibilité du saccharose, en plus du gène de résistance aux antibiotiques, a été recombinée de la le chromosome et sur le plasmide. En conséquence de l'échange par recombinaison, les régions de flanquement et des ADN entre eux sont insérés dans le chromosome.
Nous avons utilisé avec succès ces méthodes pour générer de multiples mutations chromosomiques dans la même souche de Synechocystis sp. PCC6803 (ci - après dénommé Synechocystis) 13,14, pour introduire des mutations ponctuelles dans un gène d'intérêt et 13 pour l' expression de cassettes de gènes. Alors que la génération de KO non marqués a été démontrée avant notre travail dans Synechocystis 15,16, une méthode détaillée, aidé parune présentation visuelle des étapes critiques, ne sont pas accessibles au public. Nous avons également appliqué la même méthode pour la production de KO marquées dans un autre modèle cyanobactérie, Synechococcus sp. PCC7002 (ci - après dénommé Synechococcus). Ce protocole offre une méthode claire et simple pour générer des mutants et un protocole rapide pour la validation et le stockage de ces souches.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Préparation de la culture des médias
- Préparer le milieu BG11 selon Castenholz 1988 17.
- Préparer des solutions mères de BG11 100x, oligo - éléments et fer stock (tableau 1).
- Préparer des solutions séparées de stocks de phosphate, Na 2 CO 3 actions, N - [Tris (hydroxyméthyl) méthyl] -2-aminoéthanesulfonique (TES) tampon et NaHCO 3 (Tableau 1).
- Autoclave le phosphate et Na 2 CO 3 actions. Filtre-stériliser le tampon TES et NaHCO 3 avec 0,2 um filtres.
- Préparer BG11 en combinant 976 ml d'eau, 10 ml de 100x BG11, 1 ml d'oligo-éléments et 1 ml de fer stock et autoclave la solution. Après cette solution est refroidie à la température ambiante, ajouter 1 ml de bouillon de phosphate, 1 ml de Na 2 CO 3 stock et 10 ml de NaHCO 3.
- Pour BG11 milieu solide, ajouter 15 g d'agar-agar et 700 ml d'eau à un flask. Pour le second flacon, ajouter 3 g de Na 2 S 2 O 3, 226 ml d'eau, 10 ml de 100x BG11, 1 ml d'oligo - éléments et 1 ml de fer stock. Autoclave les deux solutions. Après ces solutions sont refroidies à la température ambiante, mélanger et ajouter 1 ml de stock de phosphate, 1 ml de Na 2 CO 3 actions, 10 ml de tampon TES, et 10 ml de NaHCO 3.
Remarque: Les solutions sont préparées séparément pour éviter la précipitation de certains sels.
- Pour la sélection de saccharose, de préparer une 50% (p / v) de saccharose. Filtre stériliser la solution avec 0,2 um filtres et ajouter à BG11 (100 ml de saccharose à 50% à 900 ml de BG11) pour produire BG11 / 5% des plaques de saccharose.
Remarque: Ne pas ajouter de NaHCO 3 à BG11 / 5% des plaques d'agar saccharose. Ajouter Na 2 CO 3 comme normale. - Pour la culture de Synechococcus ajouter 10 ml de 1 M de 4- (2-hydroxyéthyl) pipérazine-1-éthanesulfonique , l' acide N - (2-hydroxyéthyl) pipérazine NR42, - (2-éthanesulfonique) (HEPES) et 1 ml de vitamine B 12 (tableau 1) à 1 litre de milieu BG11.
Note: Transformation de souches cultivées en vente dans le commerce des médias de BG11 est nettement moins efficace que dans les recettes des médias de BG11 décrits ici et est donc pas recommandé.
2. Croissance de cyanobactérielles Souches
- Les souches de la culture dans 100 ml de flacons coniques ayant un volume maximum de 50 ml et agiter à 120 tours par minute. Sceller plaques BG11 avec Parafilm et ponction trois petits trous dans le côté de la plaque pour permettre l'échange de gaz. Incuber toutes les souches à 30 ° C sous les ampoules fluorescentes dans un photobioréacteur à une intensité lumineuse entre 20-40 pmol photons m -2 s -1.
- Utiliser les meilleures techniques stériles. Manipuler toutes les souches de cyanobactéries dans une hotte à flux laminaire.
Note: Ceci est particulièrement important lorsque les souches sont mises en culture avec un milieu contenant du saccharose, qui peut être facilement contaminATED.
3. Génération du plasmide Constructs
- Conception ensembles d'amorces, y compris les sites d'enzymes de restriction requis, en utilisant un logiciel de conception d'amorce tels que Primer3 (http://frodo.wi.mit.edu/primer3/), pour amplifier deux ~ 1 régions kb 5 'et 3' de la gène d'intérêt. Consulter la séquence du génome de l'espèce cyanobactériens via Cyanobase (http://genome.kazusa.or.jp/cyanobase). Voir le tableau 2 pour toutes les amorces utilisées ici. Lors de la conception des amorces considèrent les facteurs suivants:
- Veiller à ce que les régions amplifiées comprennent 5 'et 3' du gène qui sera muté, par exemple la figure 1.
- Ne pas muter les régions intergéniques pour éviter la mutation involontaire d'antisens et des ARN non codants. Pour la génération de mutants dans Synechocystis, reportez - vous à la liste des sites de départ de transcription documentés dans Mitschke et al., 2011 18, afin d'éviter la mutation du antisensou des ARN non codants.
- Lorsque des régions flanquant le choix ne comprennent pas l'ensemble du cadre de lecture ouvert de gènes adjacents comme l' expression de ces gènes dans Escherichia coli peuvent interférer avec le clonage.
