

Introduction
シアノバクテリアは、地球上のほぼすべての自然環境の中で見つかった細菌の進化的に古代と多様な門です。海洋生態系において、それらは特に豊富であり、炭素固定1の約半分を、海洋における窒素固定2と炭化水素生産3のトン数百万の大半は毎年を占め、多くの栄養素のサイクルで重要な役割を果たしています。葉緑体は、真核藻類や植物の光合成を担う細胞小器官は、宿主生物4に包まれたシアノバクテリアから進化した可能性があります。シアノバクテリアは、その多くが植物に保存されている、光合成、電子輸送5および生化学的経路の研究のための有用なモデル生物を証明しています。付加シアノバクテリアにおいてますます食品、バイオ6、電気7及びそれらのハイによる産業用化合物8の製造に使用されています太陽エネルギー9を用いて、バイオマスへの水とCO 2のghly効率的に変換します。多くの種はシアノバクテリアが潜在的に農業生産に影響を与えることなく、大規模に成長させることができたことを示唆し、最小限の栄養素と海水と非耕地で栽培することができます。特定の種も抗真菌、抗菌性及び抗癌化合物10,11を含む天然物の源です。
変異体を生成する能力は、シアノバクテリアの光合成、生化学および生理学、および産業用菌株の開発に不可欠なを理解するための鍵です。公表された研究の大部分は、目的の部位に抗生物質耐性カセットの挿入によって遺伝的に改変された株を生成します。これは、いくつかの抗生物質耐性カセットは、シアノバクテリアにおける使用のために利用可能であるように、株に導入され得る変異の数を制限します。抗生物質の再付与する遺伝子を含む株sistanceは、バイオ燃料および他の低価値製品12を製造するための唯一の費用効果的な手段である可能性が高い開放池の工業生産のために使用することができません。マークされていない変異体の生成は、これらの制限を克服します。マークされていない変異体は、意図的に含まれない限り、外来DNAを含有しない、複数回操作することができます。したがって、所望のように株のように多くの変更を生成することができます。また、修飾部位の下流の遺伝子上の極性効果は、生体13のより正確な修正を可能にする、最小限にすることができます。
変異株、削除対象の遺伝子に隣接するシアノバクテリアの染色体における領域と同一の2つのDNA断片を含有する自殺プラスミド(隣接領域の5 'および3'と呼ばれる)を生成するために最初に構成されています。二つの遺伝子は、これらの隣接領域間に挿入されます。これらの一つは、抗生物質耐性タンパク質をコードします。第二は、SacBを、PRODを符号化レバンスクラーゼuces、化合物は、スクロースに対する感受性を付与します。プロセスの最初の段階では、マークされた変異体は、いくつかの外来DNAを含む、すなわち株は、生成されます。プラスミド構築物は、シアノバクテリア細胞と混合し、DNAは、生物によって天然に取り込まれます。形質転換体は、適切な抗生物質およびPCRにより確認変異体の遺伝子型を含有する寒天プレート上での増殖によって選択されます。自殺プラスミドは、目的の菌株内で複製することはできません。したがって、任意の抗生物質耐性コロニーは、目的の遺伝子が染色体に挿入することにより組換え事象から生じます。マークされていない変異体を生成するために、マークされた変異体は、その後、ちょうど5 'および3'フランキング領域を含む第二の自殺プラスミドと混合されます。外来DNAの挿入が必要な場合は、これらのDNA断片の間に挿入された目的の遺伝子を含有するカセットを有する領域に隣接する5 'および3'からなるプラスミドを用いることができます。セレctionは、ショ糖を含有する寒天プレート上での成長によるものです。 sacB遺伝子産物が発現されたときに、スクロースを細胞に致死的であるように、生存細胞のみがショ糖感受性遺伝子は、抗生物質耐性遺伝子に加えて、外に再結合されていることにより第2の組換え事象が発生したものであり、染色体とプラスミド上に。組換え交換の結果として、隣接する領域と、それらの間のいずれかのDNAが染色体に挿入されています。
我々は成功しシネコシスティスの同じ系統で複数の染色体の変異を生成するために、これらのメソッドを使用しています。 (以下、 シネコシスティスという)PCC6803 13,14は 、関心対象13の遺伝子に、遺伝子カセットの発現のための単一点突然変異を導入します。マークされていないノックアウトの生成はシネコシスティス 15,16、詳細な方法で私たちの仕事の前に実証されてきたが、によって支援重要なステップのビジュアルプレゼンテーションは、公開されていません。また、別のモデルのラン藻、 シネココッカス属にマークされたノックアウトを生成するために同じ方法を適用しました。 PCC7002(以下、 シネココッカスと呼ばれます)。このプロトコルは、変異体およびこれらの株を検証し、保存するための迅速なプロトコルを生成するための明確な、簡単な方法を提供します。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
培養培地の調製
- Castenholz、1988年17に従ってBG11培地を準備します。
- 要素と鉄ストック( 表1)をトレースし、100×BG11のストック溶液を準備します。
- リン酸ストックの別々の溶液を用意し 、Na 2 CO 3株、N - [トリス(ヒドロキシメチル)メチル] -2-アミノエタンスルホン酸(TES)バッファーおよびNaHCO 3( 表1)。
- リン酸およびNa 2 CO 3銘柄をオートクレーブ。フィルター滅菌するTESは、0.2μmのフィルターでバッファリングし、NaHCO 3。
