

Introduction
Cyanobakterier er en evolusjonært gammel og mangfoldig phylum av bakterier som finnes i nesten alle naturlige miljøet på jorda. I marine økosystemer er de spesielt rikt og spiller en sentral rolle i mange næringssykluser, sto for omtrent halvparten av karbonfiksering 1, flertallet av nitrogenfiksering 2 og hundrevis av millioner av tonn med hydrokarbonproduksjon tre i havet årlig. Kloroplaster, organeller ansvarlig for fotosyntesen i eukaryote alger og planter, er sannsynlig å ha utviklet seg fra en cyanobacterium som ble oppslukt av en vertsorganisme 4. Cyanobakterier har vist seg å være nyttige modellorganismer for studiet av fotosyntese, elektrontransport 5 og biokjemiske veier, mange av dem er konservert i planter. I tillegg cyanobakterier i økende grad brukes til produksjon av mat, biodrivstoff 6, elektrisitet 7 og industrielle forbindelser 8, på grunn av deres highly effektiv konvertering av vann og CO 2 til biomasse ved hjelp av solenergi 9. kan dyrkes mange arter være på ikke-dyrkbar jord med minimal næringsstoffer og sjøvann, noe som tyder på at cyanobakterier potensielt kan dyrkes i stor skala uten at det påvirker landbruksproduksjon. Visse arter er også kilder for naturlige produkter, blant annet soppdrepende, antibakterielle og anti-cancerforbindelsene 10,11.
Evnen til å generere mutanter er nøkkelen til å forstå cyanobacterial fotosyntese, biokjemi og fysiologi, og essensielt for utvikling av stammer til industrielle formål. Flertallet av publiserte studier generere genmodifiserte stammer ved innsetting av en antibiotikaresistens kassett inn i området av interesse. Dette begrenser antall mutasjoner som kan innføres i en stamme, som bare noen få antibiotikaresistens kassetter er tilgjengelig for bruk i cyanobakterier. Stammer som inneholder gener som gir antibiotikaresistens remotstands kan ikke brukes til industriell produksjon i åpne dammer, noe som sannsynligvis vil være den eneste kostnadseffektiv måte å produsere biodrivstoff og andre lav verdi produkter 12. Den generasjonen av umerkede mutanter overvinner disse begrensningene. Umerkede mutanter ikke inneholder noe fremmed DNA, hvis hensikt er inkludert, og kan manipuleres flere ganger. Derfor er det mulig å generere så mange endringer i en belastning som ønsket. I tillegg kan polare effekter på gener nedstrøms av modifikasjonen området reduseres til et minimum, slik at mer nøyaktig endring av organismen 13.
For å generere mutantstammer, selvmords plasmider som inneholder to DNA-fragmenter identisk til regioner i den cyanobacterial kromosomet som flankerer genet som skal fjernes (betegnet 5 'og 3' flankerende regioner) blir først konstruert. To gener blir så innsatt mellom disse flankerende områder. En av disse koder for en antibiotisk resistens-protein; den andre koder SACB, som prodUCES levansukrase, en forbindelse overdragelse følsomhet for sukrose. I det første trinn av fremgangsmåten, som er merket mutanter, dvs. stammer inneholdende noe fremmed DNA, blir generert. Plasmidkonstruksjonen blandes med de cyanobakterielle celler og DNA tas opp naturligvis av organismen. Transformanter blir utvalgt ved vekst på agarplater inneholdende det passende antibiotikum og den mutante genotype bekreftet ved PCR. Selvmords plasmider kan ikke gjenskape innenfor belastningen av interesse. Derfor noen antibiotikaresistente kolonier vil resultere fra en rekombinasjon, hvorved genet av interesse inn i kromosomet. For å generere umerkede mutanter, er den markerte mutant deretter blandet med en andre selvmord plasmid som inneholder bare de 5 'og 3' flankerende regioner. Men hvis innskudd av fremmed DNA er nødvendig, et plasmid bestående av den 5 'og 3' flankerende regioner med en kassett som inneholder gener av interesse er satt inn mellom disse DNA-fragmenter, kan anvendes. SeleDette skjer er via vekst på agarplater inneholdende sukrose. Som sukrose er dødelig for cellene når det SACB genproduktet blir uttrykt, er de eneste cellene som overlever er de hvor en andre rekombinerings-hendelse har inntruffet, hvorved sukrose følsomhet genet, i tillegg til det antibiotiske resistensgenet, er blitt rekombinert ut av kromosom og på plasmidet. Som en konsekvens av den recombinational utvekslingen kan de flankerende områder og en hvilken som helst DNA mellom dem satt inn i kromosomet.
Vi har med hell brukt disse metodene for å generere flere kromosommutasjoner i samme stamme av Synechocystis sp. PCC6803 (heretter referert til som Synechocystis) 13,14, for å innføre enkeltpunktmutasjoner i et gen av interesse 13 og for ekspresjon av genet kassetter. Mens generasjonen av umerkede knockouts har blitt vist før vårt arbeid i Synechocystis 15,16, en detaljert metode, hjulpet aven visuell presentasjon av de kritiske trinn, er ikke offentlig tilgjengelig. Vi har også søkt samme metode for generering av merkede knockouts i en annen modell cyanobacterium, Synechococcus sp. PCC7002 (heretter referert til som Synechococcus). Denne protokollen gir en klar og enkel metode for å generere mutanter og en hurtig protokoll for validering og lagring av disse stammene.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Utarbeidelse av Culture Media
- Forbered BG11 medium ifølge Castenholz 1988 17.
- Forbered lager løsninger av 100x BG11, sporstoffer og jern lager (tabell 1).
- Fremstille separate oppløsninger av fosfat lager, Na 2 CO 3 lager, N - [tris (hydroksymetyl) metyl] -2-aminoetansulfonsyre (TES) buffer og NaHCO3 (tabell 1).
- Autoklaver fosfat og Na 2 CO 3 aksjer. Filter-sterilisere TES buffer og NaHCO3 med 0,2 mikrometer filtre.
