

Introduction
I cianobatteri sono un phylum evolutivamente antica e diversificata di batteri che si trovano in quasi tutti gli ambienti naturali della Terra. Negli ecosistemi marini sono particolarmente abbondanti e svolgono un ruolo chiave in molti cicli degli elementi nutritivi, che rappresentano circa la metà di fissazione del carbonio 1, la maggior parte dell'azoto fissazione 2 e centinaia di milioni di tonnellate di produzione di idrocarburi 3 negli oceani ogni anno. Cloroplasti, l'organello responsabile per la fotosintesi delle alghe e delle piante eucariotiche, è probabile che si sono evoluti da un cianobatterio che è stato inghiottito da un organismo ospite 4. Cianobatteri sono dimostrati utili organismi modello per lo studio della fotosintesi, trasporto di elettroni 5 e percorsi biochimici, molti dei quali sono conservati nelle piante. Inoltre cianobatteri sono sempre più utilizzati per la produzione di alimenti, biocarburanti 6, elettricità 7 e industriali composti 8, a causa della loro altaghly efficiente conversione di acqua e CO 2 alla biomassa utilizzando l'energia solare 9. Molte specie possono essere coltivate su terreni non coltivabili con sostanze nutritive minime e acqua di mare, il che suggerisce che i cianobatteri potenzialmente potrebbero essere coltivate su larga scala senza compromettere la produzione agricola. Alcune specie sono anche fonti di prodotti naturali, tra cui antifungini, antibatteriche e anti-cancro composti 10,11.
La capacità di generare mutanti è la chiave per comprendere la fotosintesi cianobatteri, biochimica e fisiologia, ed essenziale per lo sviluppo di ceppi per uso industriale. La maggior parte degli studi pubblicati generi geneticamente ceppi modificati mediante inserimento di una cassetta di resistenza antibiotica nel sito di interesse. Ciò limita il numero di mutazioni che possono essere introdotte in un ceppo, come solo alcune cassette di resistenza agli antibiotici sono disponibili per l'uso in cianobatteri. I ceppi che contengono geni che conferiscono re antibioticoresistenza non può essere utilizzato per la produzione industriale in vasche aperte, che rischia di essere l'unico mezzo di costo-efficacia per la produzione di biocarburanti e di altri prodotti a basso valore 12. La generazione di mutanti non marcate supera queste limitazioni. mutanti non marcate non contengono DNA estraneo, se non intenzionalmente incluso, e può essere manipolato più volte. Pertanto è possibile generare come molte modifiche in un ceppo lo desideri. Inoltre, gli effetti polari geni a valle del sito di modifica possono essere minimizzati, consentendo la modifica più precisa dell'organismo 13.
Per generare ceppi mutanti, plasmidi suicidi contenenti due frammenti di DNA identiche alle regioni nel cromosoma cianobatteri che fiancheggiano il gene da cancellare (definito il 5 'e 3' regioni fiancheggianti) sono prima costruiti. Due geni sono poi inseriti tra le regioni fiancheggianti. Uno di questi codifica una proteina resistenza agli antibiotici; il secondo codifica SACB, che produces levansucrasi, un composto che conferisce sensibilità al saccarosio. Nella prima fase del processo, mutanti marcati, cioè ceppi contenenti alcune DNA estraneo, vengono generati. Il costrutto plasmidico è mescolato con le cellule di cianobatteri e il DNA è occupato naturalmente dall'organismo. I trasformanti sono selezionati dalla crescita su piastre di agar contenenti l'antibiotico appropriato e il genotipo mutante verificata mediante PCR. plasmidi suicidio non può replicare entro il ceppo di interesse. Pertanto eventuali colonie resistenti agli antibiotici risulteranno da un evento di ricombinazione per cui il gene di interesse inserito nel cromosoma. Per generare mutanti non marcati, il mutante contrassegnata viene poi mescolato con un secondo plasmide suicida contenente solo i 5 'e 3' regioni fiancheggianti. Tuttavia, se è necessario l'inserimento di DNA estraneo, un plasmide costituito dal 5 'e 3' delle regioni fiancheggianti con cassette contenenti i geni di interesse inserito tra questi frammenti di DNA, può essere utilizzato. Selection è attraverso la crescita su piastre di agar contenenti saccarosio. Come saccarosio è letale per le cellule quando il prodotto genico SACB è espresso, le uniche cellule che sopravvivono sono quelli in cui si è verificato un secondo evento di ricombinazione, per cui il gene sensibilità saccarosio, oltre al gene di resistenza agli antibiotici, è stato ricombinato fuori cromosoma e sul plasmide. Come conseguenza dello scambio ricombinazione, le regioni fiancheggianti e tutte DNA tra loro sono inseriti nel cromosoma.
Abbiamo usato con successo questi metodi per generare mutazioni multiple cromosomiche nello stesso ceppo di Synechocystis sp. PCC6803 (di seguito denominato Synechocystis) 13,14, di introdurre mutazioni puntiformi singoli in un gene di interesse e 13 per l'espressione di cassette geniche. Mentre la generazione di fori non marcati è stata dimostrata prima del nostro lavoro in Synechocystis 15,16, un metodo dettagliato, aiutato dauna presentazione visiva delle fasi critiche, non è a disposizione del pubblico. Abbiamo inoltre applicato lo stesso metodo per la generazione di fori evidenziati in un altro modello cianobatterio, Synechococcus sp. PCC7002 (di seguito denominato Synechococcus). Questo protocollo fornisce una chiara, semplice metodo per la generazione di mutanti e un rapido protocollo per la validazione e la memorizzazione di questi ceppi.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Preparazione della cultura dei media
- Preparare medio BG11 secondo le Castenholz 1988 17.
- Preparare soluzioni di BG11 100x, elementi e ferro stock (tabella 1) tracciare.
- Preparare soluzioni separate di azioni di fosfato, Na 2 CO 3 stock, N - [Tris (idrossimetil) metil] -2-aminoethanesulfonic (TES) tampone e NaHCO 3 (Tabella 1).
- Autoclave il fosfato e Na 2 CO 3 scorte. Filtro-sterilizzare TES buffer e NaHCO 3 con 0,2 micron filtri.