- 水976ミリリットル、100倍BG11の10ミリリットル、鉄の株式の1微量元素のミリリットルと1ミリリットルを組み合わせることにより、BG11を準備し、溶液をオートクレーブ。この溶液を室温まで冷却した後、リン酸株式の1ミリリットルをNa 2 CO 3株式の1ミリリットルおよびNaHCO 3の10ミリリットルを追加します。
- BG11固形培地については、寒天の15グラムと1 FLAへの水の700ミリリットルを追加します。SK。第二のフラスコに、のNa 2 S 2 O 3、水226ミリリットル、100倍BG11の10ミリリットル、微量元素と鉄の株式の1ミリリットルの1ミリリットルの3グラムを追加します。両方の溶液をオートクレーブ。これらのソリューションは、室温まで冷却した後、それらを組み合わせて、1リン酸株式のミリリットルし 、Na 2 CO 3株式の1ミリリットル、TES緩衝液の10ミリリットル、およびNaHCO 3の10ミリリットルを追加します。
注:ソリューションは、特定の塩の沈殿を回避するために、別々に調製されます。
- ショ糖の選択については、ショ糖溶液(w / v)の50%を準備します。フィルタは、0.2μmのフィルターで溶液を滅菌し、BG11 / 5%ショ糖プレートを製造するために(BG11の900ミリリットルに50%ショ糖の100ミリリットル)をBG11に追加します。
注:BG11 / 5%ショ糖寒天プレートに炭酸水素ナトリウムを追加しないでください。通常通りのNa 2 CO 3を追加します。 - シネココッカスの培養10mlの1Mの4-(2-ヒドロキシエチル)ピペラジン-1-エタンスルホン酸、Nの追加- (2-ヒドロキシエチル)ピペラジンNRを42; - (2-エタンスルホン酸)(HEPES)及びBG11培地1 LのビタミンB 12( 表1)の1 mlです。
注意:市販のBG11培地で培養株の形質転換は、ここで説明BG11メディアのレシピに比べて大幅に少ない効率的であるため、お勧めできません。
シアノバクテリア株の2.成長
- 培養は、50ミリリットルの最大容量で100ミリリットル三角フラスコに株および120 rpmで振とうします。パラフィルムでシールBG11プレートとガス交換を可能にするために、プレートの側面に三つの小さな穴を穿刺。 20-40マイクロモル光子メートル-2秒-1との間の光強度で光バイオリアクター内の蛍光灯の下で、30℃ですべての株を培養します。
- 最高の滅菌技術を使用してください。層流フード内のすべてのシアノバクテリアの株を扱います。
注:株は簡単にcontaminすることができ、ショ糖を含む培地と培養した場合、これは特に重要ですated。
プラスミド構築物の3世代
- 二〜1kbの領域5 'および3'を増幅するために、例えば、プライマー3(http://frodo.wi.mit.edu/primer3/)などのプライマー設計ソフトウェアを使用して、必要な制限酵素部位を含むプライマーの設計セット、目的の遺伝子。 Cyanobase(http://genome.kazusa.or.jp/cyanobase)を介してシアノバクテリア種のゲノム配列を参照してください。ここで使用される全てのプライマーについては表2を参照してください。設計する際のプライマーは、以下の要因を考慮してください。
- その増幅された領域5を含む確保'および3'変異される遺伝子、 例えば、 図1の領域。
- アンチセンスおよび非コードRNAの意図しない変異を回避するために、遺伝子間領域を変異しないでください。 シネコシスティスにおける変異体の生成のために、Mitschke らに記載の転写開始部位のリストを参照してください。、2011 18、アンチセンスの変異を避けるためにまたはRNAを非コード。
- 隣接領域を選択する際、大腸菌におけるこれらの遺伝子の発現などに隣接した遺伝子のオープンリーディングフレーム全体がクローニングを妨害する可能性が含まれていません。
- 製造業者の指示に従って、高忠実度DNAポリメラーゼを用いてPCRにより産物を増幅します。
注:我々の経験では、この酵素は、いくつかのエラーを生成します。- HFバッファーとDMSOの0、1.5または3μlのいずれかを含有する50μlのPCR反応を設定します。反応あたり100ngのゲノムDNAを使用してください。 72℃の最終伸長工程に続いて、30秒、10秒、30秒30秒67°C、72°C、98°Cの35ラウンドのために、98℃の最初の変性工程からなるプログラムを使用5分間、C。これは、典型的には、一貫性のある製品を提供します。
- ゲル電気泳動により正しいサイズのためのエンドヌクレアーゼ酵素で消化したPCR産物とサンプルを確認してください。 (w / v)アガロースゲルを0.02%を含有する1%の実行100 Vで45分間(v / v)の臭化エチジウム
注意:エチジウムブロマイドは、潜在的な変異原であり、適切な保護を処理する必要があります。 - 製造業者の説明書に従ってDNA精製キットを用いてPCR産物を精製します。また、アガロースゲルから切り出し片を含む、プラスミド断片の精製のために、このキットを使用します。水の14μlの精製されたDNAを溶出させます。
- クローニング工程のために、製造者の指示に従って30μlの総体積で> 37℃で1時間制限酵素反応混合物をインキュベートします。
- 精製された消化されたプラスミドの5μlを、精製された消化インサートの12μlを、バッファおよびリガーゼの1μlと2μLを含む全量20μlで> 1時間、室温でライゲーション工程、ライゲーションDNAフラグメントのために。