- Amplifier produits par PCR en utilisant une haute fidélité de l'ADN polymérase selon les instructions du fabricant.
Remarque: Dans notre expérience cette enzyme produit quelques erreurs.- Mettre en place des réactions de PCR 50 ul contenant du tampon HF et 0, 1,5 ou 3 pi de DMSO. En utilisant 100 ng d'ADN génomique par réaction. Utiliser un programme consistant en une étape de dénaturation initiale de 98 ° C pendant 30 secondes, 35 cycles de 98 ° C pendant 10 s, 67 ° C pendant 30 secondes, 72 ° C pendant 30 secondes, suivie d'une étape d'extension finale de 72 ° C pendant 5 min. Cela donne généralement des produits homogènes.
- Vérifier les produits de PCR et les échantillons digérés avec des endonucléases pour la bonne taille par électrophorèse sur gel. Exécuter 1% (p / v) de gels d'agarose contenant 0,02%(V / v) de bromure d'éthidium pendant 45 min à 100 V.
ATTENTION: Le bromure d'éthidium est un mutagène potentiel et doit être manipulé avec une protection appropriée. - On purifie les produits de PCR en utilisant un kit de purification d'ADN selon les instructions du fabricant. également utiliser ce kit pour la purification de fragments de plasmides, y compris des pièces découpées à partir de gels d'agarose. Eluer ADN purifié dans 14 pi d'eau.
- Pour les étapes de clonage, incuber les endonucléases de restriction mélanges réactionnels à 37 ° C pendant> 1 heure dans un volume total de 30 ul conformément aux instructions du fabricant.
- Pour les étapes de ligature, des fragments d'ADN ligaturer à la température ambiante pendant> 1 heure dans un volume total de 20 ul, contenant 5 ul de plasmide digéré purifié, 12 ul d'insert digéré purifié, 2 pi de tampon et 1 ul de ligase.
- Préparer des cellules transformées d' Escherichia coli DH5a selon la méthode suivante.
- Cultivez un E. nuit coli
- Inoculer 400 ml de LB dans un flacon conique de 1 L contenant 6 ml de 1 M de MgCl2 (tableau 1) avec 1 ml de culture du jour au lendemain.
- Cultiver la culture à 37 ° C à 220 rpm pendant environ 4 heures ou jusqu'à ce que la DO 600nm atteint 0,4-0,6.
- Placez les cellules sur la glace pendant 1 h.
- Centrifugeuse à 2.800 xg pendant 10 min pour sédimenter les cellules à 4 ° C.
- Retirer le surnageant et remettre en suspension dans 160 ml de solution A (tableau 1) et on incube sur de la glace pendant 20 minutes.
- Centrifugeuse à 2.800 xg pendant 10 min pour sédimenter les cellules à 4 ° C.
- Retirer le surnageant et remettre en suspension dans 4 ml de solution A + glycérol (tableau 1).
- Préparer 50 aliquotes, geler en N 2 liquide, conserver à -80 ° C.
- Mélanger 5 pl de mélange de ligature avec 50 ul de cellules compétentes et on incube pendant 1 heure sur de la glace.
- Choc thermique des cellules à 42 ° C pendant 90 secondes, follomené par incubation sur la glace pendant 2 min.
- Ajouter 950 pi de milieu LB (tableau 1) et on incube à 37 ° C pendant 1 heure.
- Aliquote 50 et 200 pi sur des plaques avec l'antibiotique approprié, que ce soit à l'ampicilline (100 pg / ml) et / ou de la kanamycine (30 pg / ml).
ATTENTION: Les deux kanamycine et à l'ampicilline sont toxiques et doivent être manipulés avec une protection appropriée. - Choisissez et incuber des colonies isolées dans 2 ml de milieu LB inoculés avec l'antibiotique approprié.
- On purifie tous les plasmides en utilisant un kit de purification de plasmide miniprep selon les instructions du fabricant.
- Générer des plasmides, dans cet exemple spécifique pour frapper les gènes cpcC1C2, selon les étapes suivantes.
- Amplifier la région 1012 pb 5 'flanquant (fragment gauche) en utilisant des amorces cpcC1C2leftfor et cpcC1C2leftrev (Voir l' étape 3.2, tableau 2). Retirer une petite quantité de la réaction PCR et confirmer si leproduit de taille correcte a été amplifié par électrophorèse sur gel (étape 3.3). Digest ce fragment et pUC19 avec Xba I et Bam HI (étape 3.5).
- Purifier les deux préparations (étape 3.4), ligaturer (étape 3.6), transformer (étape 3.7) et mis en place quatre 2 ml LB cultures liquides avec ampicilline (100 pg / ml) à partir de colonies distinctes pour la purification de plasmide via minipreps (étape 3.8).
- Vérifier l' insertion du fragment dans pUC19 par Xba I / Bam HI digestion et l' électrophorèse sur gel (étape 3.3). Des bandes de 2660 pb et 1012 pb indiquent l'introduction correcte de l'insert dans le plasmide.
- Amplifier la région 1.016 pb 3 'flanquant (fragment à droite) en utilisant des amorces cpcC1C2rightfor et cpcC1C2rightrev (Voir l' étape 3.2, tableau 2). Retirer une petite quantité de la réaction PCR et confirmer si le produit de la taille correcte a été amplifié par électrophorèse sur gel (étape 3.3). Digest ce fragment et pUC19 avec Sac I et Eco RI (step 3.5).
- Purifier les deux préparations (étape 3.4), ligaturer (étape 3.6), transformer (étape 3.7) et mis en place quatre 2 ml LB cultures liquides avec ampicilline (100 pg / ml) à partir de colonies distinctes pour la purification de plasmide via minipreps (étape 3.8).