- Preparare BG11 combinando 976 ml di acqua, 10 ml di 100x BG11, 1 ml di oligoelementi e 1 ml di ferro stock e autoclave la soluzione. Dopo questa soluzione è stata raffreddata a temperatura ambiente, aggiungere 1 ml di brodo di fosfato, 1 ml di Na 2 CO 3 stock e 10 ml di NaHCO 3.
- Per mezzo solido BG11, aggiungere 15 g di agar e 700 ml di acqua ad uno flask. Per il secondo pallone, aggiungere 3 g di Na 2 S 2 O 3, 226 ml di acqua, 10 ml di 100x BG11, 1 ml di oligoelementi e 1 ml di ferro stock. Autoclave entrambe le soluzioni. Dopo queste soluzioni sono raffreddati a temperatura ambiente, combinarli e aggiungere 1 ml di brodo di fosfato, 1 ml di Na 2 CO 3 stock, 10 ml di tampone TES e 10 mi di NaHCO 3.
Nota: Le soluzioni sono preparati separatamente per evitare la precipitazione di alcuni sali.
- Per la selezione sul saccarosio, preparare un 50% (w / v) di saccarosio. Filtro sterilizzare la soluzione con 0,2 micron filtri e aggiungere a BG11 (100 ml di 50% di saccarosio per 900 ml di BG11) per la produzione di BG11 / 5% di saccarosio piastre.
Nota: Non aggiungere NaHCO 3 per BG11 / 5% di saccarosio piastre di agar. Aggiungere Na 2 CO 3 come normale. - Per la coltura di Synechococcus aggiungere 10 ml di 1 M 4- (2-idrossietil) piperazina-1-etansolfonico acido N - (2-idrossietil) piperazine- NR42; - (acido 2-etansolfonico) (HEPES) e 1 ml di vitamina B 12 (Tabella 1) a 1 L di mezzo BG11.
Nota: Trasformazione di ceppi coltivate in mezzi BG11 commercialmente disponibile è significativamente meno efficiente nelle ricette multimediali BG11 qui descritte e pertanto non è raccomandato.
2. Crescita di cianobatteri Ceppi
- Culture ceppi in 100 ml beute con un volume massimo di 50 ml e agitare a 120 rpm. Sigillare piastre BG11 con parafilm e puntura tre piccoli fori nel lato della piastra per consentire lo scambio di gas. Incubare tutti i ceppi a 30 ° C sotto lampade fluorescenti in un fotobioreattore ad una intensità di luce tra 20-40 mmol fotoni m-2 sec -1.
- Utilizzare migliori tecniche sterili. Maniglia tutti i ceppi di cianobatteri in una cappa a flusso laminare.
Nota: Questo è particolarmente importante quando i ceppi sono coltivate con terreni contenenti saccarosio, che può essere facilmente Contaminated.
3. Generazione di plasmidi costrutti
- set di progettazione di primer, tra cui i siti di enzima di restrizione richiesti, utilizzando software di progettazione di primer, come Primer3 (http://frodo.wi.mit.edu/primer3/), per amplificare due ~ 1 regioni kb 5 'e 3' del gene di interesse. Consultare la sequenza del genoma della specie di cianobatteri via Cyanobase (http://genome.kazusa.or.jp/cyanobase). Vedere la Tabella 2 per tutti i primer utilizzati qui. Nel progettare primer prendere in considerazione i seguenti fattori:
- Assicurarsi che le regioni amplificate includono 5 'e 3' regioni del gene che verrà mutato, ad esempio figura 1.
- Non mutare regioni intergeniche per evitare la mutazione involontaria di antisenso e RNA non codificanti. Per la generazione di mutanti in Synechocystis, consultare l'elenco dei siti di inizio della trascrizione documentati in Mitschke et al. 2011 18, al fine di evitare mutazione del antisensoo RNA non codificanti.
- Quando si sceglie regioni fiancheggianti non comprendono l'intera griglia di lettura aperta di geni adiacenti come espressione di questi geni in Escherichia coli può interferire con la clonazione.
- Amplificare prodotti di PCR utilizzando ad alta fedeltà DNA polimerasi in base alle istruzioni del produttore.
Nota: Nella nostra esperienza questo enzima produce pochi errori.- Impostare reazioni PCR 50 ml contenente tampone HF e uno 0, 1.5 o 3 ml di DMSO. Utilizzare 100 ng di DNA genomico per reazione. Utilizzare un programma di base di una fase iniziale di denaturazione di 98 ° C per 30 sec, 35 cicli di 98 ° C per 10 sec, 67 ° C per 30 sec, 72 ° C per 30 sec, seguita da una fase di estensione finale di 72 ° C per 5 min. Questo dà in genere prodotti coerenti.
- Verificare prodotti di PCR e campioni digeriti con gli enzimi endonucleasi per la dimensione corretta tramite elettroforesi su gel. Eseguire 1% (w / v) gel di agarosio contenente 0,02%(V / v) bromuro di etidio per 45 minuti a 100 V.
ATTENZIONE: etidio bromuro è un potenziale mutageno e deve essere maneggiato con una protezione adeguata. - Purificare prodotti PCR utilizzando un kit di purificazione del DNA secondo le istruzioni del produttore. Anche utilizzare questo kit per la purificazione di frammenti di plasmidi, tra cui pezzi tagliati dal gel di agarosio. Eluire DNA purificato in 14 ml di acqua.
- Per la procedura di clonazione, incubare miscele di reazione di restrizione che a 37 ° C per> 1 ora in un volume totale di 30 microlitri secondo le istruzioni del produttore.
- Per la procedura legatura, frammenti di DNA legare a temperatura ambiente per> 1 ora in un volume totale di 20 microlitri, contenente 5 ml di plasmide digerito purificato, 12 ml di inserto digerito purificato, 2 microlitri di tampone e 1 ml di ligasi.
- Preparare cellule trasformanti Escherichia coli DH5α secondo il seguente metodo.
- Crescere un E. durante la notte coli
- Seminare 400 ml LB in una beuta da 1 L contenente 6 ml 1 M MgCl 2 (Tabella 1) con 1 ml di coltura durante la notte.