- 以下の方法に従って大腸菌 DH5αの形質転換体細胞を準備します。
- 一夜E.を育てます大腸菌
- 一晩培養物1mlられた6 ML 1 MのMgCl 2( 表1)を含有する 1Lの三角フラスコ中400 mlのLBを接種します。
- 約4時間、またはOD 600nmでは 0.4から0.6に達するまで、220 rpmで37℃で培養物を成長させます。
- 氷上で1時間細胞を置きます。
- 4℃で細胞をペレット化するために10分間、2800×gで遠心分離します。
- 160ミリリットルの溶液A( 表1)に上清および再懸濁を削除し、20分間氷上でインキュベートします。
- 4℃で細胞をペレット化するために10分間、2800×gで遠心分離します。
- 4ミリリットル溶液A +グリセロール( 表1)に上清および再懸濁を削除します。
- 50μlのアリコートを準備し、液体N 2中で凍結し、-80℃で保存。
- コンピテントセル50μlの連結混合物の5μLを混合し、氷上で1時間インキュベートします。
- 熱は90秒、follo 42℃で細胞を衝撃2分間氷上でインキュベートすることによって結婚。
- LB培地( 表1)の950μLを加え、37℃で1時間インキュベートします。
- 適切な抗生物質を含むプレート上にアリコート50と200μlの、アンピシリン(100μg/ ml)および/またはカナマイシン(30μgの/ ml)のいずれか。
注意:カナマイシンおよびアンピシリンの両方は有毒であり、適切な保護を処理する必要があります。 - 適切な抗生物質を接種した2ミリリットルのLB培地で単一コロニーを選択し、インキュベートします。
- 製造者の指示に従ってミニプレッププラスミド精製キットを使用して、すべてのプラスミドを精製します。
- 以下の手順に従って、cpcC1C2遺伝子をノックアウトするためのこの特定の例では、プラスミドを生成します。
- (ステップ3.2、 表2を参照してください)プライマーcpcC1C2leftforとcpcC1C2leftrevを使用して、1012 bpの5 '隣接領域(左フラグメント)を増幅。 PCR反応の少量を削除しているかどうかを確認正しいサイズの産物はゲル電気泳動(ステップ3.3)を介して増幅されました。 Xba IおよびBam HI(ステップ3.5)で、このフラグメントとのpUC19を消化。
- 両方の製剤(ステップ3.4)、ライゲーション(ステップ3.6)を精製、(ステップ3.7)およびミニプレップ(ステップ3.8)を介してプラスミド精製のための独立したコロニーからアンピシリン(100μg/ ml)と4 2ミリリットルのLB液体培地を設定を変換します。
- Xba I / BamHIで消化し 、ゲル電気泳動(ステップ3.3)を介してpUC19にフラグメントの挿入を確認してください。 2660 bpおよび1012 bpでのバンドは、プラスミドへの挿入の正しい導入を示しています。
- (ステップ3.2、 表2を参照してください)プライマーcpcC1C2rightforとcpcC1C2rightrevを使用して、1016 bpの3 'フランキング領域(右フラグメント)を増幅。 PCR反応の少量を除去し、正確なサイズの産物はゲル電気泳動(ステップ3.3)を介して増幅されたかどうかを確認します。 (STをSacIおよびEcoRIでこの断片とpUC19のダイジェストEP 3.5)。
- 両方の製剤(ステップ3.4)、ライゲーション(ステップ3.6)を精製、(ステップ3.7)およびミニプレップ(ステップ3.8)を介してプラスミド精製のための独立したコロニーからアンピシリン(100μg/ ml)と4 2ミリリットルのLB液体培地を設定を変換します。
- サック I / エコ RI消化(ステップ3.5)、ゲル電気泳動(ステップ3.3)を介してpUC19にフラグメントの挿入を確認してください。 2660 bpおよび1016 bpでのバンドは、プラスミドへの挿入の正しい導入を示しています。
注:pUC19に地域の3のクローニングのための領域とのSac I / エコ RI 5 'のクローニングのためのXba I / バム HIサイトは可能な限り使用されています。実現可能な場合は、常にその後でクローニング手順を実行するのが容易であることを確認するために5 '領域または3のためのフォワードプライマー'領域のリバースプライマー上のBam HI部位を含みます。 - 配列は、例えば挿入部位、M1にわたるプライマーを使用して正しいかどうかの両方のインサート配列を決定します3フォワードおよびM13リバース( 表2)。シーケンスは、エラーが隣接領域に導入されていない確実にするために正確でなければなりません。
- Xba I / BamHIで消化を介してのpUC19から左の断片を切り出し。 Xba I / バム HI(ステップ3.5)としたpUC19 +右の断片を消化。
- 手術用メスの刃を用いてDNAの切除を介して、アガロースゲル(ステップ3.3)から1012塩基対左フラグメントおよび3676塩基対のpUC19 +右フラグメントを精製します。
- 両方の製剤(ステップ3.4)、ライゲーション(ステップ3.6)を精製、(ステップ3.7)およびミニプレップ(ステップ3.8)を介してプラスミド精製のための独立したコロニーからアンピシリン(100μg/ ml)と4 2ミリリットルのLB液体培地を設定を変換します。
- Xba I / BamHIで消化(ステップ3.5)、ゲル電気泳動(ステップ3.3)を介したpUC19 +右フラグメントに断片を挿入するために確認してください。 3676 bpおよび1012 bpでのバンドは(プラスミドBとしてこれを参照してください)プラスミドにインサートの正しい挿入を示しています。