- Forbered BG11 ved å kombinere 976 ml vann, 10 ml 100x BG11, 1 ml av sporstoffer og 1 ml jern lager og autoklaver løsningen. Etter denne løsningen er avkjølt til romtemperatur, tilsett 1 ml av fosfat lager, 1 ml Na 2 CO 3 aksjer og 10 ml av NaHCO3.
- For BG11 fast medium, tilsett 15 g agar og 700 ml vann til en flask. Til den andre kolben, tilsettes 3 g Na 2 S 2 O 3, 226 ml vann, 10 ml av 100 x BG11, 1 ml sporelementer og 1 ml av jern lager. Autoklav begge løsninger. Etter at disse løsningene er avkjølt til romtemperatur, kombinere dem og tilsett 1 ml fosfat lager, 1 ml Na2CO 3 lager, 10 ml TES-buffer, og 10 ml av NaHCO3.
Note: Solutions er forberedt separat for å unngå utfelling av visse salter.
- For valg av sukrose, fremstille en 50% (vekt / volum) sukrose-løsning. Filter sterilisere løsningen med 0,2 um filtre og legge til BG11 (100 ml 50% sukrose til 900 ml BG11) for å frembringe BG11 / 5% sukrose platene.
Merk: Ikke legg NaHCO3 til BG11 / 5% sukrose agarskåler. Legg Na 2 CO 3 som normalt. - For dyrkning av Synechococcus tilsett 10 ml 1 M 4- (2-hydroksyetyl) piperazin-1-etansulfonsyre, N - (2-hydroksyetyl) piperazin-NR42, - (2-etansulfonsyre) (HEPES) og 1 ml vitamin B 12 (tabell 1) til 1 liter BG11 medium.
Merk: Transformasjon av stammer dyrket i kommersielt tilgjengelig BG11 media er betydelig mindre effektiv enn i BG11 medie oppskrifter som er beskrevet her, og anbefales derfor ikke.
2. Vekst av cyanobacterial Stammer
- Kultur-stammene i 100 ml koniske kolber med et maksimalt volum på 50 ml og riste ved 120 opm. Forsegl BG11 plater med Parafilm og punkterings tre små hull i siden av platen for å tillate gassutveksling. Inkuber alle stammer ved 30 ° C under lysrør i en fotobioreaktor med lett intensitet mellom 20-40 mikromol fotoner m -2 sek -1.
- Bruk beste sterile teknikker. Håndter alle cyanobakterietoksiner stammer i en laminær hette.
Merk: Dette er spesielt viktig når påkjenningen er dyrket med medier som inneholder sukrose, som lett kan contaminrerte.
3. generasjon plasmidkonstruksjoner
- Design sett av primere, inkludert de nødvendige restriksjonsenzymseter, ved hjelp av primer design programvare som Primer3 (http://frodo.wi.mit.edu/primer3/), for å forsterke to ~ 1 kb områder 5 'og 3' i gen av interesse. Ta kontakt med genomsekvens av cyanobakterietoksiner arter via Cyanobase (http://genome.kazusa.or.jp/cyanobase). Se tabell 2 for alle primere som brukes her. Ved utforming primere vurdere følgende faktorer:
- Sikre at forsterkede regionene består av 5 'og 3' regioner av genet som skal muteres, for eksempel figur 1.
- Ikke mutere intergeniske regioner for å unngå utilsiktet mutasjon av antisense og ikke-kodende RNA. For generasjonen av mutanter i Synechocystis, se listen over transkripsjonsstartsider dokumentert i Mitschke et al., 2011 18, for å unngå mutasjon av antisenseeller ikke-kodende RNA.
- Ved valg av flankeregioner ikke inneholder hele den åpne leseramme av tilstøtende gener som ekspresjon av disse gener i Escherichia coli kan forstyrre kloning.
- Forsterke produkter ved PCR ved hjelp av high fidelity DNA polymerase i henhold til produsentens instruksjoner.
Merk: I vår erfaring er dette enzymet produserer feil.- Sett opp 50 mL PCR reaksjoner inneholder HF buffer og enten 0, 1,5 eller 3 pl DMSO. Bruk 100 ng genomisk DNA pr reaksjon. Bruker et program som består av en innledende denatureringstrinn på 98 ° C i 30 sek, 35 runder 98 ° C i 10 sek, 67 ° C i 30 sek, 72 ° C i 30 sekunder, etterfulgt av en avsluttende forlengelsestrinn på 72 ° C i 5 min. Dette gir vanligvis konsekvent produkter.
- Kontroller PCR produktene og prøver fordøyd med endonuklease enzymer for riktig størrelse via gel elektroforese. Forsøk 1% (w / v) agarose-geler inneholdende 0,02%(V / v) etidiumbromid i 45 minutter ved 100 V.
FORSIKTIG: Etidiumbromid er en potensiell mutagen og bør håndteres med passende beskyttelse. - Rense PCR produkter ved hjelp av en DNA rensing kit i henhold til produsentens instruksjoner. Bruk også dette settet for rensing av plasmid fragmenter, inkludert stykker kuttet fra agarosegeler. Eluere renset DNA i 14 ul vann.
- For kloningstrinn, inkuber restriksjonsendonuklease reaksjonsblandinger ved 37 ° C for> 1 time i et totalvolum på 30 ul i henhold til produsentens instruksjoner.
- For ligeringstrinn, ligere DNA-fragmenter ved værelsestemperatur i> 1 time i et totalvolum på 20 ul, inneholdende 5 ul av renset spaltet plasmid, 12 pl av renset spaltet innsatsen, 2 ul av buffer og 1 pl ligase.
- Forbered Escherichia coli DH5a transformante celler i henhold til den følgende metode.
- Grow en overnatting E. coli
- Inokulere 400 ml LB i en 1-liters konisk kolbe inneholdende 6 ml 1 M MgCl2 (tabell 1) med 1 ml av over natten-kultur.
