

Introduction
Цианобактерии являются эволюционно древней и разнообразной филюм бактерий, обнаруженных в почти каждой природной среды на Земле. В морских экосистемах они особенно обильны и играют ключевую роль во многих питательных циклов, что составляет примерно половину фиксации углерода 1, большинство фиксации азота 2 и сотни миллионов тонн добычи углеводородов 3 в океане ежегодно. Хлоропласты, органелл отвечает за фотосинтез в эукариотических водорослей и растений, вероятно, произошли от цианобактерии , которая была охвачена организма - хозяина 4. Цианобактерии доказали полезные модели организмов , предназначенных для изучения фотосинтеза, транспорта электронов 5 и биохимических путей, многие из которых сохраняется в растениях. Для этого цианобактерий все чаще используются для производства пищевых продуктов, различных видов биотоплива 6, электричество 7 и промышленных соединений 8, из - за их приветльное эффективное преобразование воды и CO 2 на биомассу с использованием солнечной энергии 9. Многие виды могут быть выращены на не пахотной земли с минимальными питательными веществами и морскую воду, предполагая, что цианобактерии потенциально можно выращивать в больших масштабах, не затрагивая сельскохозяйственного производства. Некоторые виды также являются источниками натуральных продуктов, в том числе противогрибковыми, антибактериальными и противоопухолевыми соединениями 10,11.
Способность генерировать мутантов является ключом к пониманию цианобактерий фотосинтез, биохимии и физиологии, а также важное значение для развития штаммов для промышленных целей. Большинство опубликованных исследований генерируют генетически модифицированные штаммы путем введения антибиотика кассеты сопротивления в месте интереса. Это ограничивает число мутаций, которые могут быть введены в штамм, а всего лишь несколько антибиотических кассет сопротивления доступны для использования в цианобактерии. Штаммы, содержащие гены, придающие повторно антибиотиктивление не может быть использован для промышленного производства в открытых водоемах, который, вероятно, будет единственным экономически эффективным средством для производства биотоплива и других продуктов с низким значение 12. Поколение немаркированных мутантов преодолевает эти ограничения. Немаркированные мутанты не содержат чужеродной ДНК, если намеренно не включены, и ими можно манипулировать несколько раз. Поэтому можно создавать столько изменений в штамме по своему желанию. Кроме того, полярные эффекты на гены ниже сайта модификации , может быть сведено к минимуму, что позволяет более точно модификации организма 13.
Для создания мутантных штаммов, самоубийство плазмид, содержащих два фрагмента ДНК идентичны областям в цианобактерий хромосомы, фланкирующих ген должен быть удален (называется 5 'и 3' фланкирующие области) сначала построены. Два гена, которые затем вставляются между этими фланкирующих. Один из них кодирует антибиотик белок устойчивости; вторая кодирует SACB, который Продлевансахаразу спам, соединение придания чувствительности к сахарозе. На первом этапе процесса, отмечены мутанты, т.е. штаммы , содержащие некоторую чужеродную ДНК, образуются. Плазмидной конструкции смешивают с цианобактерий клеток и ДНК ресуспендировали естественным организмом. Трансформанты отбирали по росту на чашках, содержащих соответствующий антибиотик, и мутантный генотип проверены с помощью ПЦР. Суицид плазмид не может реплицировать в пределах требуемого штамма. Поэтому любые резистентные к антибиотикам колоний будет результатом рекомбинации в результате чего ген, представляющий интерес в вставленную в хромосому. Для генерации немаркированных мутантов, отмеченный мутант затем смешивают со вторым самоубийцы плазмидой, содержащий только 5 'и 3' фланкирующие области. Тем не менее, если введение чужеродной ДНК требуется, плазмиду, состоящую из 5 'и 3' фланкирующие области с кассетой, содержащей гены, представляющие интерес, вставленной между этими фрагментами ДНК, могут быть использованы. Селефикция это с помощью роста на чашках с агаром, содержащей сахарозу. Поскольку сахароза является летальной для клеток , когда продукт гена SACB выражается, только клетки , которые выживают , являются те , в которых произошло второе событие рекомбинации, в результате чего ген сахарозы чувствительность, в дополнение к гену устойчивости к антибиотику, был рекомбинируют из ряда хромосомы и на плазмиды. Как следствие рекомбинацнонного обмена, фланкирующие области и любую ДНК между ними вставлены в хромосому.