- Vérifier l' insertion du fragment dans pUC19 par Sac I / Eco RI de digestion (étape 3.5) et l' électrophorèse sur gel (étape 3.3). Des bandes de 2660 pb et 1016 pb indiquent l'introduction correcte de l'insert dans le plasmide.
Remarque: Xba I / Bam HI sites de clonage de la région 5 'et Sac I / Eco RI pour le clonage de la région 3' pUC19 sont utilisées dans la mesure du possible. Si possible, toujours inclure un site Bam HI sur l'amorce inverse pour la «région ou l'amorce directe pour la 3 '5 région pour veiller à ce que les étapes de clonage plus tard sont plus faciles à réaliser. - Séquence deux inserts pour déterminer si la séquence est correcte en utilisant des amorces couvrant le site d'insertion, par exemple M13 vers l' avant et inverse M13 ( voir le tableau 2). La séquence doit être correcte pour assurer qu'aucune erreur ne sont introduits dans des régions adjacentes.
- Exciser le fragment de gauche pUC19 par Xba I / Bam HI digestion. Digest pUC19 + fragment droit avec Xba I / Bam HI (étape 3.5).
- On purifie le fragment de 1 012 pb et 3676 pb gauche pUC19 + fragment de droite à partir d'un gel d'agarose (étape 3.3) par l'intermédiaire de l'excision de l'ADN en utilisant une lame de scalpel.
- Purifier les deux préparations (étape 3.4), ligaturer (étape 3.6), transformer (étape 3.7) et mis en place quatre 2 ml LB cultures liquides avec ampicilline (100 pg / ml) à partir de colonies distinctes pour la purification de plasmide via minipreps (étape 3.8).
- Vérifiez l' insertion du fragment dans pUC19 + fragment droit via Xba I / Bam HI digestion (étape 3.5) et électrophorèse sur gel (étape 3.3). Des bandes de 3676 pb et 1012 pb indiquent une insertion correcte de l'insert dans le plasmide (voir à ce que le plasmide B).
Remarque: La cassette de NPT1 / sacB n'a pas besoin d'être purifiés à partir de gels d'agarose depuis pUM24cm code pour une protéine conférant une résistance au chloramphénicol. Par conséquent , si les colonies sont cultivées sur LB / ampicilline / kanamycine plaques de gélose , la seule combinaison possible qui conduira à des colonies résistantes est l' incorporation de la NPT1 / Cassette sacB dans le plasmide B. - Purifier les deux préparations (étape 3.4), ligaturer (étape 3.6), à transformer (étape 3.7) et mis en place quatre 2 ml de LB cultures en milieu liquide avec de l'ampicilline (100 pg / ml) et de kanamycine (30 pg / ml) à partir des colonies distinctes pour la purification de plasmides via minipreps (étape 3.8).
- Vérifier l'insertion de la NPT1 / la cassette sacB dans le plasmide B par digestion BamHI (étape 3.5) et l' électrophorèse sur gel (étape 3.3). Des bandes de 4688 pb et 3894 pb indiquent une insertion correcte de the insérer dans le plasmide (voir à ce que le plasmide A).
- Alternativement, l'extrémité émoussée NPT1 / Cassette sacB et clone dans un site d'endonucléase de restriction différente entre les fragments gauche et à droite dans le plasmide B. Le NPT1 / Cassette sacB doit être cloné entre les fragments gauche et droite.
Remarque: Si l'expression d'une cassette étrangère est nécessaire, cela devrait être inséré entre les fragments gauche et droite du plasmide B. Ce plasmide est ensuite utilisé dans les étapes knockout non marquées.
4. Génération de signalisés Mutants Synechocystis et Synechococcus
- Mettre en place une culture fraîche en inoculant une boucle complète de cellules dans 30-50 ml de milieu de BG11. Cultiver la culture pendant 2-3 jours pour DO 750nm = 0,2 à 0,6.
Remarque: En règle générale des colonies individuelles sont trop petites à utiliser pour l'inoculation et l'exposition des cellules individuelles à même les niveaux de lumière faible se traduira par photoinhibition et la sélectionpour les mutants résistants à la lumière. - Centrifugeuse 1-2 ml de la culture à 2300 g pendant 5 minutes et éliminer le surnageant. Ne pas centrifuger des cultures de cyanobactéries à> 2,300 xg car cela pourrait endommager les cellules. Laver le culot une fois avec un milieu de BG11.
Note: Ne pas remettre en suspension les cellules par tourbillonnement, car cela pourrait entraîner une perte de pili qui sont essentiels pour l'absorption d'ADN. Resuspendre les cellules par pipetage doux. - Ajouter au milieu BG11 à un volume final de 100 ul. Transfert des cellules dans un tube à fond rond de 14 ml.
- Ajouter 1 pg de plasmide A aux cellules et mélanger en tapotant doucement. Ajouter <10 ul de plasmide.
Remarque: De préférence, le plasmide doit être à une concentration de> 100 ng / pl, mais des concentrations plus faibles que celui-ci sont suffisantes pour une transformation réussie. - Lay tubes à l'horizontale dans l'incubateur. Incuber cultures pour 4-6 heures.
Remarque: Les cellules peuvent être brièvement mélangés en tapant tous les 1-2 heures, mais ce ne sont pas essentiels. Les échantillons peuvent être placés dans unsecouant incubateur bien que cela n'améliore pas de manière significative l'efficacité. - Étaler aliquotes du mélange d'ADN de culture cellulaire / plasmide sur des plaques d'agar sans antibiotiques BG11. Typiquement 20 pi et 80 aliquotes sont étalées sur des plaques séparées.