- Crescere la coltura a 37 ° C a 220 rpm per circa 4 ore o fino a OD 600nm raggiunge 0,4-0,6.
- Mettere le cellule in ghiaccio per 1 ora.
- Centrifugare a 2.800 xg per 10 minuti per sedimentare le cellule a 4 ° C.
- Rimuovere il surnatante e risospendere in 160 ml di soluzione A (Tabella 1) e incubare su ghiaccio per 20 min.
- Centrifugare a 2.800 xg per 10 minuti per sedimentare le cellule a 4 ° C.
- Rimuovere il surnatante e risospendere in 4 ml Soluzione A + glicerolo (Tabella 1).
- Preparare aliquote di 50 microlitri, congelare in N liquido 2, conservare a -80 ° C.
- Mescolare 5 ml di miscela di ligasi con 50 ml di cellule competenti e incubare per 1 ora su ghiaccio.
- Heat Shock le cellule a 42 ° C per 90 sec, Folloa capo incubazione in ghiaccio per 2 min.
- Aggiungere 950 ml di LB supporti (Tabella 1) e incubare a 37 ° C per 1 ora.
- Aliquotare 50 e 200 microlitri su piastre con l'antibiotico appropriato, sia ampicillina (100 ug / ml) e / o kanamicina (30 mcg / ml).
ATTENZIONE: Sia kanamicina e ampicillina sono tossici e devono essere maneggiati con una protezione adeguata. - Pick and incubare singole colonie in 2 ml di mezzi LB inoculati con l'antibiotico appropriato.
- Purificare tutti i plasmidi utilizzando un kit di purificazione miniprep plasmide secondo le istruzioni del produttore.
- Genera plasmidi, in questo esempio specifico per buttare giù i geni cpcC1C2, secondo la seguente procedura.
- Amplificare la regione fiancheggiante 1.012 bp 5 '(frammento a sinistra) usando primer cpcC1C2leftfor e cpcC1C2leftrev (vedere il punto 3.2, Tabella 2). Rimuovere una piccola quantità della reazione PCR e confermare se laprodotto dimensione corretta è stato amplificato tramite elettroforesi su gel (passo 3,3). Digerire questo frammento e pUC19 con Xba I e BamHI (passo 3,5).
- Purificare entrambe le preparazioni (punto 3.4), legare (passo 3,6), la trasformazione (passo 3,7) e impostare quattro 2 ml di LB colture liquide con ampicillina (100 mg / ml) da colonie separate per la purificazione plasmide via minipreps (passo 3,8).
- Controllare l'inserimento del frammento in pUC19 via Xba I / Bam HI digestione e elettroforesi su gel (fase 3.3). Bande di 2.660 bp e 1.012 bp indicano corretta introduzione dell'inserto nel plasmide.
- Amplificare la regione fiancheggiante 1.016 bp 3 '(frammento a destra) usando primer cpcC1C2rightfor e cpcC1C2rightrev (vedere il punto 3.2, Tabella 2). Rimuovere una piccola quantità della reazione PCR e confermare se il prodotto formato corretto è stato amplificato tramite elettroforesi su gel (fase 3.3). Digerire questo frammento e pUC19 con Sac I e Eco RI (step 3.5).
- Purificare entrambe le preparazioni (punto 3.4), legare (passo 3,6), la trasformazione (passo 3,7) e impostare quattro 2 ml di LB colture liquide con ampicillina (100 mg / ml) da colonie separate per la purificazione plasmide via minipreps (passo 3,8).
- Controllare l'inserimento del frammento in pUC19 via Sac I / Eco RI digestione (passo 3.5) e gel elettroforesi (passo 3.3). Bande di 2.660 bp e 1.016 bp indicano corretta introduzione dell'inserto nel plasmide.
Nota: siti Xba I / Bam HI per clonazione della regione 5 'e Sac I / Eco RI per la clonazione della regione 3' in pUC19 sono utilizzati ove possibile. Se possibile, sempre includere un sito di Bam HI sul primer reverse per la 'regione o il primer forward per il 3' 5 regione per garantire che passi dopo clonazione sono più facili da eseguire. - Sequenza entrambi gli inserti per determinare se la sequenza è corretta utilizzando primer che attraversa il sito di inserimento, ad esempio M13 in avanti e retromarcia M13 (Tabella 2). La sequenza deve essere corretta per garantire l'assenza di errori vengono introdotti in regioni fiancheggianti.
- Asportare il frammento di sinistra da pUC19 via Xba I / Bam HI digestione. Digest pUC19 + frammento scorretto con Xba I / Bam HI (passo 3,5).
- Purificare il frammento di sinistra 1.012 bp e 3.676 bp pUC19 + frammento di destra da un gel (passo 3,3) attraverso l'escissione del DNA utilizzando un bisturi.
- Purificare entrambe le preparazioni (punto 3.4), legare (passo 3,6), la trasformazione (passo 3,7) e impostare quattro 2 ml di LB colture liquide con ampicillina (100 mg / ml) da colonie separate per la purificazione plasmide via minipreps (passo 3,8).
- Controllare l'inserimento del frammento in pUC19 + frammento destra via Xba I / Bam HI digestione (passo 3.5) e gel elettroforesi (passo 3.3). Bande di 3.676 bp e 1.012 bp indicano corretto inserimento dell'inserto nel plasmide (fare riferimento a questo come plasmide B).
Nota: La cassetta NPT1 / SACB non deve essere purificato da gel di agarosio dal pUM24cm codifica una proteina che conferisce resistenza al cloramfenicolo. Quindi, se le colonie sono coltivate su LB / ampicillina / kanamicina piastre di agar l'unica combinazione possibile che porterà alla colonie resistenti è incorporazione della NPT1 / cassetta SACB in plasmide B. - Purificare entrambe le preparazioni (punto 3.4), legare (passo 3,6), trasformare (passo 3,7) e impostare quattro 2 ml di LB colture liquide con ampicillina (100 mg / ml) e kanamicina (30 mg / ml) da colonie separate per la purificazione plasmide via minipreps (passo 3,8).