Мы успешно использовали эти методы , чтобы генерировать множественные мутации в хромосомных того же штамма Synechocystis зр. PCC6803 (далее именуемые как Synechocystis) 13,14, ввести единые точечные мутации в ген интереса 13 и для экспрессии гена кассет. В то время как поколение немаркированных нокаутов было продемонстрировано до нашей работы в Synechocystis 15,16, подробный метод, которому помогаетвизуальное представление важных шагов, не является общедоступной. Кроме того, мы применили тот же метод для генерации отмеченными нокаутов в другой модели цианобактерии, Synechococcus зр. PCC7002 (далее именуемой Synechococcus). Этот протокол обеспечивает четкий, простой способ для генерации мутантов и быстрый протокол для проверки и хранения этих штаммов.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Подготовка медиа-культуры
- Подготовка BG11 среды в соответствии с Castenholz, 1988 17.
- Подготовить исходные растворы 100x BG11, микроэлементов и железа запасов (таблица 1).
- Подготовка отдельных растворов фосфата запаса, Na 2 CO 3 сток, N - [трис (гидроксиметил) метил] -2-аминоэтансульфоновая кислота (TES) , буфер и раствором NaHCO 3 (Таблица 1).
- Автоклава фосфат и Na 2 CO 3 запасов. Фильтр-стерилизовать TES буфера и NaHCO 3 с 0,2 мкм фильтров.
- Готовят BG11 путем объединения 976 мл воды, 10 мл 100х BG11 1 мл микроэлементов и 1 мл железа запаса и автоклав раствор. После этого раствор остынет до комнатной температуры, добавляют 1 мл фосфатного запаса 1 мл Na 2 CO 3 акций и 10 мл NaHCO 3.
- Для BG11 твердой среды, добавляют 15 г агара и 700 мл воды на одну FLAпестрый Ко второй колбу, добавляют 3 г Na 2 S 2 O 3, 226 мл воды, 10 мл 100х BG11, 1 мл микроэлементов и 1 мл железа складе. Автоклав оба решения. После того, как эти решения охлаждают до комнатной температуры, объединить их и добавить 1 мл фосфатного запаса 1 мл Na 2 CO 3, запас 10 мл буфера TES, и 10 мл NaHCO 3.
Примечание: Растворы получают отдельно, чтобы избежать осаждения некоторых солей.
- Для селекции на сахарозе, приготовить 50% (вес / объем) раствор сахарозы. Фильтр стерилизовать раствор с 0,2 мкм-фильтры и добавить к BG11 (100 мл 50% сахарозы в 900 мл BG11) для получения BG11 / 5% пластины сахарозы.
Примечание: Не добавляйте NaHCO 3 к BG11 / 5% сахарозы чашки с агаром. Добавить Na 2 CO 3 в нормальном режиме. - Для культивирования Synechococcus добавляют 10 мл 1 М 4- (2-гидроксиэтил) пиперазин-1-этансульфоновой кислоты, N - (2-гидроксиэтил) пиперазин - NR42; - (2-этансульфоновой кислоты) (HEPES) и 1 мл витамина В 12 (таблица 1) до 1 л BG11 среды.
Примечание: Трансформация штаммов, культивируемых в коммерчески доступных BG11 средах значительно менее эффективно, чем в рецептах BG11 средств, описанных здесь, и, следовательно, не рекомендуется.
2. Рост цианобактерий Штаммы
- Культура напрягает в 100 мл конические колбы с максимальным объемом 50 мл и встряхивают при 120 оборотах в минуту. Уплотнение BG11 пластины с парафильмом и колотых трех небольших отверстий в стороне пластины, чтобы обеспечить газообмен. Выдержите все штаммы при 30 ° C под лампами дневного света в фотобиореактора при интенсивности света между 20-40 мкмоль фотонов м -2 сек -1.
- Используйте лучшие стерильные методы. Обращайтесь со всеми цианобактериальными напряжения в ламинарном потоке.
Примечание: Это особенно важно, когда штаммы культивируют со средами, содержащими сахарозы, которые могут быть легко contaminованные.
3. Генерация плазмидной конструктов
- Конструктивные наборы праймеров, в том числе требуемых участков фермента рестрикции, с использованием разработки программного обеспечения праймера, таких как Primer3 (http://frodo.wi.mit.edu/primer3/~~HEAD=pobj), чтобы усилить два ~ 1 килобайта областей 5 'и 3' Представляющий интерес ген. Обратитесь к последовательности генома цианобактерии видов с помощью Cyanobase (http://genome.kazusa.or.jp/cyanobase). Приведены в таблице 2 для всех праймеров , используемых здесь. При проектировании праймеров следует учитывать следующие факторы:
- Убедитесь , что амплифицированные регионы включают 5 'и 3' области гена , который будет мутировать, например , рис 1.
- Не мутировать межгенные области, чтобы избежать непреднамеренного мутации антисмысловых и некодирующих РНК. Для генерации мутантов в Synechocystis, обратитесь к списку транскрипционных стартовых сайтов документированных в Mitschke и др., 2011 18, с тем чтобы избежать мутации антисмысловыхили некодирующие РНК.