- Dyrk kulturen ved 37 ° C ved 220 rpm i ca 4 timer eller inntil OD-600 nm når 0,4 til 0,6.
- Plassere cellene på is i 1 time.
- Sentrifuger ved 2800 xg i 10 minutter for å pelletere cellene ved 4 ° C.
- Fjern supernatant og resuspender i 160 ml oppløsning A (tabell 1) og inkuberes på is i 20 min.
- Sentrifuger ved 2800 xg i 10 minutter for å pelletere cellene ved 4 ° C.
- Fjern supernatant og resuspender i 4 ml løsning A + glycerol (tabell 1).
- Forbered 50 ul porsjoner, fryse i flytende N2, lagre ved -80 ° C.
- Bland 5 pl av ligeringsblandingen med 50 ul kompetente celler og inkuberes i 1 time på is.
- Varmesjokk cellene ved 42 ° C i 90 sek, Followed ved inkubasjon på is i 2 min.
- Legg 950 pl LB-medium (tabell 1) og inkuberes ved 37 ° C i 1 time.
- Delmengde 50 og 200 ul på platene med det passende antibiotikum, enten ampicillin (100 ug / ml) og / eller kanamycin (30 ug / ml).
FORSIKTIG: Både kanamycin og ampicillin er giftig og bør håndteres med passende beskyttelse. - Plukk og inkuberes enkeltkolonier i 2 ml LB media inokulert med riktig antibiotika.
- Rens alle plasmider ved hjelp av en miniprepp plasmid rensing kit i henhold til produsentens instruksjoner.
- Generer plasmider, i dette konkrete eksempelet for å slå ut de cpcC1C2 gener, i henhold til de neste trinnene.
- Forsterke 1012 bp 5 'flankerende område (venstre fragment) ved hjelp av primere cpcC1C2leftfor og cpcC1C2leftrev (Se trinn 3.2, tabell 2). Fjerne en liten mengde av PCR-reaksjonen og bekrefte hvorvidtriktig størrelse produktet har blitt amplifisert via gelelektroforese (trinn 3.3). Fordøye dette fragment og pUC19 med Xbal og BamHI (trinn 3.5).
- Rens begge preparater (trinn 3.4), ligate (trinn 3.6), forvandle (trinn 3.7) og sette opp fire 2 ml LB flytende kulturer med ampicillin (100 pg / ml) fra separate kolonier for plasmid rensing via minipreps (trinn 3.8).
- Sjekk for innføring av fragmentet inn i pUC19 via XbaI / BamHI fordøyelse og gelelektroforese (trinn 3.3). Band av 2660 bp og 1012 bp indikere korrekt innføring av innsatsen i plasmidet.
- Forsterke 1016 bp 3 'flankerende område (høyre fragment) ved hjelp av primere cpcC1C2rightfor og cpcC1C2rightrev (Se trinn 3.2, tabell 2). Fjerne en liten mengde av PCR-reaksjonen og bekrefte om den korrekte størrelse produktet har blitt amplifisert via gelelektroforese (trinn 3.3). Fordøye dette fragmentet og pUC19 med Sac I og Eco RI (step 3.5).
- Rens begge preparater (trinn 3.4), ligate (trinn 3.6), forvandle (trinn 3.7) og sette opp fire 2 ml LB flytende kulturer med ampicillin (100 pg / ml) fra separate kolonier for plasmid rensing via minipreps (trinn 3.8).
- Sjekk for innføring av fragmentet inn i pUC19 via SacI / Eco RI spaltning (trinn 3.5) og gelelektroforese (trinn 3.3). Band av 2660 bp og 1016 bp indikere korrekt innføring av innsatsen i plasmidet.
Merk: Xbal / BamHI-seter for kloning av 5'-regionen og Sac I / Eco RI for kloning av 3'-regionen til pUC19 brukes der det er mulig. Hvis mulig, omfatter alltid et BamHI-sete på revers primer for 5'-regionen eller foroverprimeren for 3'-regionen for å sikre at senere kloningstrinn er lettere å utføre. - Sekvens begge innsatser for å bestemme om sekvensen er riktig ved anvendelse av primere som spenner over innføringsstedet, f.eks M13 forover og M13 revers (tabell 2). Sekvensen må være riktig for å sikre at ingen feil blir introdusert inn flankerende regioner.
- Eksisere venstre fragment fra pUC19 via XbaI / Bam HI fordøyelsen. Fordøye pUC19 + riktig fragment med Xbal / BamHI (trinn 3.5).
- Rens 1012 bp venstre fragment og 3676 bp pUC19 + høyre fragment fra en agarosegel (trinn 3.3) via fjerning av DNA ved hjelp av en skalpell blad.
- Rens begge preparater (trinn 3.4), ligate (trinn 3.6), forvandle (trinn 3.7) og sette opp fire 2 ml LB flytende kulturer med ampicillin (100 pg / ml) fra separate kolonier for plasmid rensing via minipreps (trinn 3.8).
- Sjekk for innføring av fragmentet inn i pUC19 + riktig fragment via Xbal / BamHI-nedbrytning (trinn 3.5) og gelelektroforese (trinn 3.3). Band av 3676 bp og 1012 bp indikere riktig innsetting av innsatsen i plasmidet (se dette som plasmid B).
Merk: npt1 / SACB kassetten trenger ikke å bli renset fra agarosegeler siden pUM24cm koder for et protein overdragelse kloramfenikol-resistens. Derfor, dersom kolonier blir dyrket på LB / ampicillin / kanamycin-agarplater den eneste mulige kombinasjoner som vil føre til resistente kolonier er inkorporering av npt1 / SACB kassetten inn i plasmid B. - Rens begge preparater (trinn 3.4), ligate (trinn 3.6), transform (trinn 3.7) og satt opp fire 2 ml LB-væskekulturer med ampicillin (100 ug / ml) og kanamycin (30 ug / ml) fra separate kolonier for plasmid rensing via minipreps (trinn 3.8).