- Controllare inserimento della NPT1 / cassetta SACB nel plasmide B con Bam HI digestione (passo 3.5) e gel elettroforesi (passo 3.3). Bande di 4.688 bp e 3.894 bp indicano il corretto inserimento di The inserire nel plasmide (fare riferimento a questo come plasmide A).
- In alternativa, l'estremità smussata NPT1 / cassetta SACB e clone in un diverso sito di restrizione endonucleasi tra i frammenti di sinistra e di destra nel plasmide B. La / cassette SACB NPT1 deve essere clonato tra i frammenti di destra e di sinistra.
Nota: Se è necessaria espressione di una cassetta straniera allora questo dovrebbe essere inserito tra i frammenti sinistro e destro del plasmide B. Questo plasmide viene poi utilizzato nelle fasi ad eliminazione diretta non marcati.
4. Generazione dei mutanti Synechocystis e Synechococcus tracciato
- Impostare una coltura fresca inoculando un ciclo completo di cellule in 30-50 ml di terreno BG11. Far crescere la coltura per 2-3 giorni a OD 750 nm = 0.2 a 0.6.
Nota: Tipicamente le singole colonie sono troppo piccoli per usare per l'inoculazione e orientamento delle singole celle anche bassi livelli di luce si tradurrà in fotoinibizione e selezioneper i mutanti resistenti alla luce. - Centrifuga 1-2 ml di cultura a 2.300 xg per 5 minuti e scartare il surnatante. Non centrifugare alcun culture cianobatteri a> 2.300 xg per evitare di danneggiare le cellule. Lavare il pellet una volta con il mezzo BG11.
Nota: Non sospendere nuovamente le cellule vortex in quanto ciò potrebbe comportare la perdita di Pili che sono essenziali per l'assorbimento del DNA. Risospendere le cellule delicatamente pipettando. - Aggiungere mezzo BG11 ad un volume finale di 100 microlitri. Trasferire le cellule in una provetta 14 ml a fondo rotondo.
- Aggiungere 1 mg di plasmide A alle cellule e mescolare delicatamente intercettazioni. Aggiungere <10 ml di plasmide.
Nota: Preferibilmente il plasmide dovrebbe essere ad una concentrazione di> 100 ng / ml, ma concentrazioni inferiori questo sono adeguati per trasformazione di successo. - Lay tubi in orizzontale in incubatrice. Incubare culture per 4-6 ore.
Nota: Le celle possono essere mescolati brevemente toccando ogni 1-2 ore, ma questo non è essenziale. I campioni possono essere collocati in unscuotendo incubatore anche se questo non migliora significativamente l'efficienza. - Distribuire aliquote della miscela di DNA coltura cellulare / plasmide su piastre di agar BG11 senza antibiotici. Tipicamente 20 ml e 80 aliquote microlitri sono distribuite su piatti separati.
- ~ 24 ore dopo, aggiungere 2,5-3 ml di 0,6% soluzione di agar in acqua contenente kanamicina (per 20 ml: 0,12 g di agar, 100 microlitri di 100 mg / ml kanamicina) alla piastra di agar. Raffreddare questa soluzione a ~ 42 ° C e aggiungere al bordo della piastra di agar. Inclinare la piastra in modo che la soluzione si forma uno strato uniforme 'superiore agar' sulla superficie.
- Incubare le piastre di agar per un ulteriore periodo di tempo. Le colonie dovrebbero essere visibili dopo circa 7 giorni.
Nota: piastre di agar possono essere impilati 3 alta in un incubatore. Tipicamente centinaia di colonie sono ottenuti per trasformazione. - Streak colonie individuali su BG11 + kanamicina (30 mg / ml) piastre di agar. Dividere la piastra di agar in 6 settori e utilizzare uno stuzzicadenti punta smussata a striscia fuorile colonie più di ogni singolo settore. Ottenere singole colonie non è importante, solo la crescita dei trasformanti.
- Confermare marcata knockout mediante PCR utilizzando Taq polimerasi del DNA secondo le istruzioni del produttore. Aggiungere 2 ml di MgCl 2 (25 mM) per reazione.
- Rimuovere una piccola percentuale delle cellule e trasferire in una provetta contenente 50 ml di acqua e 20 ~ 425-600 micron perle di vetro. Agitare in un vibratore per 5 min a ~ 2,000 rpm. Centrifugare a 15.700 xg per 5 minuti e utilizzare 5 ml di surnatante per 50 microlitri reazione PCR.
Nota: Non risospendere la soluzione. La detriti cellulari deve rimanere sul fondo della provetta.
- Rimuovere una piccola percentuale delle cellule e trasferire in una provetta contenente 50 ml di acqua e 20 ~ 425-600 micron perle di vetro. Agitare in un vibratore per 5 min a ~ 2,000 rpm. Centrifugare a 15.700 xg per 5 minuti e utilizzare 5 ml di surnatante per 50 microlitri reazione PCR.
- Convalida mutanti
- primer design che abbracciano la regione ad eliminazione diretta utilizzando software di progettazione di primer (come Primer3). primers disegno da ~ 200 bp lati della regione knockout.
Nota: Primer per verificare il mutante cpcC1C2 sono riportati nella tabella 2e sono definiti cpcC1C2for e cpcC1C2rev. - Amplificare prodotti usando un programma base di una fase iniziale di denaturazione a 95 ° C per 2 min, 35 cicli di 95 ° C per 1 min, 60 ° C per 1 min, 72 ° C per 1 min per kb di sequenza, seguita da una step estensione finale di 72 ° C per 5 min. Includere un controllo wild-type. Questo dà in genere prodotti coerenti.
- Verificare il genotipo tramite elettroforesi su gel. Trasformanti knockout delimitate mostra una striscia di ~ 4 kb (0,2 kb sia dal frammenti sinistro e destro più il NPT1 / cassette SACB) e l'assenza della banda wild-type (Figura 2).
Nota: In alcuni casi ~ 4 kb band non è osservata nel marcato mutante causa della grande dimensione di questo prodotto di PCR. Tuttavia, se una banda corrispondente alla dimensione prevista del wild-type non viene rispettato, in genere questo ceppo è un ko marcata.