- При выборе фланкирующие области не включают в себя всю открытую рамку считывания смежных генов как экспрессии этих генов в кишечной палочки могут помешать клонирования.
- Amplify продуктов ПЦР с использованием высокой точности воспроизведения ДНК-полимеразы в соответствии с инструкциями изготовителя.
Примечание: В нашем опыте этот фермент производит несколько ошибок.- Установка 50 мкл ПЦР-реакций, содержащие HF буфер и либо 0, 1,5 или 3 мкл ДМСО. С помощью 100 нг геномной ДНК на реакцию. Используйте программу, состоящую из начальной стадии денатурации 98 ° С в течение 30 сек, 35 раундов 98 ° С в течение 10 сек, 67 ° C в течение 30 сек, 72 ° С в течение 30 сек, с последующим конечным стадией удлинения в 72 ° С в течение 5 мин. Как правило, это дает последовательные продукты.
- Проверка продуктов ПЦР и образцы перевариваются с эндонуклеазы ферментами для правильного размера с помощью гель-электрофореза. Выполнить 1% (вес / объем) агарозном геле, содержащем 0,02%(Об / об) этидий бромида в течение 45 мин при 100 В.
ВНИМАНИЕ: этидий бромид является потенциальным мутагенным и должны быть обработаны с соответствующей защитой. - Очисти продуктов ПЦР с использованием набора для очистки ДНК в соответствии с инструкциями изготовителя. Также используйте этот набор для очистки плазмид фрагментов, в том числе кусочки, вырезанные из агарозных гелей. Элюции очищенную ДНК в 14 мкл воды.
- Для стадий клонирования, инкубировать реакционных смесей эндонуклеаз рестрикции при 37 ° С в течение 1 ч> в общем объеме 30 мкл в соответствии с инструкциями изготовителя.
- Для стадий лигирования, лигирование ДНК-фрагменты, при комнатной температуре в течение> 1 часа в общем объеме 20 мкл, содержащем 5 мкл очищенного переваренной плазмидой 12 мкл очищенного переваренной вставки, 2 мкл буфера и 1 мкл лигазы.
- Приготовьте трансформантов клетки кишечной палочки DH5 & alpha ; в соответствии со следующим способом.
- Вырасти на ночь E. палочки
- Инокулируйте 400 мл LB в 1 л коническую колбу , содержащую 6 мл 1 М MgCl 2 (таблица 1) с 1 мл ночной культуры.
- Grow культуру при 37 ° C при 220 оборотах в минуту в течение примерно 4 ч или до OD 600 нм достигает 0,4-0,6.
- Поместите клетки на льду в течение 1 часа.
- Центрифуга при 2800 х г в течение 10 мин для осаждения клеток при 4 ° С.
- Удалить супернатант и ресуспендируют в 160 мл раствора А (таблица 1) и инкубировать на льду в течение 20 мин.
- Центрифуга при 2800 х г в течение 10 мин для осаждения клеток при 4 ° С.
- Удалить супернатант и ресуспендируют в 4 мл раствора A + глицерин (таблица 1).
- Готовят 50 мкл аликвоты, замораживать в жидком N 2, хранить при температуре -80 ° С.
- Смешайте 5 мкл смеси лигировани с 50 мкл компетентных клеток и инкубируют в течение 1 ч на льду.
- Теплового шока клетки при 42 ° С в течение 90 сек, FOLLOср инкубацией на льду в течение 2 мин.
- Добавить 950 мкл LB сред (таблица 1) и инкубируют при температуре 37 ° С в течение 1 часа.
- Аликвоты 50 и 200 мкл на чашки с соответствующим антибиотиком, либо ампициллин (100 мкг / мл) и / или канамицин (30 мкг / мл).
ВНИМАНИЕ: Оба канамицин и ампициллин являются токсичными и должны быть обработаны с соответствующей защитой. - Комплектование и инкубировать отдельных колоний в 2 мл LB-средах, привитых соответствующий антибиотик.
- Очищают все плазмиды с использованием набора для очистки Минипрепарат плазмиды в соответствии с инструкциями изготовителя.
- Сформировать плазмид, в этом конкретном примере для выбивания генов cpcC1C2, в соответствии со следующими этапами.
- Amplify фланговые область 1,012 п.о. 5 '(левый фрагмент) с использованием праймеров cpcC1C2leftfor и cpcC1C2leftrev (шаг 3.2, таблица 2). Удалить небольшое количество ПЦР-реакции и подтвердить, является липравильный продукт размер был усилен с помощью гель-электрофореза (шаг 3.3). Обработайте этот фрагмент и pUC19 с Xba I и Bam HI (шаг 3.5).
- Очищают оба препарата (этап 3.4), перевязывать (шаг 3.6), преобразование (этап 3.7) и установить четыре 2 мл БЛ суспензионных культур с ампициллином (100 мкг / мл) из отдельных колоний для очистки плазмиды с помощью минипрепараты (этап 3.8).