- Se etter innsetting av npt1 / SACB kassetten inn plasmid B via Bam HI fordøyelse (trinn 3.5) og gel elektroforese (trinn 3.3). Band av 4688 bp og 3894 bp indikere riktig innsetting av the sette inn i plasmidet (se dette som plasmid A).
- Alternativt kan den butte enden npt1 / SACB kassett og klonet inn i en annen restriksjonsendonukleasesete mellom de venstre og høyre fragmenter i plasmid B. npt1 / SACB kassett må bli klonet mellom de venstre og høyre fragmenter.
Merk: Hvis det kreves uttrykk for en utenlandsk kassett så dette bør settes inn mellom venstre og høyre fragmenter av plasmid B. Dette plasmidet blir deretter brukt i umerkede knockout trinn.
4. generasjon Merkede Synechocystis og Synechococcus Mutants
- Sett opp en ny kultur ved inokulering en løkke full av celler i 30-50 ml BG11 medium. Grow kulturen i 2-3 dager til OD 750nm = 0,2 til 0,6.
Merk: Typisk individuelle kolonier er for små til å bruke for inokulering og eksponering av de enkelte cellene til og med lave nivåer av lys vil resultere i fotoinhibering og seleksjonfor lette resistente mutanter. - Sentrifuge 1-2 ml av kulturen ved 2300 xg i 5 min og supernatanten kastes. Ikke sentrifuger noen cyanobakterietoksiner kulturer ved> 2300 xg da dette kan skade cellene. Vask pellet gang med BG11 medium.
Merk: Ikke resuspender celler ved å virvle da dette kan føre til tap av pili som er avgjørende for DNA-opptak. Suspender cellene ved forsiktig pipettering. - Legg BG11 medium til et sluttvolum på 100 ul. Overfør cellene til en 14 ml rundbunnet rør.
- Tilsett 1 ug av plasmid A til cellene og blandes ved forsiktig banking. Legg til <10 mL av plasmidet.
Merk: Fortrinnsvis bør plasmidet være ved en konsentrasjon på> 100 ng / mL, men konsentrasjoner lavere enn dette er tilstrekkelig for vellykket transformasjon. - Legg rør ned horisontalt i kuvøse. Inkuber kulturer i 4-6 timer.
Merk: Celler kan kort blandes ved å banke hver 1-2 timer, men dette er ikke avgjørende. Prøvene kan plasseres i enristeinkubator selv om dette ikke i betydelig grad øke effektiviteten. - Spre alikvoter av cellekulturen / plasmid-DNA-blandingen på BG11 agarplater uten antibiotika. Vanligvis 20 mL og 80 pl prøver er spredt på separate plater.
- ~ 24 timer senere, tilsett 2,5 til 3 ml av 0,6% Agar oppløsning i vann inneholdende kanamycin (per 20 ml: 0,12 g agar, 100 pl 100 mg / ml kanamycin) til agarplaten. Avkjøl denne løsningen til ~ 42 ° C, og legge til kanten av agar plate. Vipp plate så løsningen danner en enda 'top agar "lag på overflaten.
- Inkuber agarplater for en ytterligere periode. Kolonier bør være synlig etter ca 7 dager.
Merk: agarskåler kan stables 3 i høyden i en inkubator. Vanligvis hundrevis av kolonier oppnås per transformasjon. - Stripe individuelle kolonier på BG11 + kanamycin (30 ug / ml) agarplater. Del agar plate inn i 6 sektorer og bruke en butt ende tannpirker til strek utkoloniene enn hver enkelt sektor. Innhenting av enkeltkolonier er ikke viktig, bare vekst av transformantene.
- Bekreft markert knockout ved PCR ved bruk Taq DNA polymerase i henhold til produsentens instruksjoner. Tilsett 2 mL av MgCl2 (25 mM) pr reaksjon.
- Fjerne en liten andel av cellene og overfør til et rør inneholdende 50 mL vann og ~ 20 425-600 pm glassperler. Rist i en vibrator i 5 minutter ved ~ 2000 rpm. Sentrifuger ved 15700 xg i 5 minutter og bruke 5 ul supernatant per 50 pl PCR-reaksjon.
Merk: Ikke suspen løsningen. Celleavfallet behov for å holde på bunnen av røret.
- Fjerne en liten andel av cellene og overfør til et rør inneholdende 50 mL vann og ~ 20 425-600 pm glassperler. Rist i en vibrator i 5 minutter ved ~ 2000 rpm. Sentrifuger ved 15700 xg i 5 minutter og bruke 5 ul supernatant per 50 pl PCR-reaksjon.
- Valider mutanter
- Design primere som spenner knockout regionen bruker primer design programvare (for eksempel Primer3). Design primere som starter på ~ 200 bp hver side av knockout regionen.
Merk: Grunning for å verifisere cpcC1C2 mutant er skissert i tabell 2og kalles cpcC1C2for og cpcC1C2rev. - Forsterke produkter ved hjelp av et program som består av en innledende denatureringstrinn på 95 ° C i 2 minutter, 35 runder 95 ° C i 1 min, 60 ° C i 1 minutt, 72 ° C i 1 min per kb av sekvensen, fulgt av en endelig forlengelsestrinn på 72 ° C i 5 minutter. Inkluder en vill-type kontroll. Dette gir vanligvis konsekvent produkter.
- Kontroller genotype via gel elektroforese. Merkede knockout transformanter viser et band av ~ 4 kb (0,2 kb fra både venstre og høyre fragmenter pluss npt1 / SACB kassett) og fravær av villtype bånd (figur 2).
Merk: I visse tilfeller en ~ 4 kb bandet ikke er observert i den markerte mutant grunn av den store størrelsen på dette PCR-produktet. Imidlertid, hvis et bånd som tilsvarer den forventede størrelse av villtype ikke er observert da typisk denne stammen er en markert utstansing.