- primer design che abbracciano la regione ad eliminazione diretta utilizzando software di progettazione di primer (come Primer3). primers disegno da ~ 200 bp lati della regione knockout.
- Se un wild-type band è ancora presente poi ri-striscia la tensione su unBG11 fresco + kanamicina (30 mg / ml) piastra di agar e ripetere la PCR. Ripetere il processo di ri-striature fino a quando il mutante è segregato in modo che nessun wild-type banda si osserva nella reazione di PCR.
Nota: Aumentando la quantità di kanamicina ad una concentrazione di 50 ug / ml, quindi 100 mg / ml è talvolta indispensabile per separare completamente un mutante marcata. - Se il ceppo mostra un profilo mutante marcata tramite PCR, poi ri-striscia su un fresco BG11 + kanamicina (30 mg / ml) piastra di agar. Utilizzare questo ceppo per generare il ko non marcato.
Nota: Il protocollo può essere utilizzato per generare mutanti marcati con solo una cassetta di resistenza antibiotica cioè sostituendo la cassetta NPT1 / SACB solo con la cassetta NPT1 da pUC18K 20 tra frammenti sinistro e destro..
5. Generazione di non contrassegnati Synechocystis Mutanti
- Impostare una cultura fresca di eliminazione diretta segnata inoculando un ciclo completo di cells in 30-50 ml di terreno BG11. Far crescere la coltura per 2-3 giorni a OD 750 nm = 0.2 a 0.6.
- Centrifuga 10 ml della cultura a 2.300 xg per 5 minuti e scartare il surnatante. Lavare una volta con il mezzo BG11.
Nota: Non sospendere nuovamente le cellule vortex in quanto ciò potrebbe comportare la perdita di Pili che sono essenziali per l'assorbimento del DNA. Risospendere le cellule delicatamente pipettando. - Aggiungere BG11 ad un volume finale di 200 microlitri. Trasferire le cellule in una provetta 14 ml a fondo rotondo.
- Aggiungere 1 mg di DNA plasmide B alle cellule e mescolare delicatamente intercettazioni.
- Incubare i campioni per 4-6 ore. Lay tubi in orizzontale.
Nota: Le celle possono essere mescolati brevemente toccando ogni 1-2 ore, ma questo non è essenziale. I campioni possono essere collocati in un incubatore scuotendo anche se questo non migliora l'efficienza. - Aggiungere 1,8 ml di terreno BG11 e incubare campioni per un totale di 4 giorni con agitazione. Questo è un tempo sufficiente per consentire la ricombinazione a verificarsi in molteplici copie cromosomiche.
- aliquote piastra della miscela di trasformazione su BG11 / 5% di saccarosio piastre di agar. Piatto 50 ml, 10 ml e 1 ml per piastra di agar. Se un prato colonia appare su tutte queste piastre di agar diluire la soluzione più e aliquota su piatti freschi. Le colonie dovrebbero essere visibili dopo circa 7 giorni.
- Patch 30-50 colonie individuali su BG11 + kanamicina (30 mcg / ml) piastre di agar prime e BG11 / 5% di saccarosio piastre di agar secondo, utilizzando uno stuzzicadenti punta smussata. Tutti i batteri che crescono su BG11 / 5% di saccarosio piatti ma piatti non BG11 + kanamicina sono potenziali fori non marcati. Batteri che crescono su entrambe le piastre sono probabilmente saccarosio resistenti a causa di una mutazione nel gene SACB.
- Verificare ko non marcate utilizzando gli stessi primer e metodo è stato utilizzato per verificare i fori marcati. Es cpcC1C2for e cpcC1C2rev (Tabella 2) per verificare il ko non marcato cpcC1C2. Un ko non marcato mostra una band su un gel di agarosio corrispondente to le dimensioni wild-type minus regione soppresso (Figura 2).
- Se il ceppo mostra un profilo marcato mutante tramite PCR (passo 4.11.2) e elettroforesi su gel (Figura 2), poi ri-striscia su una piastra di agar BG11 fresca senza antibiotici.
6. conservazione a lungo termine di ceppi
- Impostare una coltura fresca del ceppo inoculando un ciclo completo di cellule in 30-50 ml di terreno BG11. Far crescere la coltura per 3-4 giorni a OD 750 nm = 0,4-0,7.
- Lavare le cellule una volta con BG11 e risospendere in ~ 2 ml di BG11.
- Aggiungere 0,8 ml di cellule concentrate a un tubo. Quindi aggiungere 0,2 ml di 80% del filtro sterilizzato glicerolo.
- Opzionale: Aggiungere 0,93 ml di cellule concentrate a un altro tubo. Aggiungere 0,07 ml di DMSO a questo tubo.
ATTENZIONE: DMSO è tossico e deve essere maneggiato con una protezione adeguata. - Conservare entrambi i tubi a -80 ° C. Per rilanciare ceppi rimuovere il tubo e raschiare via alcune cellule con un dente smussatoscegliere su una piastra di agar senza antibiotici. Streak come normale utilizzando un'ansa sterile.

Figura 1: costruzione di plasmidi per la generazione di fori marcati e non marcati, ad esempio cpcC1 e cpcC2 a Synechocystis (A) Regione del genoma Synechocystis dove si trovano (B) cpcC1 e cpcC2 e geni adiacenti.. Evidenziata in nero è la regione del genoma da cancellare nel mutante. (C) Siti del genoma che sono amplificato mediante PCR. La 'regione fiancheggiante (indicato in blu) e 3' 5 fiancheggiante regione (indicata in rosso) sono amplificati con i siti di restrizione endonucleasi di clonazione in pUC19. Il 5 '(o 3') fiancheggiante regione è escisse di pUC19 e inserito nel pUC19 + 3 '(o 59;) fiancheggiante regione plasmide per generare plasmide B. (D) La NPT1 / cassette SACB da pUM24 viene asportato tramite Bam HI digestione e inserito tra il 5 'e 3' regioni fiancheggianti per generare plasmidi A. Si prega di cliccare qui per visualizzare un più grande versione di questa figura.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Disegno plasmide è fondamentale per la generazione di successo di entrambi i mutanti marcati e non marcati. Figura 1 è riportato un esempio di plasmide A e B utilizzato per generare una delezione mutanti nei geni Synechocystis cpcC1 e cpcC2 13. In ogni caso le 5 'e 3' regioni fiancheggianti sono circa 900-1.000 bp. regioni fiancheggianti ridotti possono essere utilizzati anche se la più piccola che abbiamo trialed successo è stato di circa 500 bp. Plasmid B può anche contenere una cassetta genica tra il 5 'e 3' ~ 1 kb regioni fiancheggianti o una versione modificata della sequenza del gene nativo.