- Проверка для вставки фрагмента в pUC 19 через Xba I / Bam HI переваривания и гель - электрофореза (этап 3.3). Полосы 2660 п.н. и 1,012 пар оснований указывают правильное введение вставки в плазмиду.
- Amplify фланговые область 1016 п.н. 3 '(справа фрагмент) с использованием праймеров cpcC1C2rightfor и cpcC1C2rightrev (шаг 3.2, таблица 2). Удалить небольшое количество ПЦР-реакции и подтвердить ли правильный продукт размер был усилен с помощью гель-электрофореза (этап 3.3). Обработайте этот фрагмент и pUC19 с Sac I и Eco RI (улер 3.5).
- Очищают оба препарата (этап 3.4), перевязывать (шаг 3.6), преобразование (этап 3.7) и установить четыре 2 мл БЛ суспензионных культур с ампициллином (100 мкг / мл) из отдельных колоний для очистки плазмиды с помощью минипрепараты (этап 3.8).
- Проверка для вставки фрагмента в pUC 19 посредством Sac I / Eco RI переваривания (этап 3.5) и гель - электрофореза (этап 3.3). Полосы 2660 п.н. и 1016 п.н. указывают правильное введение вставки в плазмиду.
Примечание: Xba I / Bam HI сайты для клонирования 5' - области и Sac I / Eco RI для клонирования 3' - области в pUC19 используются везде , где это возможно. Если это возможно, всегда включают в себя сайт Bam HI на обратного праймера для "области или прямого праймера для 3 '5 региона для обеспечения того , позже Клонирование шаги легче выполнить. - Последовательность обе вставки , чтобы определить , является ли правильность последовательности с использованием праймеров , охватывающих место введения, например M13 вперед и М13 обратным (Таблица 2). Последовательность должна быть правильной, чтобы гарантировать, что никакие ошибки не будут введены в фланкирующих.
- Вырежьте левый фрагмент из pUC19 с помощью Xba I / Bam HI пищеварения. Гидролизуют pUC19 + правый фрагмент с Xba I / Bam HI (шаг 3.5).
- Очищают 1,012 п.о. левый фрагмент и 3676 п.н. pUC19 + правый фрагмент из агарозного геля (шаг 3.3) с помощью иссечения ДНК, используя лезвие скальпеля.
- Очищают оба препарата (этап 3.4), перевязывать (шаг 3.6), преобразование (этап 3.7) и установить четыре 2 мл БЛ суспензионных культур с ампициллином (100 мкг / мл) из отдельных колоний для очистки плазмиды с помощью минипрепараты (этап 3.8).
- Проверьте для вставки фрагмента в pUC19 + правый фрагмент через Xba I / Bam HI пищеварения (этап 3.5) и гель - электрофореза (шаг 3.3). Полосы 3676 п.н. и 1,012 пар оснований указывают на правильность установки вставки в плазмиду (относятся к этому как плазмиды B).
Примечание: NPT1 / SACB кассета не должны быть очищены от агарозных гелей , поскольку pUM24cm кодирует белок , придающий устойчивость к хлорамфениколу. Поэтому , если колонии выращивают на LB / ампициллин / канамицину агаром единственно возможная комбинация , которая приведет к резистентных колоний включение NPT1 / SACB кассеты в плазмиду В. - Очищают оба препарата (шаг 3.4), перевязывать (шаг 3.6), преобразование (шаг 3.7) и установить четыре 2 мл LB жидких культур с ампициллином (100 мкг / мл) и канамицин (30 мкг / мл) из отдельных колоний для очистки плазмиды с помощью минипрепараты (шаг 3.8).
- Проверка для вставки NPT1 / SACB кассеты в плазмиду B через Bam HI пищеварении (этап 3.5) и гель - электрофореза (этап 3.3). Полосы 4,688 п.о. и 3,894 пар оснований указывают на правильную вставку-гое вставки в плазмиду (см это как плазмида A).
- В качестве альтернативы, тупой конец NPT1 / SACB кассета и клон в другой сайт рестрикции эндонуклеазы между левым и правым фрагментами в плазмиде В. NPT1 / SACB кассета должна быть клонирована между левым и правым фрагментами.
Примечание: Если требуется экспрессии чужеродного кассеты, то это должно быть вставлено между левым и правым фрагментами плазмиды B. Эту плазмиду затем используют в немаркированных шагов выбивки.
4. Генерация маркированных Synechocystis и Synechococcus Мутанты
- Установить свежую культуру прививка полный цикл клеток в 30-50 мл BG11 среды. Grow культуру в течение 2-3 дней до OD 750 нм = от 0,2 до 0,6.