- Design primere som spenner knockout regionen bruker primer design programvare (for eksempel Primer3). Design primere som starter på ~ 200 bp hver side av knockout regionen.
- Hvis en villtype båndet fremdeles er til stede så re-strek belastningen på enfrisk BG11 + kanamycin (30 ug / ml) agar plate og gjenta PCR. Gjenta re-striper prosessen til mutanten er adskilt slik at det ikke vill-type bånd er observert i PCR-reaksjonen.
Merk: Øke mengden av kanamycin til en konsentrasjon på 50 ug / ml, deretter 100 pg / ml er noen ganger nødvendig for å skille en markert mutant fullt ut. - Dersom belastningen viser en markert mutant profil via PCR, deretter re-strek på en ny BG11 + kanamycin (30 ug / ml) agar plate. Bruk denne belastningen til å generere den umerkede knockout.
Merk: Protokollen kan brukes til å generere merkede mutanter med bare en antibiotisk resistens kassett dvs. ved å erstatte den npt1 / SACB kassett med bare npt1 kassetten fra pUC18K 20 mellom venstre og høyre fragmenter..
5. generasjon Umerkede Synechocystis Mutants
- Sett opp en ny kultur preget knockout ved inokulering en løkke full av calen til 30-50 ml BG11 medium. Grow kulturen i 2-3 dager til OD 750nm = 0,2 til 0,6.
- Sentrifuger 10 ml kulturen på 2300 xg i 5 min og kast supernatanten. Vask en gang med BG11 medium.
Merk: Ikke resuspender celler ved å virvle da dette kan føre til tap av pili som er avgjørende for DNA-opptak. Suspender cellene ved forsiktig pipettering. - Legg BG11 til et sluttvolum på 200 ul. Overfør cellene til en 14 ml rundbunnet rør.
- Tilsett 1 ug av plasmid B-DNA til cellene og blandes ved forsiktig banking.
- Prøvene inkuberes i 4-6 timer. Legg rør ned horisontalt.
Merk: Celler kan kort blandes ved å banke hver 1-2 timer, men dette er ikke avgjørende. Prøvene kan plasseres i en risteinkubator, selv om dette ikke forbedrer effektiviteten. - Legg 1,8 ml BG11 medium og inkubere prøvene i totalt 4 dager med rysting. Dette er tilstrekkelig tid til å tillate rekombinasjon for å forekomme i flere kromosomale kopier.
- Plate porsjoner av transformasjonsblandingen på BG11 / 5% sukrose agar-plater. Platen 50 pl, 10 pl og 1 ul pr agar plate. Hvis en koloni plen vises på alle disse agarplater fortynne løsningen ytterligere og alikvot på friske plater. Kolonier bør være synlig etter ca 7 dager.
- Patch 30-50 individuelle kolonier på BG11 + kanamycin (30 ug / ml) agarplater første og BG11 / 5% sukrose agar-plater sekund, ved hjelp av en butt ende tannpirker. Eventuelle bakterier som vokser på BG11 / 5% sukrose plater, men ikke BG11 + kanamycin plater er potensielle umerkede knockouts. Bakterier som vokser på begge platene er sannsynlig å være motstandsdyktig sukrose på grunn av en mutasjon i genet SACB.
- Kontroller umerkede knockouts som bruker de samme primere og metoden som ble brukt til å kontrollere de merkede dekslene. F.eks cpcC1C2for og cpcC1C2rev (tabell 2) for å verifisere cpcC1C2 umerket knockout. En umerket knockout viser et band på en agarosegel tilsvar to villtype størrelse minus slettet regionen (figur 2).
- Dersom belastningen viser en umerket mutant profil via PCR (trinn 4.11.2) og gelelektroforese (figur 2), deretter re-strek på en ny BG11 agarplate uten antibiotika.
6. Langsiktig Lagring av stammer
- Sett opp en frisk kultur av stammen ved å inokulere en løkke full av celler i 30-50 ml BG11 medium. Grow kulturen i 3-4 dager til OD 750nm = 0,4 til 0,7.
- Vask celler gang med BG11 og resuspender i ~ 2 ml BG11.
- Legg 0,8 ml konsentrert celler til en tube. Deretter legger 0,2 ml 80% glycerol filtersterilisert.
- Valgfritt: Legg 0,93 ml konsentrert celler til et annet rør. Legg 0,07 ml DMSO til dette røret.
FORSIKTIG: DMSO er giftig og bør håndteres med passende beskyttelse. - Lagre begge rør ved -80 ° C. Å gjenopplive stammer fjerne røret og skrape av noen celler med en stump tannplukke bort på en agarplate uten antibiotika. Strek ut som vanlig ved hjelp av en steril løkke.

Figur 1: Plasmid konstruksjon for generering av merkede og umerkede knockouts, f.eks cpcC1 og cpcC2 i Synechocystis (A) Region av Synechocystis genomet der (B) cpcC1 og cpcC2 og tilstøtende genene er plassert.. Uthevet i svart er den regionen i genomet som skal slettes i mutant. (C) områder av genomet som blir amplifisert ved PCR. 5 'flankerende område (angitt i blått) og 3' flankerende område (markert med rødt) blir forsterket med restriksjonsendonukleaseseter for kloning i pUC19. Den 5 '(eller 3') flankerende region er skåret ut av pUC19 og innsatt i pUC19 + 3 '(eller 59;) flankerende område plasmidet å generere plasmid B. (D) npt1 / SACB kassett fra pUM24 skjæres via Bam HI fordøyelsen og inn mellom 5 'og 3' flankerende områder å generere Plasmid A. Klikk her for å se et større versjon av denne figuren.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Plasmid utforming er avgjørende for vellykket dannelse av både merkede og umerkede mutanter. Figur 1 gir et eksempel på plasmid A og B som brukes for å generere en delesjonsmutant i den Synechocystis genene cpcC1 og cpcC2 13. I hvert tilfelle ble de 5 'og 3' flankerende regioner er tilnærmet 900-1,000 bp. Redusert flankerer regioner kan brukes selv om den minste vi har lykkes trialed har vært om lag 500 bp. Plasmid B kan også inneholde et gen kassett mellom 5 'og 3' ~ 1 kb flankerende regioner eller en modifisert versjon av det native gen-sekvensen.