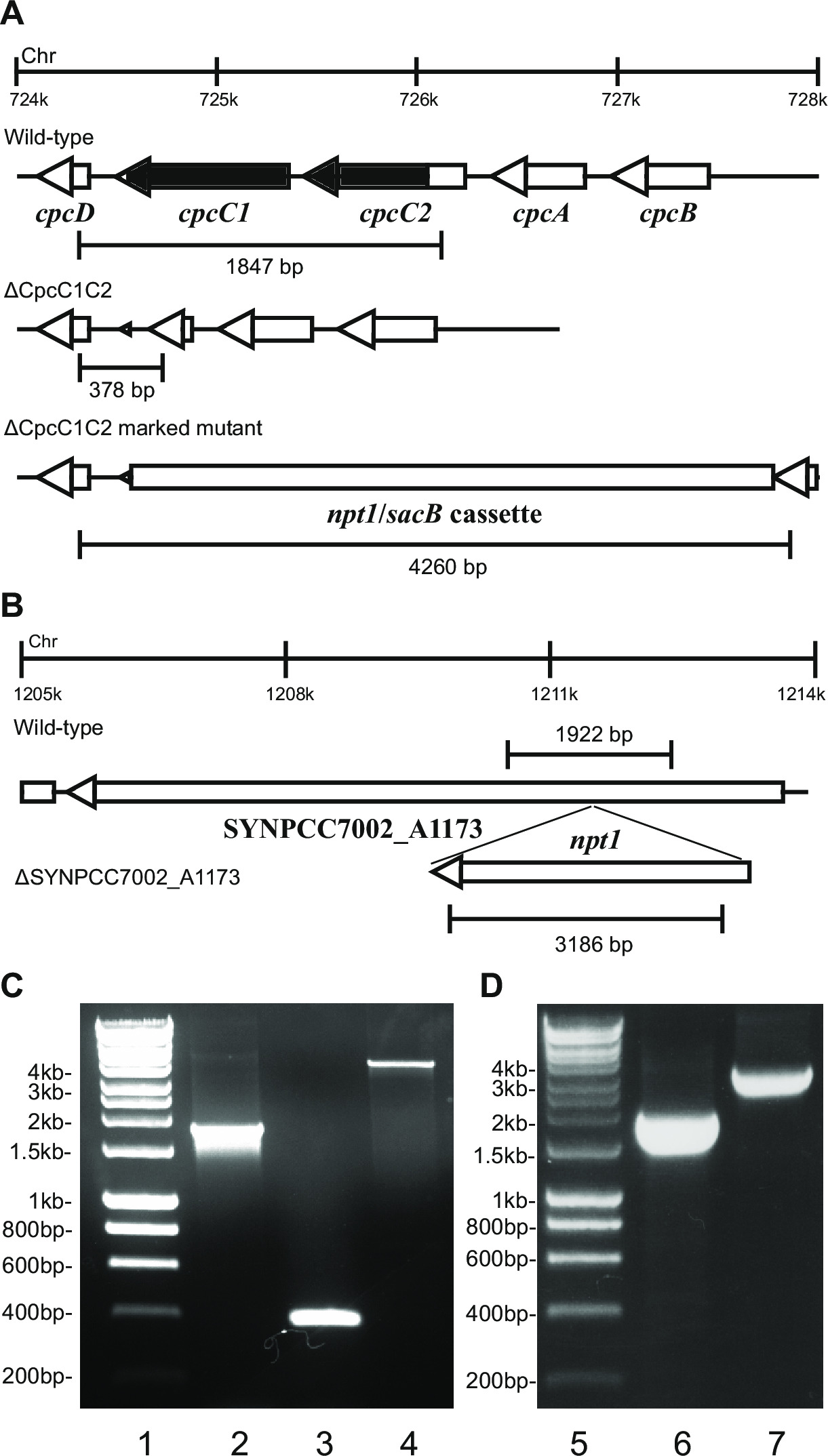
Figura 2: Verifica dei mutanti marcati e non marcati, ad esempio cpcC1 / cpcC2
Dopo trasformazione di plasmide A nelle cellule, tipicamente diverse centinaia di colonie appariranno su un piatto dopo circa 7-10 giorni. Le colonie sono <1 mm di diametro e non aumentano di dimensione per le prossime settimane. Pertanto è fondamentale utilizzare uno stuzzicadenti punta smussata per rimuovere la colonia e strisciare su un BG11 + kanamicina piastra di agar fresco. Circa la metà delle colonie ri-striato cresceranno dopo 4-6 giorni. Se i geni sono non essenziali e mutanti mostrano una crescita simile al ceppo wild-type sotto la luce continua di 20-40 micromoli fotoni m -2 sec -1 (ad esempio terminali mutanti ossidasi a Lea-Smith et al. 2013 14) (Figura 3), allora tutti i cromosomi dovrebbero contenere una copia di t egli NPT1 / cassette SACB sequenza inserita, come determinato tramite PCR. Se i geni sono non essenziale e mutanti mostrano un fenotipo lenta crescita sotto luce continua di 20-40 mmol fotoni m -2 sec -1 (es ficobilisoma mutanti carenti in Lea-Smith et al. 2014 13) (Figura 3), quindi vari cicli di ri-striature su piastre di agar BG11 con graduale aumento della quantità di kanamicina sono essenziali al fine di ottenere un mutante marcata segregazione. Una volta che un mutante segregato si ottiene questo dovrebbe essere ri-striato su un BG11 più kanamicina piastra di agar fresco per garantire che la separazione è stata completata. Se ripetuti giri di striature non diano luogo ad un segregato marcata mutante allora il gene è probabile essenziale per la sopravvivenza. La figura 4 fornisce una descrizione sommaria delle fasi sperimentali coinvolti nella generazione mutante non marcato.