Примечание: Обычно отдельные колонии слишком малы, чтобы использовать для инокуляции и воздействия отдельных клеток даже низких уровней света приведет к фотоингибированию и отбордля легких устойчивых мутантов. - Центрифуга 1-2 мл культуры при 2,300 мкг в течение 5 мин и отбросить супернатант. Не центрифугировать никаких цианобактериальными культур при> 2.300 XG, так как это может привести к повреждению клеток. Вымойте гранул один раз с BG11 средой.
Примечание: Не ресуспендирования клетки встряхиванием, так как это может привести к потере ворсинок, которые имеют важное значение для поглощения ДНК. Ресуспендируют клеток осторожно пипеткой. - Добавить BG11 среды до конечного объема 100 мкл. Передача клеток в 14 мл с круглым дном трубки.
- Добавьте 1 мкг плазмиды A к клеткам и перемешать путем осторожного постукивания. Добавить <10 мкл плазмиды.
Примечание: Предпочтительно плазмида должна быть в концентрации> 100 нг / мкл, но концентрации ниже этого являются достаточными для успешной трансформации. - Lay трубы горизонтально в инкубаторе. Инкубируйте культур в течение 4-6 ч.
Примечание: Клетки могут быть кратко смешаны, нажав каждые 1-2 ч, но это не является существенным. Образцы могут быть помещены ввстряхивая инкубатор, хотя это не приводит к существенному повышению эффективности. - Распространение аликвот смеси ДНК клеточной культуры / плазмиды на BG11 чашки с агаром без антибиотиков. Как правило, 20 мкл и 80 мкл аликвоты распространяются на отдельные пластины.
- ~ 24 ч позже, добавляют 2,5-3 мл 0,6% раствора агара в воде, содержащей канамицин (на 20 мл раствора: 0,12 г агар, 100 мкл 100 мг / мл канамицина) на агаровой пластине. Охлаждают этот раствор ~ 42 ° С, и добавить к краю чашки с агаром. Наклон пластины таким образом раствор образует ровный слой 'верхнего агара' на поверхности.
- Выдержите агаром на дополнительный период времени. Колонии должны быть видны примерно через 7 дней.
Примечание: Чашки с агаром могут быть сложены 3 высоко в инкубаторе. Обычно сотни колоний получают за трансформации. - Трековые отдельные колонии на BG11 + канамицин (30 мкг / мл) чашки с агаром. Разделите агаризованной на 6 секторов и использовать тупым концом зубочистки полосу изколонии над каждым отдельным сектором. Получение отдельных колоний не важно, только рост трансформантов.
- Подтвердите выраженное нокаута методом ПЦР с использованием Taq ДНК-полимеразы в соответствии с инструкциями изготовителя. Добавьте 2 мкл MgCl 2 (25 мМ) на реакцию.
- Удалите небольшую часть клеток и переносят в пробирку, содержащую 50 мкл воды и 20 ~ 425-600 мкм стеклянные бусы. Взболтать в вибраторе в течение 5 мин при температуре ~ 2000 оборотов в минуту. Центрифуга при 15700 х г в течение 5 мин и используют 5 мкл супернатанта в 50 мкл ПЦР-реакции.
Примечание: Не ресуспендируют решение. Клеточный дебрис должна оставаться в нижней части трубы.
- Удалите небольшую часть клеток и переносят в пробирку, содержащую 50 мкл воды и 20 ~ 425-600 мкм стеклянные бусы. Взболтать в вибраторе в течение 5 мин при температуре ~ 2000 оборотов в минуту. Центрифуга при 15700 х г в течение 5 мин и используют 5 мкл супернатанта в 50 мкл ПЦР-реакции.
- Проверка мутантов
- Дизайн праймеров, которые охватывают область нокаута с использованием праймера разработки программного обеспечения (например, Primer3). Дизайн праймеров, начиная с ~ 200 п.н. обе стороны от области выбивки.
Примечание: Грунтовки для проверки мутант cpcC1C2 приведены в Таблице 2и называются cpcC1C2for и cpcC1C2rev. - Amplify продукты, используя программу, состоящую из начальной стадии денатурации 95 ° С в течение 2 мин, 35 циклов 95 & deg; С в течение 1 мин, 60 ° С в течение 1 мин, 72 ° С в течение 1 мин на т.п.н. последовательности, за которым следует Последний шаг расширение 72 ° С в течение 5 мин. Включите контроль дикого типа. Как правило, это дает последовательные продукты.
- Проверьте генотип с помощью гель-электрофореза. Отмеченные нокаут трансформанты покажет полосу ~ 4 кб (0,2 кб из левого и правого фрагментов плюс NPT1 / SACB кассете) и отсутствие дикого типа полосы (рисунок 2).