- ~ 24 h plus tard, ajouter 2,5 à 3 ml de solution de gélose à 0,6% dans de l'eau contenant de la kanamycine (par 20 ml: 0,12 g d'agar, 100 ul de 100 mg / ml de kanamycine) à la plaque de gélose. Refroidir cette solution à ~ 42 ° C, et ajouter au bord de la plaque de gélose. Inclinez la plaque de sorte que la solution forme un 'agar top' couche uniforme sur la surface.
- Incuber plaques d'agar pour une nouvelle période de temps. Colonies devraient être visibles après environ 7 jours.
Remarque: Les plaques de gélose peuvent être empilés 3 élevée dans un incubateur. Typiquement, des centaines de colonies sont obtenues par transformation. - Strie colonies individuelles sur BG11 + kanamycine (30 ug / ml) des plaques de gélose. Divisez la plaque de gélose en 6 secteurs et utiliser un cure-dent fin émoussée série surles colonies sur chaque secteur. L'obtention de colonies isolées est pas importante croissance, juste des transformants.
- Confirmer KO marquée par PCR en utilisant l'ADN polymérase Taq selon les instructions du fabricant. Ajouter 2 ul de MgCl2 (25 mM) par réaction.
- Retirer une petite proportion des cellules et transférer dans un tube contenant 50 pi d'eau et 20 ~ 425-600 perles de verre um. Agiter dans un vibreur pendant 5 min à ~ 2000 tours par minute. Centrifuger à 15700 xg pendant 5 min et utiliser 5 pi de surnageant par 50 pi réaction PCR.
Note: Ne pas remettre en suspension la solution. Les débris cellulaires doit rester au fond du tube.
- Retirer une petite proportion des cellules et transférer dans un tube contenant 50 pi d'eau et 20 ~ 425-600 perles de verre um. Agiter dans un vibreur pendant 5 min à ~ 2000 tours par minute. Centrifuger à 15700 xg pendant 5 min et utiliser 5 pi de surnageant par 50 pi réaction PCR.
- Validez mutants
- amorces de conception qui couvrent la région à élimination directe en utilisant un logiciel de conception d'amorce (comme Primer3). Concevoir des amorces à partir de ~ 200 pb de chaque côté de la région de knock-out.
Note: Amorces pour vérifier le mutant cpcC1C2 sont décrits dans le tableau 2et sont appelés cpcC1C2for et cpcC1C2rev. - Amplifier les produits à l'aide d'un programme comprenant une étape de dénaturation initiale de 95 ° C pendant 2 min, 35 cycles de 95 ° C pendant 1 min, 60 ° C pendant 1 min, 72 ° C pendant 1 minute par kb de la séquence, suivi d'un étape d'extension finale de 72 ° C pendant 5 min. Inclure un contrôle de type sauvage. Cela donne généralement des produits homogènes.
- Vérifiez le génotype par électrophorèse sur gel. Transformants knockout signalisés montreront une bande de ~ 4 kb (0,2 kb à partir de deux fragments gauche et droit ainsi que le NPT1 / Cassette sacB) et l'absence de la bande de type sauvage (Figure 2).
Remarque: Dans certains cas, une bande de 4 kb ~ est pas observée dans le mutant marqué en raison de la grande taille de ce produit de PCR. Toutefois, si une bande correspondant à la taille attendue de type sauvage n'a pas observé alors typiquement cette souche est un KO marquée.
- amorces de conception qui couvrent la région à élimination directe en utilisant un logiciel de conception d'amorce (comme Primer3). Concevoir des amorces à partir de ~ 200 pb de chaque côté de la région de knock-out.
- Si une bande de type sauvage est toujours présent puis re-strie la souche sur unBG11 frais + kanamycine (30 pg / ml) plaque de gélose et répéter la PCR. Répétez le processus de re-strie jusqu'à ce que le mutant est distinct de sorte qu'aucune bande de type sauvage est observée dans la réaction PCR.
Note: L'augmentation de la quantité de kanamycine à une concentration de 50 pg / ml, puis 100 ug / ml est parfois indispensable pour isoler un mutant marqué pleinement. - Si la souche présente un profil mutant marqué par PCR, puis ré-série sur un BG11 frais + kanamycine (30 pg / ml) de plaque de gélose. Utilisez cette souche pour générer le knock-out non marqué.
Remarque: Le protocole peut être utilisé pour générer des mutants marqués avec seulement une cassette de résistance aux antibiotiques -à- dire en remplaçant la NPT1 / la cassette sacB avec seulement la cassette de NPT1 pUC18K 20 entre les fragments de gauche et de droite..
5. Génération de Unmarked Synechocystis Mutants
- Mettre en place une nouvelle culture de l'élimination directe marquée par l'inoculation d'une boucle complète de caunes dans 30-50 ml de milieu de BG11. Cultiver la culture pendant 2-3 jours pour DO 750nm = 0,2 à 0,6.
- Centrifuger de 10 ml de la culture à 2300 g pendant 5 minutes et éliminer le surnageant. Laver une fois avec un milieu de BG11.
Note: Ne pas remettre en suspension les cellules par tourbillonnement, car cela pourrait entraîner une perte de pili qui sont essentiels pour l'absorption d'ADN. Resuspendre les cellules par pipetage doux. - BG11 ajouter à un volume final de 200 ul. Transfert des cellules dans un tube à fond rond de 14 ml.
- Ajouter 1 pg d'ADN plasmidique B aux cellules et mélanger en tapotant doucement.
- Incuber les échantillons pendant 4-6 heures. Poser les tubes à l'horizontale.