注:pUM24cmは、クロラムフェニコール耐性を付与するタンパク質をコードしているので、NPT1 /のsacBカセットをアガロースゲルから精製する必要はありません。コロニーをLB /アンピシリン/カナマイシン寒天プレート上で増殖させている場合はそのため耐性コロニーにつながる唯一の可能な組み合わせは、プラスミドBにNPT1 /のsacBカセットの組み込みであります - 両方の調製物を精製(ステップ3.4)、ライゲーション(ステップ3.6)、(ステップ3.7)とは、プラスミド精製のための独立したコロニーからアンピシリン(100μg/ ml)およびカナマイシン(30μgの/ ml)で4 2ミリリットルのLB液体培地を設定変換しますミニプレップを経由して(ステップ3.8)。
- BamHI消化(ステップ3.5)、ゲル電気泳動(ステップ3.3)を介して、プラスミドBにNPT1 /のsacBカセットの挿入を確認してください。 4688 bpおよび3894 bpでのバンドは番目の正しい挿入を示しています電子プラスミド(プラスミドAとしてこれを参照)に挿入します。
- また、左右のフラグメント間の異なる制限エンドヌクレアーゼ部位に平滑末端NPT1 /のsacBカセットおよびクローンプラスミドBにNPT1 /のsacBカセットは、左右の断片の間にクローン化されなければなりません。
注:外国カセットの発現が必要とされる場合、これは、このプラスミドは、次にマークされていないノックアウト工程で使用されるプラスミドBの左右の断片の間に挿入されるべきです。
マークシネコシスティスおよびシネココッカス変異体の4世代
- BG11培地の30〜50ミリリットルへの細胞の完全なループを接種することにより、新鮮な培養を設定します。 0.6のOD 750nmで = 0.2に2〜3日間培養を成長させます。
注意:一般的に個々のコロニーは、光のたとえ低レベルの光阻害と選択になりますするために個々の細胞の接種および露光に使用するには小さすぎます光耐性変異体のために。 - 5分間、2300×gで文化の遠心分離機1-2ミリリットル、上清を捨てます。これは、細胞に損傷を与える可能性がある> 2300×gで任意のシアノバクテリアの培養物を遠心しないでください。 BG11培地で1回ペレットを洗浄。
注:これはDNA取り込みのために必須である線毛が失われる可能性がありますようにボルテックスして細胞を再懸濁しないでください。穏やかなピペッティングにより細胞を再懸濁します。 - 100μlの最終体積にBG11培地を追加します。 14ミリリットル丸底チューブに細胞を移します。
- 細胞へのプラスミドA1μgのを追加し、穏やかなタッピングによって混合します。 <プラスミドの10μLを加えます。
注:好ましくは、プラスミドは> 100 ngの/μlの濃度であるべきであるが、これよりも低い濃度が成功した形質転換のために適切です。 - インキュベーター中で水平にチューブを下にして置きます。 4-6時間培養をインキュベートします。
注:細胞は、簡単にすべての1~2時間をタップして混合することができるが、これは必須ではありません。サンプルが中に配置することができこれは大幅に効率を改善しないが、振盪インキュベーター。 - 抗生物質を含まないBG11寒天プレート上での細胞培養/プラスミドDNA混合物のアリコートを広げます。典型的には、20μlの80μlのアリコートを別々のプレート上に広げています。
- 寒天プレートに:〜24時間後に、(寒天の0.12グラム、100 mg / mlでのカナマイシン100μlの20ミリリットルあたり)カナマイシンを含有する水中の0.6%寒天溶液を2.5〜3ミリリットルを追加します。このソリューションへ〜42℃に冷却し、寒天プレートの端に追加します。解決策は、表面上であっても「トップアガー」層を形成するようにプレートを傾けます。
- さらなる時間のために寒天プレートをインキュベートします。コロニーは約7日後に表示されるはずです。
注:寒天プレートをインキュベーターで高3を積層することができます。典型的には、コロニーの数百人が転換ごとに得られます。 - BG11 +カナマイシン上のストリーク個々のコロニー(30 / mlの)寒天プレート。 6セクタに寒天プレートを分割し、ストリークに平滑末端爪楊枝を使います各個々のセクターの上にコロニー。単一コロニーを得ることは、形質転換体のちょうど成長重要ではありません。
- 製造元の指示に従って、Taq DNAポリメラーゼを用いたPCRによって顕著なノックアウトを確認してください。反応ごとのMgCl 2(25mMの)の2μlを添加します。
- 細胞の小さな割合を削除し、50μlの水および〜20 425から600ミクロンのガラスビーズを含むチューブに移します。 〜2,000 rpmで5分間バイブレータに振ります。 5分間、15,700×gで遠心分離し、50μlのPCR反応あたり上清の5μLを使用しています。
注意:ソリューションを再懸濁しないでください。細胞片は、チューブの底に滞在する必要があります。
- 細胞の小さな割合を削除し、50μlの水および〜20 425から600ミクロンのガラスビーズを含むチューブに移します。 〜2,000 rpmで5分間バイブレータに振ります。 5分間、15,700×gで遠心分離し、50μlのPCR反応あたり上清の5μLを使用しています。
- 変異体を検証