Примечание: В некоторых случаях т.п.н. полоса ~ 4 не наблюдается в размеченных мутанта из-за большого размера этого продукта ПЦР. Тем не менее, если полоса, соответствующая размеру ожидаемого дикого типа не наблюдается, то, как правило, этот штамм отмеченное нокаута.
- Дизайн праймеров, которые охватывают область нокаута с использованием праймера разработки программного обеспечения (например, Primer3). Дизайн праймеров, начиная с ~ 200 п.н. обе стороны от области выбивки.
- Если дикого типа группа все еще присутствует, то повторно Streak нагрузку насвежий BG11 + канамицин (30 мкг / мл) агар пластины и повторить ПЦР. Повторите процесс повторного полосатость, пока мутант не сегрегация, так что нет дикого типа полоса не наблюдается в реакции ПЦР.
Примечание: Увеличение количества канамицин в концентрации 50 мкг / мл, а затем 100 мкг / мл, иногда необходимо для того, чтобы полностью сегрегируют заметное мутанта. - Если данный штамм демонстрирует выраженный мутантный профиль методом ПЦР, а затем вновь полосу на свежую BG11 + канамицин (30 мкг / мл) чашки с агаром. Используйте этот штамм для генерации опознавательных знаков нокаутом.
Примечание: Протокол может быть использован для генерирования отмеченных мутантов с помощью всего антибиотику кассетой сопротивления т.е. путем замены NPT1 / SACB кассету с только NPT1 кассету из pUC18K 20 между левым и правым фрагментами..
5. Поколение немаркированных Synechocystis Мутанты
- Установить свежую культуру отмеченной нокаута путем инокуляции петли, полный Cгезов в 30-50 мл BG11 среды. Grow культуру в течение 2-3 дней до OD 750 нм = от 0,2 до 0,6.
- Центрифуга 10 мл культуры в 2300 мкг в течение 5 мин и отбросить супернатант. Промывают один раз BG11 средой.
Примечание: Не ресуспендирования клетки встряхиванием, так как это может привести к потере ворсинок, которые имеют важное значение для поглощения ДНК. Ресуспендируют клеток осторожно пипеткой. - Добавить BG11 до конечного объема 200 мкл. Передача клеток в 14 мл с круглым дном трубки.
- Добавьте 1 мкг ДНК плазмиду B к клеткам и перемешать путем осторожного постукивания.
- Инкубируйте образцы в течение 4-6 ч. Положите трубы вниз по горизонтали.
Примечание: Клетки могут быть кратко смешаны, нажав каждые 1-2 ч, но это не является существенным. Образцы могут быть помещены в качалке инкубаторе хотя это не улучшает эффективность. - Добавить 1,8 мл BG11 среды и инкубировать образцов в общей сложности 4 дня при встряхивании. Этого вполне достаточно времени, чтобы позволить рекомбинацию происходить в нескольких хромосомных копий.
- Пластинчатые аликвоты трансформационной смеси на BG11 / 5% сахарозы чашках с агаром. Пластина 50 мкл, 10 мкл и 1 мкл на чашку с агаром. Если газон колонии появляется на все эти агаром разбавить раствор дополнительно и аликвоты на свежих чашках. Колонии должны быть видны примерно через 7 дней.
- Patch 30-50 отдельных колоний на BG11 + канамицин (30 мкг / мл) Чашки с агаром первой и BG11 / 5% сахарозы агаром второй, используя тупой конец зубочистки. Любые бактерии, которые растут на BG11 / 5% пластин сахарозы, но не BG11 + канамицину пластины являются потенциальными немаркированных нокаутами. Бактерии , растущие на обеих пластинах могут быть сахароза устойчивы из - за мутации в гене SACB.
- Проверьте немаркированные просвечивания с использованием тех же праймеров и метод, который был использован для проверки отмеченных нокауты. Например cpcC1C2for и cpcC1C2rev (таблица 2) для проверки опознавательных знаков нокаутом cpcC1C2. Немаркированной Нокаут покажет полосу на агарозном геле соответствующий третO размер дикого типа минус удаляемая область (рисунок 2).
- Если штамм показывает немаркированную мутантный профиль с помощью ПЦР (этап 4.11.2) и гель - электрофорез (рисунок 2), а затем вновь полосу на свежей BG11 агаром без антибиотиков.
6. Долгосрочное хранение Штаммы
- Установить свежую культуру штамма путем инокуляции полный цикл клеток в 30-50 мл BG11 среды. Grow культуру в течение 3-4 дней до OD 750 нм = 0,4 до 0,7.
- Вымойте клетки один раз с BG11 и ресуспендируют в ~ 2 мл BG11.
- Добавить 0,8 мл концентрированной клеток в одну ампулу. Затем добавляют 0,2 мл 80% стерилизованной глицерина.
- Дополнительно: Добавить 0,93 мл концентрированной клеток в другую пробирку. Добавить 0,07 мл ДМСО в этой трубке.