/54001/54001fig3highres.jpg "Width =" 700 "/>
Figura 3:. La crescita di mutanti Synechocystis Esempi di mutanti che dimostrare (A) di crescita simile a wild-type e (B) una crescita più lenta rispetto wild-type. Il mutante ΔCOX manca citocromo ossidasi A causa della soppressione dei geni CtaC1D1E1. Il mutante ΔCyd manca chinolo ossidasi A causa della soppressione dei geni CydAB. Il mutante olive manca di una porzione del ficobilisoma causa della soppressione dei geni CpcABC1C2D. I campioni in (B) sono stati gorgogliare aria per facilitare la crescita. Tratto da dati pubblicati in Lea-Smith et al, 2013 14 e nel 2014 13 (www.plantphysiol.org; Copyright dell'American Society of biologi).. Clicca qui per vedere una versione più grande di questa figura.
ad esempio la figura 2. Se una cassetta gene viene inserito nel cromosoma quindi tipicamente una maggiore proporzione di kanamicina sono osservate colonie resistenti resistenti e saccarosio. Questi mutanti possono crescere sul saccarosio a causa di una mutazione nel gene SACB. Se nessun colonie resistenti sensibili e saccarosio kanamicina sono generati allora la cassetta genica è deleterio per la cellula.

Figura 4: Generazione di mutanti marcati e non marcati in Synechocystis schematica dettagli ricombinazione (A) e (B) fasi sperimentali coinvolti nella generazione mutante.. Plasmide A viene prima mescolato con le cellule. Dopo incubazione su piastre di agar contenenti kanamicina, colonie in cui si verifica un evento di ricombinazione tra il 5 'und 3 'regioni fiancheggianti (indicate in blu e rosso, rispettivamente) e la sequenza omologa nel cromosoma, sono isolati. Inoltre, la NPT1 / cassetta SACB tra il 5 'e 3' regioni fiancheggianti viene inserito nel cromosoma. A seguito di segregazione viene generato un mutante marcata. Cellule mutanti marcate vengono poi mescolati con plasmide B che può contenere sia (C) 1: 5 'e 3' regioni fiancheggianti; 2: 5 'e 3' fiancheggianti regioni con una cassetta di espressione contenente geni di interesse inserito tra queste sequenze; 3: il 5 'e 3' delle regioni fiancheggianti la sequenza wild-type con alterazioni nucleotidiche desiderate inseriti tra queste sequenze. Un secondo evento di ricombinazione omologa si verifica tra il 5 'e 3' regioni fiancheggianti e le regioni omologhe cromosoma, con conseguente rimozione del NPT1 / cassetta SACB e sia il knockout marcato o un mutante con una inserzione o alterata wild-tipe regione introdotto nel cromosoma. Clicca qui per vedere una versione più grande di questa figura.
| Ricette soluzione madre | |
| chimico | Importo (g) |
| 100x BG11 (per L) | |
| Nano 3 | 149.6 |
| MgSO 4 .7H 2 O | 7.49 |
| CaCl 2 .2H 2 O | 3.6 |
| Acido citrico | 0.6 |
| Aggiungere 1,12 ml 0,25 M Na 2 EDTA, pH 8,0 | |
| 0,25 M Na 2 EDTA, pH 8,0 (per 100 ml) | |
| na 2 | 9.3 |
| Oligoelementi (per 100 ml) | |
| H 3 BO 3 | 0,286 |
| MnCl 2 .4H 2 O | 0.181 |
| ZnSO4 7H 2 O | 0,022 |
| Na 2 Moo 4 .2H 2 O | 0,039 |
| CuSO 4 .5H 2 O | 0,008 |
| Co (NO 3) 2 6H 2 O | 0.005 |
| Ferro magazzino (per 100 ml) | |
| citrato ammonico ferrico | 1.11 |
| Fosfato magazzino (per 100 ml) | |
| K 2 HPO 4 | 3.05 |
| Na 2 CO 3 stock (per 100 ml) | |
| Na 2 CO 3 | 2 |
| Tampone TES, pH 8,2 (per 100 ml) | |
| TES | 22.9 |
| NaHCO 3 stock (per 100 ml) | |
| NaHCO 3 | 8.4 |
| HEPES, pH 8,2 (per 500 ml) | |
| HEPES | 119.15 |
| La vitamina B 12 (per 50 ml) | |
| cianocobalamina | 0.02 |
| Mezzi Luria Bertani (per 500 ml) | |
| brodo Luria Bertani | 12.5 |
| 1 M MgCl 2 (Per 100 ml) | |
| MgCl 2 .6H 2 O | 20.33 |
| MnCl 2 .4H 2 O | 0,395 |
| CaCl 2 .2H 2 O | 1.47 |
| 2- (N -Morpholino) idrato Acido etansolfonico, acido 4-Morpholineethanesulfonic (MES) | 0,4265 |
| Soluzione A + glicerolo | |
| 10 ml di soluzione A | |
| 1,5 ml di glicerolo |
Tabella 1: Le soluzioni utilizzate in questo studio.