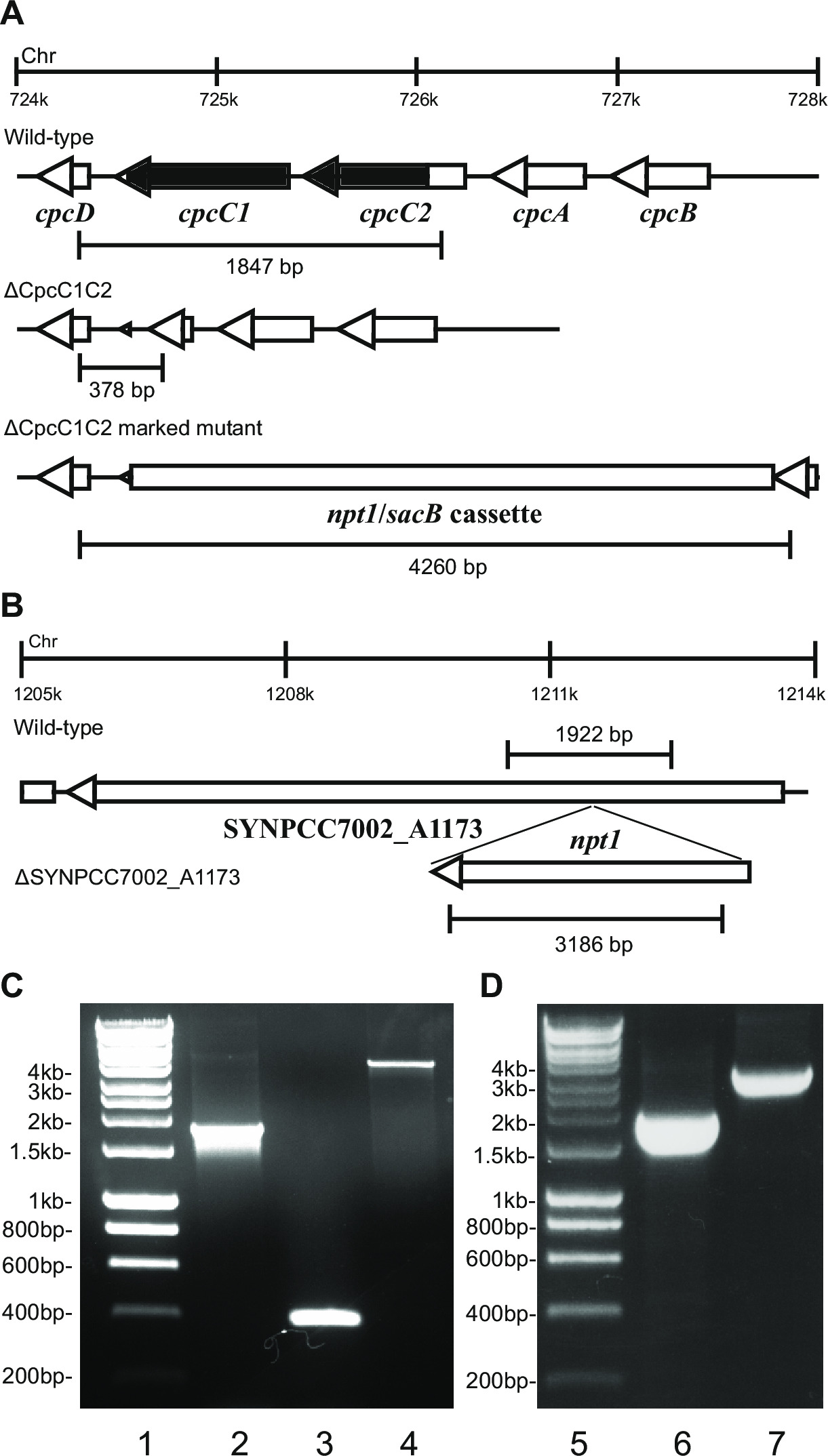
Figur 2: Verifikasjon av merkede og umerkede mutanter, f.eks cpcC1 / cpcC2
Ved transformasjon av plasmid A inn i cellene, vil typisk flere hundre kolonier vises på en plate etter ca 7-10 dager. Kolonier er <1 mm i diameter, og vil ikke øke i størrelse for de neste ukene. Derfor er det viktig å bruke en butt ende tannpirker for å fjerne kolonien og strek den på en ny BG11 + kanamycin agar plate. Omtrent halvparten av de gjen stripete kolonier vil vokse etter 4-6 dager. Hvis genene er ikke-essensielle og mutanter demonstrere vekst tilsvarende villtypestamme under kontinuerlig lys på 20-40 umol fotoner m sek -2 -1 (f.eks terminal oxidase mutanter i Lea-Smith et al., 2013 14) (figur 3), så alle kromosomene skal inneholde en kopi av t han npt1 / SACB kassett innsatte sekvensen, som bestemt ved PCR. Hvis genene er uvesentlig og mutanter viser en langsom vekst fenotype under kontinuerlig lys av 20-40 mikromol fotoner m -2 sek -1 (f.eks phycobilisome mangel mutanter i Lea-Smith et al., 2014 13) (figur 3), deretter flere runder med re-striper på BG11 agarplater med gradvis økende mengder av kanamycin er avgjørende for å oppnå en segregert markert mutant. Når en segregert mutanten oppnås dette bør re-strøket ut på en ny BG11 pluss kanamycin agar plate for å sikre at segregering er fullført. Ved gjentatte runder med striper ikke resulterer i et isolert merket mutant deretter genet er sannsynligvis nødvendig for å overleve. Figur 4 gir en oversikt over de eksperimentelle trinnene involvert i umerket mutant generasjon.