ВНИМАНИЕ: ДМСО является токсичным и должны быть обработаны с соответствующей защитой. - Хранить обе пробирки при температуре -80 ° С. Возродить штаммы удалить трубку и соскоблить некоторые клетки с тупым зубвыбрать на агаровой пластине без антибиотиков. Streak, как нормальный с помощью стерильной петли.

Рисунок 1: Плазмида конструкция для генерации и немаркированных отмеченных нокаутами, например cpcC1 и cpcC2 в Synechocystis (А) область генома Synechocystis , где (В) cpcC1 и cpcC2 и смежные гены расположены.. Выделены черным цветом является областью генома, чтобы исключить в мутанта. (С) участки генома , которые амплифицируют с помощью ПЦР. 'Фланкирующей области (выделены синим цветом) и 3' 5 фланкирующей области (обозначены красным цветом) усиливаются с рестрикционных эндонуклеаз сайтов для клонирования в pUC19. 5 '(или 3') фланкирующей области вырезают из pUC19 и вставляли в pUC19 + 3 '(или 59;) фланкирующей области плазмиды для создания плазмиды B. (D) NPT1 / SACB кассета из pUM24 вырезают с помощью Bam HI переваривания и вставляется между 5 'и 3' фланкирующие области , для создания плазмид A. Пожалуйста , нажмите здесь для просмотра большего версия этой фигуры.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Конструкция плазмида имеет решающее значение для успешной генерации обоих маркированным и немаркированным мутантов. На рисунке 1 приведен пример плазмиды А и В , используется для генерации делеционного мутанта в генах Synechocystis cpcC1 и cpcC2 13. В каждом случае 5 'и 3' фланкирующие области примерно 900-1000 пар оснований. Уменьшенные фланкирующие области можно использовать, хотя наименьшее мы успешно опробована уже около 500 пар оснований. Плазмида В также может содержать ген, кассету между 5 'и 3' ~ 1 кб фланговых областей или модифицированной версии нативной последовательности гена.
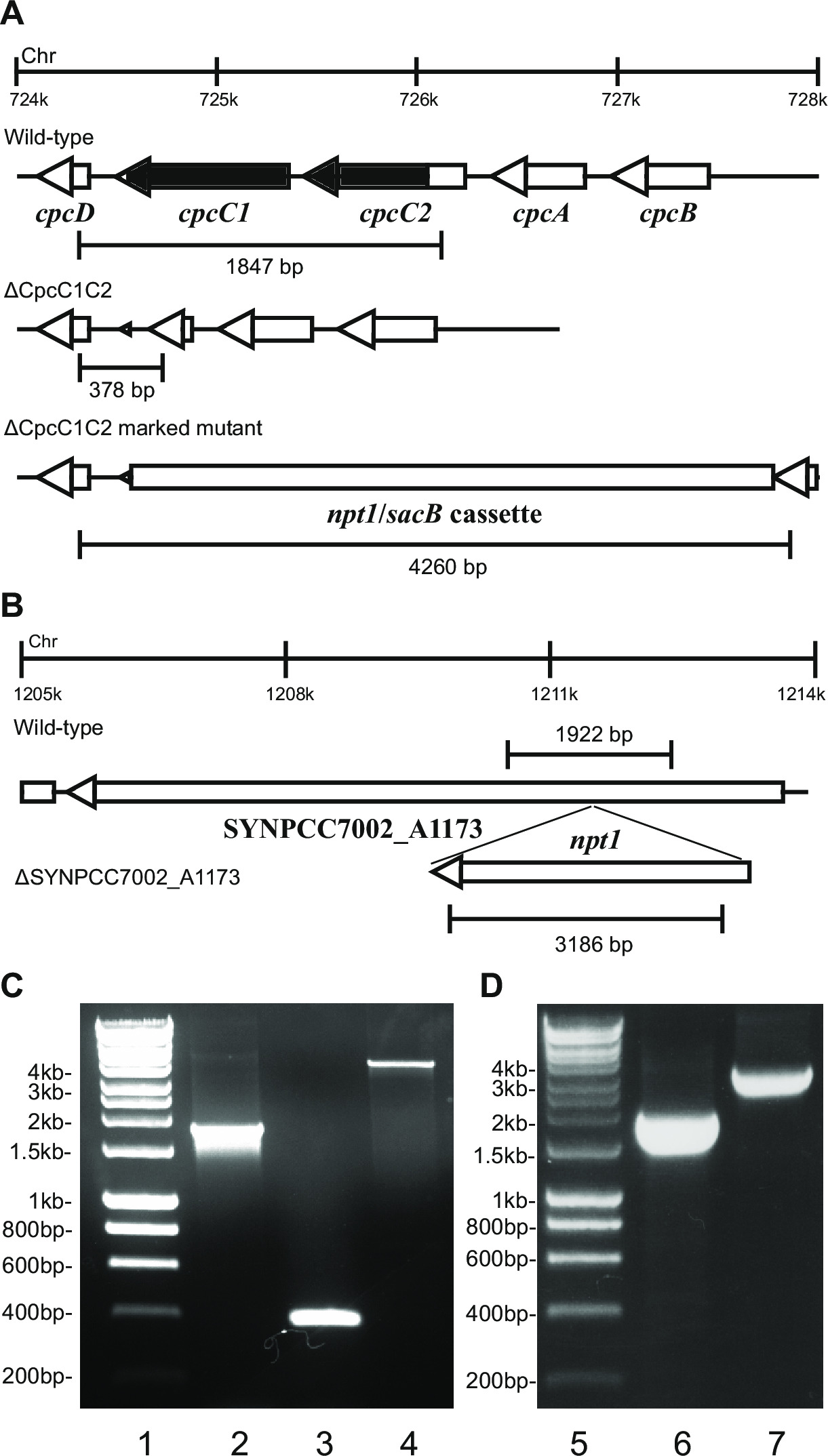
Рисунок 2: Проверка и отмеченных немаркированных мутантов, например cpcC1 / cpcC2
При преобразовании плазмиды А в клетки, как правило, несколько сотен колоний будут появляться на тарелку приблизительно через 7-10 дней. Колонии <1 мм в диаметре и не будет увеличиваться в размерах в течение следующих нескольких недель. Поэтому крайне важно, чтобы использовать тупой конец зубочистку, чтобы удалить колонию и полосу его на свежий BG11 + канамицин агаром. Приблизительно половина повторно штрихом колонии будет расти через 4-6 дней. Если гены являются несущественными и мутанты демонстрируют рост , похожий на штамме дикого типа при непрерывном освещении 20-40 мкмоль фотонов м -2 с -1 (например , оксидаза мутанты