- (このようなプライマー3など)プライマー設計ソフトウェアを使用してノックアウト領域にまたがる設計プライマー。デザインプライマーは〜200bpのノックアウト領域のいずれかの側から始まります。
注:cpcC1C2変異を確認するためのプライマーは、表2に概説されていますそして、cpcC1C2forとcpcC1C2revと呼ばれています。 - 続いて2分、1分、1分、60℃で、配列のKBあたり1分間72℃、95℃、35回、95℃の初期変性ステップからなるプログラムを用いて産物を増幅5分間72℃での最終伸長段階。野生型コントロールを含めます。これは、典型的には、一貫性のある製品を提供します。
- ゲル電気泳動により遺伝子型を確認してください。マークノックアウト形質転換体は、〜4キロバイト(左と右の断片プラスNPT1 /するsacBカセットの両方から0.2キロバイト)および野生型バンドの不在( 図2)のバンドが表示されます。
注:特定のケースで〜4キロバイトバンドが原因で、このPCR産物のサイズが大きいため著しい変異体で観察されていません。野生型の予想されるサイズに対応するバンドは、その後観察されない場合は、通常、この株は、顕著なノックアウトです。
- (このようなプライマー3など)プライマー設計ソフトウェアを使用してノックアウト領域にまたがる設計プライマー。デザインプライマーは〜200bpのノックアウト領域のいずれかの側から始まります。
- 野生型のバンドがまだ存在する場合は、上の歪みを、ストリークし直します新鮮なBG11 +カナマイシン(30μgの/ ml)を寒天プレートおよびPCRを繰り返します。何の野生型バンドをPCR反応において観察されないように、変異体が分離されるまで再ストリーキングプロセスを繰り返します。
注:を50μg/ mlの濃度になるように、カナマイシンの量を増やすと、その後100μg/ mlの完全に顕著な変異体を分離するために時々必要不可欠です。 - 株はPCRによるマークされた変異体プロファイルを示している場合は、再ストリークを新鮮なBG11 +カナマイシン(30μgの/ ml)を寒天プレート上に。無印のノックアウトを生成するには、この菌株を使用してください。
注:このプロトコルは、単に抗生物質耐性カセットとマークされた変異体を生成するために使用することができ、左右のフラグメント間pUC18K 20からわずかNPT1カセットとNPT1 /のsacBカセットを交換することによって、すなわち 。
無印シネコシスティス突然変異体の5世代
- Cの完全なループを接種することによりマークされたノックアウトの新鮮な培養を設定しますBG11培地の30〜50ミリリットルにells。 0.6のOD 750nmで = 0.2に2〜3日間培養を成長させます。
- 5分間、2300×gで文化の遠心分離機10ミリリットル、上清を捨てます。 BG11培地で1回洗浄します。
注:これはDNA取り込みのために必須である線毛が失われる可能性がありますようにボルテックスして細胞を再懸濁しないでください。穏やかなピペッティングにより細胞を再懸濁します。 - 200μlの最終容量にBG11を追加します。 14ミリリットル丸底チューブに細胞を移します。
- 細胞にプラスミドBの1μgのDNAを追加し、穏やかなタッピングによって混合します。
- 4-6時間サンプルをインキュベートします。水平にチューブを下にして置きます。
注:細胞は、簡単にすべての1~2時間をタップして混合することができるが、これは必須ではありません。これは、効率を改善しないが、試料は、振盪培養器内に配置することができます。 - BG11培地の1.8ミリリットルを加え、振とうしながら4日間の合計のためのサンプルをインキュベートします。これは、組換えが複数の染色体のコピーで起こることを可能にするのに十分な時間です。
- BG11 / 5%ショ糖寒天プレート上に形質転換混合物のプレートのアリコート。プレートを50μl、10μlの寒天プレートあたり1μlの。コロニーの芝生は、これらすべての寒天プレートに表示された場合は、新鮮なプレート上にさらなるソリューション、アリコートを希釈します。コロニーは約7日後に表示されるはずです。
- 平滑末端の爪楊枝を使用して、最初の寒天プレートBG11 +カナマイシン(30 / mlの)上の30から50個のコロニーと第二BG11 / 5%ショ糖寒天プレートにパッチを適用します。カナマイシンプレート+ BG11 BG11 / 5%スクロースプレート上ではなく、成長する任意の細菌は、潜在的なマークされていないノックアウトされています。両方のプレート上で増殖する細菌が原因のsacB遺伝子の変異に耐性ショ糖である可能性が高いです。
- マークされたノックアウトを確認するために使用されたものと同じプライマー及び方法を使用してマークされていないノックアウトを確認してください。 例えば cpcC1C2forとcpcC1C2rev( 表2)cpcC1C2無印ノックアウトを検証します。無印のノックアウトは、アガロースゲル対応トンにバンドを表示します野生型サイズOマイナス欠失領域( 図2)。
- 株はPCR(ステップ4.11.2)とゲル電気泳動( 図2)を介してマークされていない変異体プロファイルを示している場合は、抗生物質を含まない新鮮なBG11寒天プレート上-連勝を再。
株の6.長期保存
- BG11培地の30〜50ミリリットルへの細胞の完全なループを接種することにより株の新鮮な培養を設定します。 0.7のOD 750nmで = 0.4に3〜4日間培養を成長させます。
- BG11の〜2ミリリットルでBG11再懸濁して細胞を1回洗浄します。