/54001/54001fig3highres.jpg "Width =" 700 "/>
Fig. 3: Vekst av Synechocystis mutanter Eksempler på mutanter som viser (A) tilsvarende vekst til vill-type og (B) lavere vekst enn vill-type. Den ΔCOX mutant mangler cytokromoksydase grunn av sletting av CtaC1D1E1 gener. Den ΔCyd mutant mangler kinol oxidase grunn av sletting av CydAB gener. Oliven mutant mangler en del av den phycobilisome grunn av sletting av de CpcABC1C2D gener. Prøvene i (B) ble boblet med luft for å lette vekst. Gjengitt fra data publisert i Lea-Smith et al, 2013 14 og 2014 13 (www.plantphysiol.org; Copyright American Society of Plant Biologer).. Klikk her for å se en større versjon av dette tallet.
f.eks Figur 2. Når et gen kassetten blir satt inn i kromosomet da typisk en høyere andel av kanamycin resistente og sukrose-resistente kolonier er observert. Disse mutanter kan vokse på sakkarose på grunn av en mutasjon i genet SACB. Hvis ingen kanamycin følsomme og sukrose-resistente kolonier blir generert deretter genet kassetten er skadelige for cellen.

Figur 4: Generering av merkede og umerkede mutanter i Synechocystis Skjematisk detaljering (A) rekombinasjon og (B) eksperimentelle trinnene involvert i mutant generasjon.. Plasmid A blir først blandet med celler. Etter inkubering på agarplater inneholdende kanamycin, kolonier i hvilket en rekombinasjon hendelse inntreffer mellom 5'-end 3 'flankerende regioner (indikert i blått og rødt, henholdsvis), og den homologe sekvens i kromosomet, er isolert. I tillegg er det npt1 / SACB kassett mellom 5 'og 3' flankerende regioner er satt inn i kromosomet. Etter segregering en markert mutant genereres. Merkede mutante celler blir deretter blandet med plasmid B, som kan inneholde enten (C) 1: 5 'og 3' flankerende regioner; 2: 5 'og 3' flankerende regioner med en ekspresjonskassett som inneholdt gener som er av interesse er satt inn mellom disse sekvenser; 3: 5 'og 3' flankerende regioner med villtype-sekvensen med de ønskede nukleotid- forandringer innsatt mellom disse sekvenser. En annen homolog rekombinasjon finner sted mellom 5 'og 3' flankerende regioner og de homologe regionene i kromosomet, noe som resulterer i fjerning av npt1 / SACB kassett og enten den umerkede knockout eller en mutant med en innføring eller endret vill-type region innført i kromosomet. Klikk her for å se en større versjon av dette tallet.
| Stock løsning oppskrifter | |
| Kjemisk | Beløp (g) |
| 100x BG11 (per L) | |
| nano 3 | 149,6 |
| MgSO4 .7H 2 O | 7,49 |
| CaCl 2 .2H 2 O | 3.6 |
| Sitronsyre | 0.6 |
| Legg 1,12 ml 0,25 M Na2EDTA, pH 8,0 | |
| 0,25 M Na2EDTA, pH 8,0 (per 100 ml) | |
| Na2 | 9.3 |
| Sporstoffer (per 100 ml) | |
| H 3 BO 3 | 0,286 |
| MnCl2 .4H 2 O | 0.181 |
| ZnSO4 .7H 2 O | 0,022 |
| Na 2 Moo 4 .2H 2 O | 0,039 |
| CuSO4 .5H 2 O | 0,008 |
| Co (NO 3) 2 .6H 2 O | 0,005 |
| Iron lager (per 100 ml) | |
| Ferric ammonium citrate | 1.11 |
| Fosfat lager (per 100 ml) | |
| K 2 HPO 4 | 3.05 |
| Na 2 CO 3 lager (per 100 ml) | |
| Na 2 CO 3 | 2 |
| TES-buffer, pH 8,2 (per 100 ml) | |
| TES | 22.9 |
| NaHCO3 lager (per 100 ml) | |
| NaHCO3 | 8.4 |
| HEPES, pH 8,2 (per 500 ml) | |
| HEPES | 119,15 |
| Vitamin B 12 (Per 50 ml) | |
| cyanokobalamin | 0,02 |
| Luria Bertani media (Per 500 ml) | |
| Luria Bertani kjøttkraft | 12,5 |
| 1 M MgCl2 (per 100 ml) | |
| MgCl2 .6H 2 O | 20.33 |
| MnCl2 .4H 2 O | 0,395 |
| CaCl 2 .2H 2 O | 1,47 |
| 2- (N -Morpholino) etansulfonsyre hydrat, 4-Morpholineethanesulfonic syre (MES) | 0,4265 |
| Løsning A + glyserol | |
| 10 ml oppløsning A | |
| 1,5 ml glyserol |
Tabell 1: Løsninger brukt i denne studien.