Remarque: Les cellules peuvent être brièvement mélangés en tapant tous les 1-2 heures, mais ce ne sont pas essentiels. Les échantillons peuvent être placés dans un incubateur avec agitation, bien que cela n'améliore pas l'efficacité. - Ajouter 1,8 ml de milieu de BG11 et incuber des échantillons pour un total de 4 jours avec agitation. Ceci est un temps suffisant pour permettre une recombinaison de se produire dans les multiples copies chromosomiques.
- Des aliquotes de plaque du mélange de transformation sur BG11 / 5% plaques de gélose au saccharose. Planche 50 pi, 10 pi et 1 pi par plaque de gélose. Si une pelouse de colonie apparaît sur toutes ces plaques d'agar diluer la solution plus loin et aliquote sur des plaques fraîches. Colonies devraient être visibles après environ 7 jours.
- Le patch 30-50 colonies individuelles sur BG11 + kanamycine (30 pg / ml) et les premières plaques de gélose BG11 / 5% plaques de gélose au saccharose, deuxièmement, au moyen d'un cure-dent extrémité émoussée. Les bactéries qui se développent sur BG11 / 5% des plaques de saccharose, mais pas les plaques BG11 + kanamycine sont KO banalisées potentiels. Bactéries qui se développent sur les deux plaques sont susceptibles d'être résistant à la saccharose , en raison d'une mutation dans le gène sacB.
- Vérifiez KOs non marquées en utilisant les mêmes amorces et méthode que celle utilisée pour vérifier les KO marqués. Ex cpcC1C2for et cpcC1C2rev (tableau 2) pour vérifier le knock - out non marqué cpcC1C2. Un KO non marqué montrera une bande sur un agarose t gel correspondanto la taille de type sauvage , moins la région supprimée (Figure 2).
- Si la souche présente un profil non marqué mutant par PCR (étape 4.11.2) et électrophorèse sur gel (Figure 2), puis re-série sur une plaque de gélose BG11 frais sans antibiotiques.
6. Stockage à long terme de Souches
- Mettre en place une nouvelle culture de la souche en inoculant une boucle complète de cellules dans 30-50 ml de milieu de BG11. Cultiver la culture pendant 3-4 jours à OD 750nm = 0,4 à 0,7.
- Laver les cellules une fois avec BG11 et remettre en suspension dans ~ 2 ml de BG11.
- Ajouter 0,8 ml de cellules concentrées à un tube. Ensuite, ajoutez 0,2 ml de 80% stérilisé par filtration glycérol.
- Facultatif: Ajouter 0,93 ml de cellules concentrées dans un autre tube. Ajouter 0,07 ml de DMSO à ce tube.
ATTENTION: Le DMSO est toxique et doit être manipulé avec une protection appropriée. - Conserver les deux tubes à -80 ° C. Pour relancer les souches retirer le tube et gratter quelques cellules avec une dent émousséeprendre sur une plaque de gélose sans antibiotiques. Streak comme normale en utilisant une boucle stérile.

Figure 1: Construction du plasmide pour la production de débouchures marqués et non marqués, par exemple cpcC1 et cpcC2 dans Synechocystis (A) Dans la région du génome de Synechocystis où se trouvent (B) et cpcC1 cpcC2 et les gènes adjacents.. Mis en évidence en noir est la région du génome à supprimer dans le mutant. (C) Les sites du génome qui sont amplifiés par PCR. La «région adjacente (indiqué en bleu) et 3 '5 région adjacente (indiquée en rouge) sont amplifiés avec des sites d'endonucléase de restriction pour le clonage dans pUC19. 5 '(ou 3'), région flanquante est excisé hors de pUC19 et inséré dans pUC19 + 3 '(ou 59;) région flanquante plasmide pour générer le plasmide B. (D) Le NPT1 / Cassette sacB de pUM24 est excisé par Bam HI digestion et inséré entre 5 'et 3' flanquant régions pour générer le plasmide A. S'il vous plaît cliquer ici pour voir une plus grande version de ce chiffre.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
La conception du plasmide est essentielle pour le succès de la génération des deux mutants marqués et non marqués. La figure 1 donne un exemple de plasmide A et B utilisées pour générer un mutant de deletion dans les gènes et Synechocystis cpcC1 cpcC2 13. Dans chaque cas, les régions flanquantes 5 'et 3' sont à peu près 900-1,000 pb. régions flanquant réduits peuvent être utilisées, bien que le plus petit, nous avons mis à l'essai avec succès a été d'environ 500 pb. Le plasmide B peut également contenir une cassette de gènes entre l'extrémité 5 'et 3' ~ 1 kb régions adjacentes ou une version modifiée de la séquence du gène natif.