- 1管に濃縮した細胞の0.8ミリリットルを追加します。その後、80%フィルター滅菌グリセロールの0.2ミリリットルを追加します。
- オプション:別のチューブに濃縮された細胞の0.93ミリリットルを追加します。このチューブにDMSOの0.07ミリリットルを追加します。
注意:DMSOは毒性があり、適切な保護を処理する必要があります。 - -80℃で両方の管を保管してください。株を復活させるためには、チューブを取り外し、鈍い歯といくつかの細胞をこすり落とします抗生物質を含まない寒天プレート上に選びます。通常通り、滅菌ループを用いてストリーク。

図1:マークされ、マークされていないノックアウトの生成のためのプラスミド構築、 シネコシスティスにおける例えば cpcC1とcpcC2隣接する遺伝子が配置されている(B)cpcC1とcpcC2とシネコシスティスゲノムの(A)地域。黒でハイライト表示すると、変異体では削除されるゲノムの領域です。 (C)PCRによって増幅されたゲノムのサイト。 5 '(青色で表示)隣接領域および3'(赤で表示)隣接領域は、pUC19にクローニングするための制限エンドヌクレアーゼ部位で増幅されます。領域に隣接する5 '(または3')をpUC19 + 3 '(または5にpUC19の切り出しと挿入されています9;プラスミドB.(D)pUM24からNPT1 /するsacBカセットをプラスミドAを生成するために、隣接領域の5 'および3'の間のBam HI消化を介して切除し、挿入されているを生成するために、地域のプラスミドに隣接) で拡大表示こちらをクリックしてください。この図のバージョン。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
プラスミドのデザインは、両方のマークされ、マークされていない変異体の生成に成功するために重要である。 図1は 、プラスミドAの例を示し、Bはシネコシスティス遺伝子 cpcC1とcpcC2 13に欠失変異体を生成するために使用されます。それぞれの場合において、5 '及び3'フランキング領域は約900〜1000塩基対です。我々は成功し試用している最小の約500塩基対であったものの、減少フランキング領域を使用することができます。プラスミドBは、また、隣接領域の5 'および3'の約1 KB以上天然の遺伝子配列の改変されたバージョンとの間の遺伝子カセットを含むことができます。
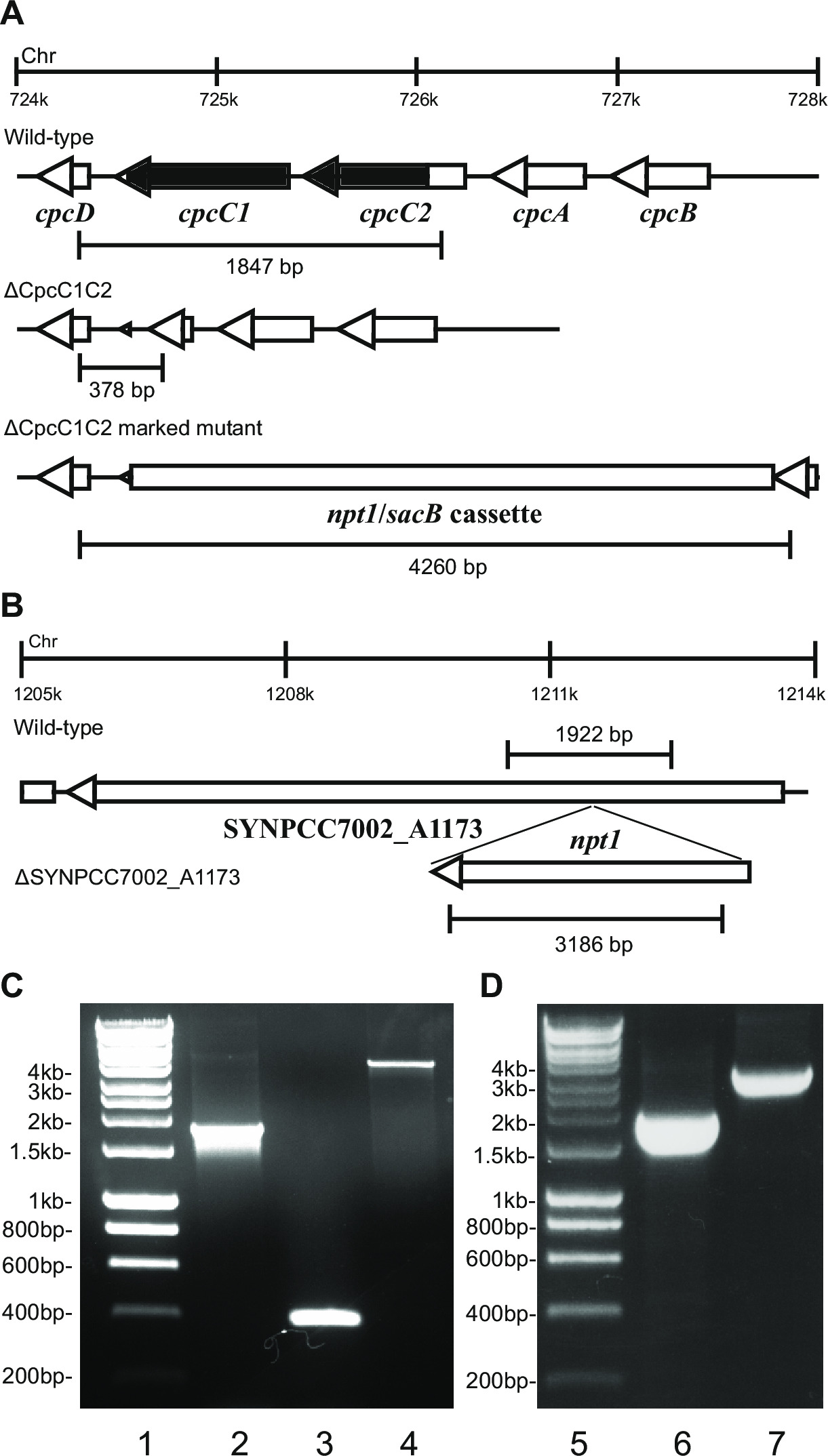
図2:マークされ、マークされていない変異体の検証、 例えば cpcC1 / cpcC2
細胞へのプラスミドAの形質転換の際に、一般的に数百個のコロニーは約7〜10日後にプレートに表示されます。コロニーは<直径1mmであり、今後数週間のサイズは増加しません。したがって、新鮮なBG11 +カナマイシン寒天プレート上のコロニーとストリークそれを除去するために平滑末端爪楊枝を使用することが重要です。約半数の再画線コロニーは、4-6日後に成長します。遺伝子は非必須であり、変異体は、20から40マイクロモル光子メートル-2秒-1の連続光下で野生型株と同様の増殖を実証している場合( 例えば 、リー・スミスら 、2013年14における末端酸化酵素変異体)( 図3)、その後、全ての染色体は、トンのコピーが含まれている必要があります PCRによって決定されるように彼は、シーケンスを挿入/するsacBカセットをNPT1。遺伝子は非必須であり、変異体は、20から40マイクロモル光子メートル-2秒-1の連続光下で低成長表現型を実証(リー・スミスらで例えばフィコビリソーム欠損変異体。、2014 13)( 図3)場合は、カナマイシン徐々に増加量のBG11寒天プレート上で再ストリーキングのいくつかのラウンドは、分離マークされた変異体を得るために不可欠です。分離された変異体が得られたら、これはその分離が完了していることを確認するために、新鮮なBG11プラスカナマイシン寒天プレート上に再ストリークしなければなりません。ストリーキングの繰り返しラウンドの変異にマーク偏析を生じない場合、遺伝子は生存に必須と思われる。 図4は、マークされていない変異体生成に関与する実験手順の概要を示します。
/54001/54001fig3highres.jpg "幅=" 700 "/>
図 3: シネコシスティス突然変異 体 の成長変異体の例としてどの野生型および(B)は、野生型よりも遅い成長への(A)同様の増殖を示しています。 ΔCOX変異体はCtaC1D1E1遺伝子の欠失によるチトクロームオキシダーゼを欠きます。 ΔCyd変異が原因CydAB遺伝子の欠失にキノールオキシダーゼを欠きます。オリーブの変異体は、CpcABC1C2D遺伝子の欠失によるフィコビリソームの一部を欠いています。 (B)中のサンプルは、成長を促進するために空気をバブリングしました。 。リー・スミスらに公表されたデータから再生し、2013年14 2014年13(www.plantphysiol.org;植物生物学者の著作権協会)。 この図の拡大版をご覧になるにはこちらをクリックしてください。
例えば、 図2 arked。遺伝子カセットが染色体に挿入されている場合には、典型的には、カナマイシンの高い割合耐性およびスクロース耐性コロニーが観察されます。これらの変異体が原因のsacB遺伝子の変異に、スクロース上で成長することができます。全くカナマイシン敏感およびスクロース耐性コロニーが生成されない場合には、遺伝子カセットは、細胞に対して有害です。