| primer | Sekvens |
| cpcC1C2leftfor | GTAC TCTAGA GCGGCTAAATGCTACGAC |
| CPCC1C2leftrev | GATC GGATCC GCGGTAATTGTTCCCTTTGA |
| cpcC1C2rightfor | GATC GAGCTC TGCACTGGTCAGTCGTTC |
| cpcC1C2rightrev | GACT GAATTC ATCGTTGCTTGAACGGTCTC |
| M13 fremover | TGTAAAACGACGGCCAGT |
| M13 revers | CAGGAAACAGCTATGAC |
| cpcC1C2for | GTTTTCATTGGCATCGGTCT |
| cpcC1C2rev | ATGTCCCAGGAACGACTGAC |
| A1173for | AGCAAACCGTTTTTGTGACC |
| A1173rev | TGCAAGGTGGCGAACTGTAT |
Tabell 2:. Primere brukt i denne studien restriksjonsendonukleaseseter, er understreket.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
De mest kritiske trinn i generering av umerkede mutanter er: 1) forsiktig plasmid utforming for å sikre at bare det aktuelle området blir endret; 2) å sikre at prøvene fortsatt axenic, spesielt når dyrket på sukrose; 3) plettetransformerte celler for merket mutant generasjon innledning på BG11 agarplater mangler antibiotika, etterfulgt av tilsetning av agar pluss antibiotika 24 timer senere; 4) dyrking merket mutanter for 4 hele dager før plating på BG11 pluss sukrose agarskåler: 5) at merket mutanter er fullt isolert og 6) grundig bekrefter genotype av mutantstammer. For dette siste trinnet, videre primere utformet for å amplifisere en del av det slettede området, kan brukes for å sikre at den har blitt fjernet. Southern blotting, mens arbeidskrevende, kan også brukes. Men vår erfaring at prosedyren er beskrevet i denne artikkelen er tilstrekkelig for riktig verifisering av mutanter. Denne fremgangsmåten har også blitt anvendt for å danne merkede mutanter i Synechococcus elongatus PCC7942. Imidlertid har gjentatte transformasjon av dette cyanobacterium vist seg utfordrende.
Hvis merket mutanter ikke kan skilles deretter ulike miljøforhold høye CO 2, lite lys (<20 mikromol fotoner m -2 sek -1) eller flere næringsstoffer (dvs. glukose) kan testes. For eksempel, er tilsetningen av glukose vesentlig for å generere photosystem II-mutanter 21. Hvis merket mutanter aldri fullt skille så genet er trolig avgjørende for levedyktigheten. Det finnes imidlertid eksempler fra litteraturen der noen forskningsmiljøer har vært ute av stand til å slå ut et gen (for eksempel Vipp i Synechocystis) 22, bare for andre grupper å vise senere at genet er ikke avgjørende 23. Dette kan skyldes forskjeller i villtype stammer eller feil plasmid design, noe som resulterer i polare effekter på tilgrensende, essensielle gener. Hvis en mutant ikke fulltsegregere vi vil anbefale at plasmidet inneholdende npt1 kassetten fra pUC18K 20 mellom venstre og høyre fragmenter bli anvendt for transformasjon. Det er lettere å bekrefte tilstedeværelsen av bånd tilsvarende villtype og mutant ved hjelp av PCR, ettersom dette fragment er omtrent 1,2 kb, sammenlignet med den 3,8 kb npt1 / SACB kassett. Dette resultatet er en viktig del av bevis som viser at genet er essensielt.
Generering av umerkede mutanter med innsatte uttrykk kassetter er generelt mer utfordrende enn utvikling av knockout stammer. Vi vanligvis uttrykke gener under kontroll av en sterk promoter cpcBAC1C2D 13. I noen tilfeller kan dette redusere sjansene for vellykket innføring av genet kassett, hvis over-ekspresjon av et protein som er skadelige for cellen. Svakere arrangører skal deretter testes. Generelt har vi observert at jo større genet kassetten er, det er desto vanskeligere å ifører det inn i genomet. Vi har ikke vært i stand til å sette inn genet kassetter større enn 5 kb. Hensyn må også tas ved valg av områder for å sette inn ekspresjonskassetter inn i genomet. Nøytrale områder som ikke påvirker cellenes levedyktighet eller vekst bør brukes. Eksempler i Synechocystis inkluderer phaAB og phaCE, som koder for proteiner som koder for polyhydroksybutyrat biosyntetiske vei 24,25. Mer nylig en omfattende liste over nøytrale områder i Synechocystis har blitt identifisert 26.
Generering av umerkede mutanter i cyanobakterier er en langsom prosess, som tar omtrent 5-7 uker hvis alle trinn gjennomfører på riktig måte. Dette er tregere enn standard metode for generering av merkede knockouts benyttes av de fleste forskningsgrupper undersøker cyanobakterier. Men fleksibiliteten til å kunne innføre ytterligere mutasjoner i umerkede mutanter delvis kompenserer for dette, siden flere plasmider fortseller innelukker en rekke kassetter som gir resistens mot forskjellige antibiotika, ikke behøver å bli konstruert. For forskningsformål evnen til å mutere multiple gener er noen ganger nødvendig for å kunne fullt ut å karakterisere funksjonelle rolle av proteiner. For eksempel har vi identifisert en skadelig fenotype kun ved delesjon av de to terminale oxidase elektron synker lokalisert til den thylakoid membranen, ettersom tap av bare ett av disse kompleksene kan bli kompensert for ved aktiviteten av den andre 14. Utvikling av en stamme for industrielle anvendelser vil også kreve flere modifikasjoner til en belastning, ikke bare for innføring av fremmede gener, men også for å øke fotosyntetisk effektivitet, lette høsting optimalisering og sletting av konkurrerende reaksjonsveier for det ønskede substrat.
Den viktigste faktoren som begrenser hastigheten på umerkede mutant generasjon er den langsomme divisjon tiden av modell cyanobakterietoksiner arter, mellom 8-20 timer avhengig av lysforhold. unDer høyere lysintensitet og CO 2 -konsentrasjoner er veksten raskere. Det er imidlertid en risiko for at mutantstammer som ikke tåler høy enten lys eller CO 2 vil bli valgt mot, eller at mutant-stammer vil gjennomgå uønskede endringer forut for fenotypisk karakterisering. Derfor er dette ikke anbefalt. Imidlertid ville det være meget fordelaktig dersom en raskere protokoll for å generere umerkede mutanter ble utviklet. Samlet sett vil dette lette utviklingen av stammer for både grunnforskning og anvendt applikasjoner. Slike stammer kunne brukes til biodrivstoff, biomasse eller kjemisk produksjon eller i å forstå mange aspekter av cyanobacterial biokjemi, genetikk og fysiologi.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Vi er takknemlige for Environmental Services Association Education Trust, den Syntetisk biologi i Cambridge SynBio fond og departementet for sosial rettferdighet og Empowerment, Government of India, for økonomisk støtte.
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. 3rd Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