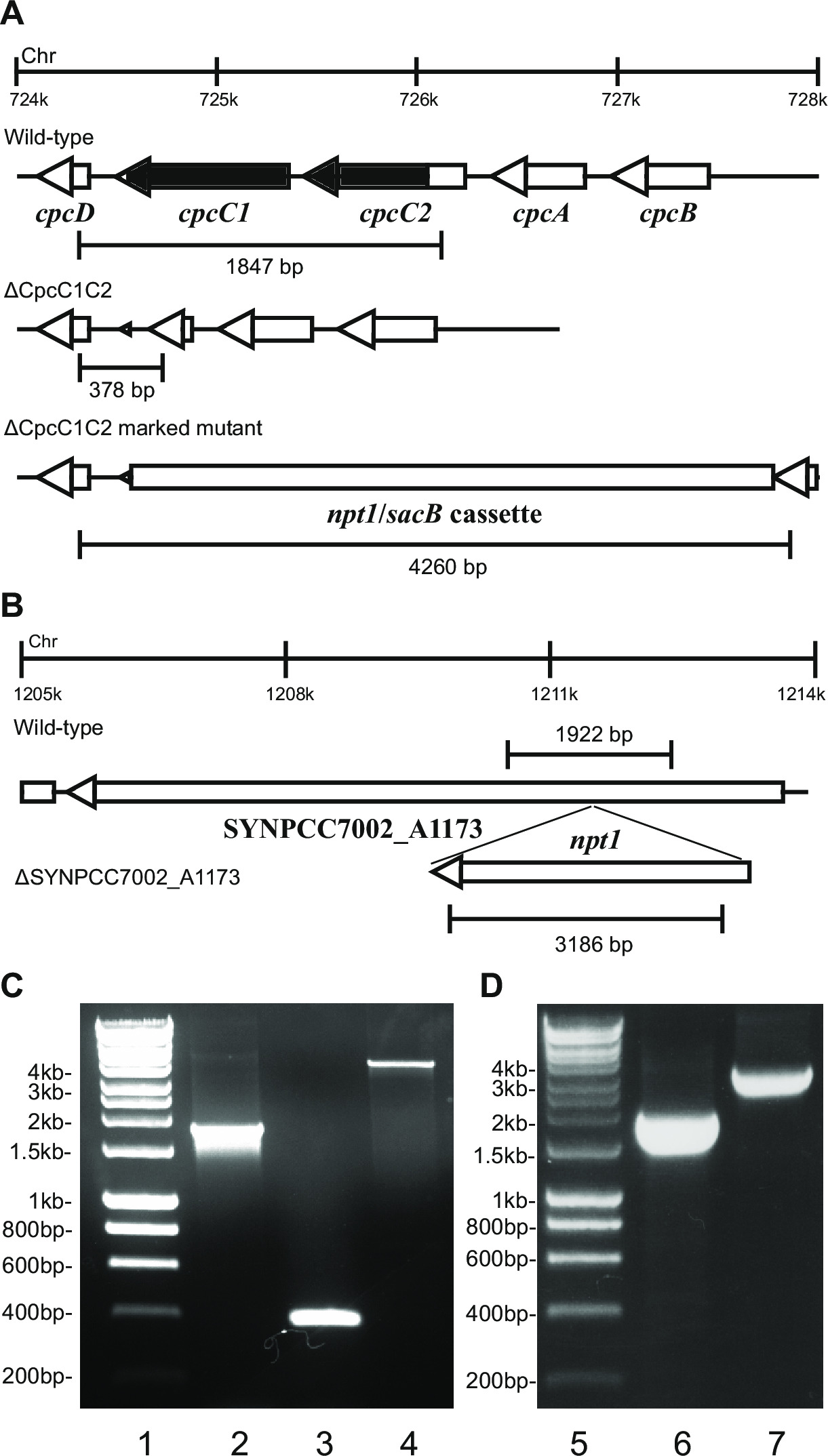
Figure 2: Vérification des mutants marqués et non marqués, par exemple cpcC1 / cpcC2
Lors de la transformation du plasmide A dans les cellules, typiquement plusieurs centaines de colonies apparaissent sur une plaque après environ 7-10 jours. Les colonies sont <1 mm de diamètre et ne seront pas augmenter en taille au cours des prochaines semaines. Par conséquent, il est essentiel d'utiliser un cure-dent fin émoussée pour éliminer la colonie et strie sur un BG11 + kanamycine plaque de gélose fraîche. Environ la moitié des colonies de re-strié va croître au bout de 4-6 jours. Si les gènes ne sont pas essentielles et mutants montrent une croissance similaire à la souche de type sauvage sous une lumière continue de 20-40 pmol photons m -2 s -1 (par exemple , des mutants d'oxydase de terminaux à Lea-Smith et al., 2013 14) (Figure 3), puis tous les chromosomes doivent contenir une copie de t il NPT1 / Cassette sacB inséré séquence, tel que déterminé par PCR. Si les gènes sont non essentiels et mutants montrent un phénotype lent de croissance sous la lumière continue de 20-40 pmol photons m -2 s -1 (par exemple phycobilisome mutants déficients dans Lea-Smith et al., 2014 13) (Figure 3), puis plusieurs cycles de re-stries sur des plaques d'agar BG11 avec une augmentation progressive des quantités de kanamycine sont essentiels afin d'obtenir un mutant marqué distinct. Une fois qu'un mutant distinct est obtenu cela devrait être re-strié sur un BG11, plus kanamycine plaque de gélose fraîche pour veiller à ce que la ségrégation est terminée. Si des cycles répétés de stries ne conduisent pas à une ségrégation marquée mutant alors le gène est susceptible essentielle pour la survie. La figure 4 donne un aperçu des étapes expérimentales impliquées dans la génération mutant non marqué.
/54001/54001fig3highres.jpg "Width =" 700 "/>
Figure 3:. La croissance des mutants Synechocystis Des exemples de mutants qui démontrent (A) une croissance similaire au type sauvage et (B) de croissance plus lent que le type sauvage. Le mutant ΔCOX manque la cytochrome oxydase en raison de la suppression des gènes CtaC1D1E1. Le mutant ΔCyd manque quinol oxydase en raison de la suppression des gènes CydAB. Le mutant d'olive manque une partie de la phycobilisome en raison de la suppression des gènes CpcABC1C2D. Les échantillons dans (B) ont été barboter avec de l'air pour faciliter la croissance. Reproduit à partir des données publiées dans Lea-Smith et al, 2013 14 et 2014 13 (www.plantphysiol.org; Droit d' auteur Société américaine des biologistes végétaux).. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
par exemple , la figure 2. Si une cassette de gène est inséré dans le chromosome alors généralement une proportion plus élevée de colonies résistantes à la kanamycine et du saccharose résistantes sont observées. Ces mutants peuvent se développer sur le saccharose en raison d'une mutation dans le gène sacB. Si aucune des colonies résistantes et sensibles à la kanamycine sont générées sucrose ensuite la cassette de gène est néfaste pour la cellule.