図4: シネコシスティス でマークされ、マークされていない変異体の生成回路図ディテール(A)組換えおよび変異体の生成に関与する(B)実験手順。。プラスミドAは、第1のセルと混合されます。組換え事象は、5 'との間に発生しているカナマイシンを含む寒天プレート、コロニー上のインキュベーション後D 3 'フランキング領域(それぞれ、青と赤で示されている)および染色体における相同配列は、単離されています。また、5 'および3'フランキング領域の間NPT1 /のsacBカセットを染色体に挿入されています。分離に続いてマークされた変異体が生成されます。マークされた変異体細胞は、(C)1含有することができるプラスミドBと混合されている:5 '及び3'フランキング領域を、 2:これらの配列の間に挿入された目的の遺伝子を含む発現カセットを有する領域に隣接する5 'および3'。 3:これらの配列の間に挿入された所望のヌクレオチド変化を有する野生型配列を有する領域をフランキング5 'および3'。 2回目の相同組換え事象は、NPT1 /のsacBカセットの除去とマークされていないノックアウトまたは挿入を有する変異体または改変された野生標準のいずれかで、その結果、5 'および3'フランキング領域と染色体における相同領域の間で起こりますE領域は、染色体に導入。 この図の拡大版をご覧になるにはこちらをクリックしてください。
| 原液のレシピ | |
| 化学物質 | 量(g) |
| 100X BG11(Lあたり) | |
| NaNO 3 | 149.6 |
| MgSO 4・7H 2 O | 7.49 |
| CaCl 2・2H 2 O | 3.6 |
| クエン酸 | 0.6 |
| 1.12ミリリットルの0.25MのNa 2 EDTA、pHが8.0を追加します。 | |
| 0.25 MのNa 2 EDTA、pHは8.0(100ミリリットルあたり) | |
| Na 2 | 9.3 |
| 微量元素(100ミリリットルあたり) | |
| H 3 BO 3 | 0.286 |
| MnCl 2・4H 2 O | 0.181 |
| ZnSO 4・7H 2 O | 0.022 |
| Na 2のMoO 4・2H 2 O | 0.039 |
| CuSO 4・5H 2 O | 0.008 |
| CO(NO 3)2・6H 2 O | 0.005 |
| 鉄の株式(100ミリリットルあたり) | |
| クエン酸第二鉄アンモニウム | 1.11 |
| リン酸株式(100ミリリットルあたり) | |
| K 2 HPO 4 | 3.05 |
| Na 2 CO 3の株式(100ミリリットルあたり) | |
| Na 2 CO 3 | 2 |
| TES緩衝液、pH 8.2、(100 1ml当たり) | |
| TES | 22.9 |
| (100ミリリットルあたり)のNaHCO 3ストック | |
| NaHCO 3 | 8.4 |
| HEPES、pHは8.2(500ミリリットルあたり) | |
| HEPES | 119.15 |
| ビタミンB 12(50ミリリットルあたり) | |
| シアノコバラミン | 0.02 |
| ルリアベルターニ培地(500ミリリットルあたり) | |
| ルリアベルターニブロス | 12.5 |
| 1 MのMgCl 2(100ミリリットルあたり) | |
| MgCl 2・6H 2 O | 20.33 |
| MnCl 2・4H 2 O | 0.395 |
| CaCl 2・2H 2 O | 1.47 |
| 2-(N-モルホリノ)エタンスルホン酸水和物、4-モルホリンエタンスルホン酸(MES) | 0.4265 |
| 溶液A +グリセロール | |
| 溶液10mlをA | |
| 1.5ミリリットルグリセロール |
表1:本研究で使用される溶液。
| プライマー | シーケンス |
| cpcC1C2leftfor | GTAC TCTAGA GCGGCTAAATGCTACGAC |
| CPCC1C2leftrev | GATC GGATCC GCGGTAATTGTTCCCTTTGA |
| cpcC1C2rightfor | GATC GAGCTC TGCACTGGTCAGTCGTTC |
| cpcC1C2rightrev | GACT GAATTC ATCGTTGCTTGAACGGTCTC |
| M13フォワード | TGTAAAACGACGGCCAGT |
| M13リバース | CAGGAAACAGCTATGAC |
| cpcC1C2for | GTTTTCATTGGCATCGGTCT |
| cpcC1C2rev | ATGTCCCAGGAACGACTGAC |
| A1173for | AGCAAACCGTTTTTGTGACC |
| A1173rev | TGCAAGGTGGCGAACTGTAT |
表2:本研究で使用したプライマーの制限エンドヌクレアーゼ部位は下線が引かれています。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
マークされていない変異体の世代の中で最も重要な手順は次のとおりです。1)慎重なプラスミドのデザインのみ標的領域が改変されていることを確認します。 2)特に、培養された場合、スクロース上のサンプルは、無菌のままであることを保証します。 3)24時間後に寒天を加えた抗生物質を添加した抗生物質を欠くBG11寒天プレート上に最初にマークされた変異体の生成のためのめっき形質転換細胞。 4)培養は、BG11プラスショ糖寒天プレート上にプレーティングする前に4日間フルのための突然変異体をマークし:5)マークされた変異体が完全に分離されていることを保証し、6)徹底的に変異株の遺伝子型を確認しました。この最後のステップのために、削除された領域の一部を増幅するために設計された追加のプライマーは、それが削除されたことを確認するために使用することができます。サザンブロット法は、面倒ながら、使用することもできます。しかし、私たちの経験では、このホワイトペーパーで説明した手順は、変異体の適切な検証のために十分であるということです。この手順はまたSynechococの著しい変異体を生成するために使用されていますelongatus PCC7942 CUS。しかし、このシアノバクテリアの繰り返し変換が困難なことが証明されています。