| Primer | Sequenza |
| cpcC1C2leftfor | GTAC TCTAGA GCGGCTAAATGCTACGAC |
| CPCC1C2leftrev | GATC GGATCC GCGGTAATTGTTCCCTTTGA |
| cpcC1C2rightfor | GATC GAGCTC TGCACTGGTCAGTCGTTC |
| cpcC1C2rightrev | GACT GAATTC ATCGTTGCTTGAACGGTCTC |
| M13 in avanti | TGTAAAACGACGGCCAGT |
| M13 inversa | CAGGAAACAGCTATGAC |
| cpcC1C2for | GTTTTCATTGGCATCGGTCT |
| cpcC1C2rev | ATGTCCCAGGAACGACTGAC |
| A1173for | AGCAAACCGTTTTTGTGACC |
| A1173rev | TGCAAGGTGGCGAACTGTAT |
Tabella 2:. Primers utilizzati nei siti di endonucleasi di restrizione questo studio sono sottolineate.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
I passi più critici nella generazione di mutanti non contrassegnate sono: 1) un'attenta progettazione plasmide per garantire solo la regione di destinazione è alterata; 2) garantire che i campioni rimangano axeniche, soprattutto quando coltivate su saccarosio; 3) placcatura cellule per la generazione di mutanti marcati trasformato inizialmente su piastre di agar BG11 privi antibiotici, seguita da aggiunta di agar più antibiotici 24 ore successive; 4) la coltura segnato mutanti per 4 giorni interi prima di placcatura su BG11 più di saccarosio piastre di agar: 5) garantire che i mutanti marcati sono completamente separati e 6) confermando a fondo il genotipo dei ceppi mutanti. Per questa ultima fase, primer aggiuntivi progettati per amplificare parte della regione eliminata, possono essere utilizzate per garantire che è stato rimosso. Southern blotting, mentre laboriosa, può anche essere usato. Tuttavia, la nostra esperienza è che la procedura descritta in questo articolo è sufficiente per una corretta verifica dei mutanti. Questa procedura è stata utilizzata anche per generare mutanti marcati in Synechococcus elongatus PCC7942. Tuttavia, ripetuto trasformazione di questo cianobatterio si è rivelato difficile.
Se mutanti selezionati non possono essere suddivise quindi diverse condizioni ambientali di alta CO 2, di scarsa luminosità (<20 mmol fotoni m-2 sec -1) o sostanze nutrienti supplementari (ad esempio glucosio) possono essere testati. Ad esempio, l'aggiunta di glucosio è essenziale per generare fotosistema II mutanti 21. Se mutanti marcati non segregano completamente allora il gene è probabilmente essenziale per la vitalità. Tuttavia, ci sono esempi tratti dalla letteratura in cui alcuni gruppi di ricerca sono stati in grado di knockout di un gene (ad esempio, Vipp in Synechocystis) 22, solo per gli altri gruppi per mostrare poi che il gene non è essenziale 23. Questo potrebbe essere dovuto a differenze nei ceppi wild-type o disegno plasmide non corretta, con conseguente effetti polari adiacenti, geni essenziali. Se un mutante non lo fa completamentesegregare si consiglia che il plasmide contenente la cassetta NPT1 da pUC18K 20 tra i frammenti di sinistra e di destra essere utilizzato per la trasformazione. È facile verificare la presenza di bande corrispondenti al wild-type e mutante mediante PCR, dal momento che questo frammento è di circa 1,2 kb, rispetto al 3,8 kb NPT1 / cassette SACB. Questo risultato è un pezzo importante di prove che dimostrano che il gene è essenziale.
Generazione di mutanti non contrassegnati con cassette di espressione inserita è in genere più impegnativo rispetto allo sviluppo di ceppi ad eliminazione diretta. In genere esprimere geni sotto il controllo del promotore forte cpcBAC1C2D 13. In alcuni casi questo può diminuire le probabilità di successo inserimento della cassetta genica, se sovra-espressione di una proteina è deleterio per la cellula. i promotori più deboli devono poi essere testati. In generale, abbiamo osservato che maggiore è la cassetta genica, più difficile è alSert è nel genoma. Non siamo stati in grado di inserire cassette geniche più grandi di 5 kb. Si deve anche essere presa nella scelta di siti da inserire cassette di espressione nel genoma. siti neutrali che non influenzano la vitalità delle cellule o la crescita dovrebbe essere usato. Esempi in Synechocystis includono phaAB e PHACE, che codificano le proteine che codificano per la biosintesi poliidrossibutirrato percorso 24,25. Più di recente un ampio elenco di siti neutri in Synechocystis è stato identificato 26.
Generazione di mutanti non marcate in cianobatteri è un processo lento, prendendo circa 5-7 settimane, se tutte le misure stanno conducendo correttamente. Questo è più lento del metodo standard di generare fori marcati utilizzati dalla maggior parte dei gruppi di ricerca che studiano cianobatteri. Tuttavia, la flessibilità di poter introdurre ulteriori mutazioni in mutanti marcati parzialmente compensa questo, poiché plasmidi aggiuntivi containing una serie di cassette che conferiscono resistenza agli antibiotici diversi, non devono essere costruiti. Per scopi di ricerca la capacità di mutare più geni volte è necessario per caratterizzare completamente il ruolo funzionale delle proteine. Ad esempio, abbiamo identificato un fenotipo deleterio solo su soppressione dei due lavandini elettroni ossidasi terminali localizzati alla membrana tilacoidi, poiché la perdita di uno solo di questi complessi potrebbe essere compensata dall'attività degli altri 14. Sviluppo di un ceppo per applicazioni industriali richiederà anche più modifiche a un ceppo, non solo per l'introduzione di geni estranei, ma anche per aumentare l'efficienza fotosintetica, ottimizzazione raccolta della luce e la cancellazione di percorsi in competizione per il substrato desiderato.
Il principale fattore limitante la velocità di generazione mutante non marcato è il tempo lento divisione della specie modello di cianobatteri, tra 8-20 ore a seconda delle condizioni di luce. under intensità di luce più elevate e concentrazioni di CO 2, la crescita è più veloce. Tuttavia, vi è il rischio che i ceppi mutanti che non tollerano né alta luce o di CO 2 sono scelti contro, o che ceppi mutanti subiranno alterazioni indesiderate prima di caratterizzazione fenotipica. Quindi questo non è raccomandato. Tuttavia, sarebbe altamente vantaggioso se un più rapido protocollo per generare mutanti marcati stato sviluppato. Nel complesso, questo faciliterebbe lo sviluppo di ceppi sia per la ricerca di base e applicazioni applicate. Tali ceppi potrebbero essere utilizzati per i biocarburanti, biomasse o di produzione chimica o nella comprensione di molti aspetti della biochimica cianobatteri, la genetica e la fisiologia.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Siamo grati alla Environmental Services Association Education Trust, la biologia sintetica a Cambridge fondo Synbio e il Ministero della giustizia sociale e responsabilizzazione, Governo dell'India, per il sostegno finanziario.
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. 3rd Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