Figure 4: Génération de mutants marqués et non marqués dans Synechocystis Schéma détaillant (A) recombinaison et (B) étapes expérimentales impliquées dans la génération mutante.. Un plasmide est d'abord mélangé avec les cellules. Après l'incubation sur des plaques d'agar contenant de la kanamycine, les colonies dans lesquelles un événement de recombinaison se produit entre l'extrémité 5 'd'und 3 'flanquant les régions (indiquées en bleu et rouge, respectivement) et la séquence homologue dans le chromosome, sont isolés. En outre, la NPT1 / la cassette sacB entre les extrémités 5 'et 3' des régions adjacentes est inséré dans le chromosome. À la suite de la séparation d'un mutant marqué est généré. Les cellules mutantes marquées sont ensuite mélangés avec le plasmide B , qui peut contenir soit (C) 1: les régions flanquantes 5 'et 3'; 2: 5 'et 3' flanquant les régions avec une cassette d'expression contenant les gènes d'intérêt inséré entre ces séquences; 3: 5 'et 3' flanquant les régions où la séquence de type sauvage avec des modifications de nucleotides souhaitées intercalés entre ces séquences. Un second événement de recombinaison homologue se produit entre les extrémités 5 'et 3' des régions adjacentes et les régions homologues dans le chromosome, ce qui entraîne l' élimination du NPT1 / la cassette sacB et soit le knock - out ou d' un mutant non marqué avec une insertion ou sauvage modifiée typrégion e introduit dans le chromosome. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
| Recettes de solution Stock | |
| Chimique | Montant (g) |
| 100x BG11 (par L) | |
| NaNO 3 | 149,6 |
| MgSO 4 7H 2 O | 7,49 |
| CaCl 2 .2H 2 O | 3.6 |
| Acide citrique | 0,6 |
| Ajouter 1,12 ml de 0,25 M de Na2EDTA, pH 8,0 | |
| 0,25 M de Na 2 EDTA, pH 8,0 (100 ml) | |
| Na 2 | 9.3 |
| Les oligo - éléments (par 100 ml) | |
| H 3 BO 3 | 0,286 |
| MnCl 2 .4H 2 O | 0,181 |
| ZnSO 4 , 7H 2 O | 0,022 |
| Na 2 MoO 4 , 2H 2 O | 0,039 |
| CuSO 4 .5H 2 O | 0,008 |
| Co (NO 3) 2 .6H 2 O | 0,005 |
| Fer stock (par 100 ml) | |
| le citrate d'ammonium ferrique | 1.11 |
| Stock Phosphate (pour 100 ml) | |
| K 2 HPO 4 | 3.05 |
| Na 2 CO 3 stock (pour 100 ml) | |
| Na 2 CO 3 | 2 |
| Tampon TES, pH 8,2 (100 ml) | |
| TES | 22,9 |
| NaHCO 3 stock (pour 100 ml) | |
| NaHCO3 | 8.4 |
| HEPES, pH 8,2 (pour 500 ml) | |
| HEPES | 119.15 |
| La vitamine B 12 (Par 50 ml) | |
| cyanocobalamin | 0,02 |
| Médias Luria Bertani (par 500 ml) | |
| bouillon Luria Bertani | 12.5 |
| 1 M de MgCl2 (pour 100 ml) | |
| MgCl 2 .6H 2 O | 20.33 |
| MnCl 2 .4H 2 O | 0,395 |
| CaCl 2 .2H 2 O | 1,47 |
| 2- (N - morpholino) l' hydrate d'acide éthanesulfonique, l' acide 4-Morpholineethanesulfonic (MES) | 0,4265 |
| Solution A + glycérol | |
| 10 ml de solution A | |
| 1,5 ml de glycerol |
Tableau 1: Les solutions utilisées dans cette étude.
| Apprêt | Séquence |
| cpcC1C2leftfor | GTAC TCTAGA GCGGCTAAATGCTACGAC |
| SCPCP1C2leftrev | GATC GGATCC GCGGTAATTGTTCCCTTTGA |
| cpcC1C2rightfor | GATC GAGCTC TGCACTGGTCAGTCGTTC |
| cpcC1C2rightrev | GACT GAATTC ATCGTTGCTTGAACGGTCTC |
| M13 avant | TGTAAAACGACGGCCAGT |
| M13 inverse | CAGGAAACAGCTATGAC |
| cpcC1C2for | GTTTTCATTGGCATCGGTCT |
| cpcC1C2rev | ATGTCCCAGGAACGACTGAC |
| A1173for | AGCAAACCGTTTTTGTGACC |
| A1173rev | TGCAAGGTGGCGAACTGTAT |
Tableau 2:. Amorces utilisées dans les sites d'endonucléase de restriction de cette étude sont soulignés.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Les étapes les plus critiques dans la production de mutants non marquées sont: 1) la conception plasmidique soin d'assurer que la région ciblée est modifiée; 2) veiller à ce que les échantillons restent axénique, en particulier lorsqu'elles sont cultivées sur le saccharose; 3) le placage des cellules pour la production mutante marquée sur BG11 transformé d'abord des plaques de gélose dépourvues d'antibiotiques, suivie par l'addition d'agar-agar, plus des antibiotiques 24 heures plus tard; 4) la culture marquée mutants pour 4 jours complets avant étalement sur BG11, plus gélose saccharose plaques: 5) veiller à ce que les mutants marqués sont entièrement séparés et 6) confirmant soigneusement le génotype des souches mutantes. Pour cette dernière étape, des amorces supplémentaires conçues pour amplifier une partie de la région supprimée, peuvent être utilisées pour s'assurer qu'il a été retiré. Transfert de Southern, alors que laborieux, peut également être utilisé. Cependant, notre expérience est que la procédure décrite dans le présent document est suffisant pour la vérification appropriée des mutants. Cette procédure a également été utilisée pour générer des mutants marqués dans Synechococcus elongatus PCC7942. Cependant, la transformation répétée de cette cyanobactérie a prouvé difficile.