マークされた変異体は、CO 2の高い、異なる環境条件を分離することができない場合は、低光(<20マイクロモル光子メートル-2秒-1)または、追加の栄養素( すなわちグルコース)を試験することができます。例えば、グルコースの添加は、光化学系II変異体21を生成するために不可欠です。マークされた変異体を完全に分離することがない場合、遺伝子は、おそらく生存に必須です。しかし、いくつかの研究グループは、遺伝子ノックアウトすることができませんでした文献からの例がある(例えば、 シネコシスティスでVIPP)22は 、他のグループは後に遺伝子が23必須ではないことを示すためだけのために。これは、隣接する、必須遺伝子上の極性効果で、その結果、野生株または不正確なプラスミドのデザインの違いによる可能性があります。変異体は完全にない場合我々は左と右のフラグメント間pUC18K 20からNPT1カセットを含むプラスミドが形質転換のために使用することをお勧めします偏析。このフラグメント3.8 kbのNPT1 /するsacBカセットに比べ、約1.2 KBのため、PCRにより、野生型および変異体に対応するバンドの存在を確認することが容易です。この結果は、遺伝子が不可欠であることを証明する証拠の重要な部分です。
挿入された発現カセットとマークされていない変異体の生成は、一般的には、ノックアウト株の開発よりも困難です。私たちは、一般的に強いcpcBAC1C2Dプロモーター 13の制御下にある遺伝子を発現します。過剰発現タンパク質の、細胞に有害である場合、いくつかのケースでは、これは、遺伝子カセットの挿入が成功の可能性を減少させることができます。弱いプロモーターを試験する必要があります。一般的に、我々は、より大きな遺伝子カセットがあることを観察した、より困難それは内にありますゲノムにそれをSERT。私たちは、5キロバイトよりも大きい遺伝子カセットを挿入することができていません。ケアはまた、ゲノムへの発現カセットを挿入するためのサイトを選択する際に注意しなければなりません。細胞の生存率や成長に影響を与えない中立のサイトを使用する必要があります。 シネコシスティスにおける例としては、ポリヒドロキシブチレート生合成経路24,25をコードするタンパク質をコードphaABとphaCEが含まれます。さらに最近ではシネコシスティスにおける中立サイトの広範なリストは26を発見されました。
シアノバクテリアでマークされていない変異体の生成は、すべてのステップが適切に実施している場合は約5-7週間を取って、ゆっくりとしたプロセスです。これは、シアノバクテリアを調査研究グループの大多数が利用するマークされたノックアウトを生成する標準的な方法よりも遅くなります。しかし、部分的にマークされていない変異体にさらなる変異を導入することができるという柔軟性が続き、追加のプラスミドので、これを補償します別の抗生物質に対する耐性を付与するカセットの範囲をaining、構築する必要はありません。研究目的のために複数の遺伝子を変異させる能力は、完全タンパク質の機能的役割を特徴付けるために時々必要です。一つだけ、これらの複合体の喪失は、他の14の活動によって補償することができるので、例えば、我々は、唯一のチラコイド膜に局在2ターミナルオキシダーゼ電子シンクの削除時に有害な表現型を同定しました。産業用アプリケーションのための株の開発はまた、外来遺伝子の導入のためだけでなく、所望の基材のための経路を競合の光収穫の最適化や削除を光合成効率を高めるだけでなく、歪みに複数の修正が必要になります。
マークされていない変異体生成速度を制限する主な要因は、光の状態に応じて8-20時間の間に、モデルのシアノバクテリア種の遅い分割時間です。国連デルより高い光強度およびCO 2濃度、成長が速くなります。しかし、ハイライト又はCO 2のいずれかに耐えることができない変異株に対して選択され、又はその変異株は、表現型の特徴付けの前に望ましくない変化を受けるおそれがあります。したがって、これはお勧めできません。マークされていない変異体を生成するために、より迅速なプロトコルが開発された場合は、それは非常に有利であろう。全体として、これは、基礎研究および応用用途の両方のための株の開発を容易にするであろう。そのような株は、バイオ燃料、バイオマスまたは化学的生産のために、またはシアノバクテリア生化学、遺伝学および生理学の多くの側面を理解するのに使用することができます。
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
私たちは、財政支援のため、インド政府環境サービス協会教育トラスト、ケンブリッジシンバイオファンドにおける合成生物学と社会正義とエンパワーメント省に感謝しています。
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. 3rd Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