Si mutants marqués ne peuvent pas être séparés , puis les différentes conditions environnementales élevées de CO 2, de faible luminosité (<20 pmol photons m -2 s -1) ou des éléments nutritifs supplémentaires (c. -à- glucose) peuvent être testés. Par exemple, l'addition de glucose est essentielle afin de générer des mutants 21 photosystème II. Si mutants marqués ne se séparent entièrement alors le gène est probablement essentielle pour la viabilité. Cependant, il existe des exemples de la littérature où certains groupes de recherche ont été incapables de KO un gène (par exemple, Vipp dans Synechocystis) 22, que pour d' autres groupes pour montrer plus tard que le gène ne soit pas essentiel 23. Cela pourrait être dû à des différences dans les souches de type sauvage ou de la conception de plasmide incorrecte, entraînant des effets polaires sur, les gènes essentiels adjacents. Si un mutant ne coupe pas complètementséparons nous recommandons que le plasmide contenant la cassette NPT1 de pUC18K 20 entre les fragments gauche et droit être utilisé pour la transformation. Il est plus facile de vérifier la présence de bandes correspondant au type sauvage et mutant par PCR, étant donné que ce fragment est d' environ 1,2 kb, par rapport au 3,8 kb NPT1 / la cassette sacB. Ce résultat est un important élément de preuve démontrant que le gène est essentiel.
Génération de mutants non marquées avec des cassettes d'expression insérées est généralement plus difficile que le développement de souches de knock-out. Nous exprimons généralement des gènes sous contrôle du promoteur cpcBAC1C2D forte 13. Dans certains cas, cela peut diminuer les risques d'insertion réussie de la cassette du gène, si la surexpression d'une protéine est nuisible à la cellule. promoteurs plus faibles devraient être testés. En général, nous avons observé que plus la cassette du gène est, plus il est difficile àSeRT dans le génome. Nous ne sommes pas en mesure d'insérer des cassettes de gènes de plus de 5 kb. Des précautions doivent également être prises dans le choix de sites pour insérer des cassettes d'expression dans le génome. des sites neutres qui ne touchent pas la viabilité cellulaire ou la croissance doit être utilisé. Exemples dans Synechocystis comprennent phaAB et PHACE, qui codent les protéines codant pour la voie de biosynthèse de polyhydroxybutyrate 24,25. Plus récemment , une liste exhaustive des sites neutres dans Synechocystis a été identifié 26.
Génération de mutants non marquées dans les cyanobactéries est un processus lent, prenant environ 5-7 semaines si toutes les étapes mènent correctement. Ceci est plus lent que la méthode standard de génération KOs marqués utilisés par la majorité des groupes de recherche enquête cyanobactérie. Toutefois, la flexibilité de pouvoir introduire des mutations non marquées dans les mutants partiellement compense cela, étant donné que les plasmides additionnels suiteaining une gamme de cassettes conférant une résistance aux différents antibiotiques, ne doivent pas être construits. Pour des fins de recherche la possibilité de muter des gènes multiples est parfois nécessaire afin de caractériser pleinement le rôle fonctionnel des protéines. Par exemple, nous avons identifié un phénotype délétère que sur la suppression des deux puits d'électrons oxydases terminales localisées sur la membrane des thylakoïdes, étant donné que la perte d'un seul de ces complexes peut être compensée par l' activité des 14 autres. Développement d'une souche pour des applications industrielles aussi nécessiter plusieurs modifications à une souche, non seulement pour l'introduction de gènes étrangers, mais aussi d'accroître l'efficacité photosynthétique, l'optimisation de la récolte de lumière et de suppression des voies en compétition pour le substrat désiré.
Le principal facteur limitant la vitesse de génération de mutant non marqué est le temps de la division lente des espèces modèle de cyanobactéries, entre 8-20 heures en fonction des conditions de lumière. ONUder intensités lumineuses plus élevées et concentrations de CO 2, la croissance est plus rapide. Cependant, il existe un risque que les souches mutantes qui ne peuvent tolérer ni lumière élevée ou CO 2 seront sélectionnés contre, ou que les souches mutantes vont subir des modifications indésirables avant la caractérisation phénotypique. Par conséquent, ce n'est pas recommandé. Cependant, il serait hautement avantageux si un protocole plus rapide pour générer des mutants non marqués a été développé. Dans l'ensemble, cela faciliterait le développement de souches pour la recherche fondamentale et les applications appliquées. De telles souches peuvent être utilisées pour les biocarburants, la biomasse ou la production de produits chimiques ou dans la compréhension de nombreux aspects de la biochimie de cyanobactéries, de la génétique et de la physiologie.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Nous sommes reconnaissants à l'Environmental Services Association Education Trust, la biologie synthétique dans le fonds Synbio Cambridge et le Ministère de la justice sociale et l'autonomisation, Gouvernement de l'Inde, pour un soutien financier.
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. 3rd Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