терминальные в Lea-Smith и др., 2013 14) (рис 3), то все хромосомы должны содержать копию Т он NPT1 / SACB вставлена кассета последовательность, как определено с помощью ПЦР. Если гены не являются существенными и мутанты демонстрируют медленный фенотип роста при непрерывном освещении 20-40 мкмоль фотонов м -2 с -1 (например , фикобилисома неудовлетворительно мутантов в Lea-Smith и др., 2014 13) (рис 3), то несколько раундов повторного полосатость на BG11 чашках с агаром с постепенным увеличением количества канамицин необходимы для того, чтобы получить сегрегированный отмеченный мутанта. После того, как сегрегированная мутант получают это должно быть повторно штрихом на свежем BG11 плюс канамицин агаром, чтобы гарантировать, что разделение завершена. Если повторные раунды полос не приводят к сегрегированная отмечен мутант , то ген, вероятно , необходим для выживания. Рисунок 4 дана схема экспериментальных операций , связанных с немаркированной мутантного поколения.
/54001/54001fig3highres.jpg "Ширина =" 700 "/>
Рис . 3: Рост Synechocystis мутантов Примеры мутантов , которые демонстрируют (А) аналогичный рост дикого типа и (В) более медленный рост , чем дикого типа. Мутант ΔCOX испытывает недостаток цитохромоксидазы из - за удаления генов CtaC1D1E1. Мутант ΔCyd испытывает недостаток хинол оксидазы из - за удаления генов CydAB. Оливково мутант не хватает часть фикобилисомой вследствие делеции генов CpcABC1C2D. Образцы в (В) продувают воздухом, чтобы способствовать росту. Воспроизводится из данных , опубликованных в Lea-Smith и др, 2013 14 и 2014 13 (www.plantphysiol.org; Авторское Американское общество Биологи растений).. Пожалуйста , нажмите здесь , чтобы посмотреть увеличенную версию этой фигуры.
например , рисунок 2. Если ген кассета вставляется в хромосому затем обычно более высокая доля канамицин наблюдаются устойчивые и сахарозу колонии , устойчивые к . Эти мутанты могут расти на сахарозе из - за мутации в гене SACB. Если ни к канамицину чувствительных и сахарозу колонии, устойчивые к не генерируются, то ген кассета безвредным для клетки.

Рисунок 4: Генерация и снимается мутантов в Synechocystis Схематическое детализации (А) рекомбинации и (В) Экспериментальные стадии , участвующие в мутантной поколения.. Плазмидную А сначала смешивают с клетками. После инкубации на чашках с агаром, содержащим канамицин, колонии, в которых происходит событие рекомбинации между 5 сверхсовременныеD 3 'фланговые области (указанные в синий и красный, соответственно) и гомологичная последовательность в хромосоме, изолированы. Кроме того, NPT1 / SACB кассета между 5 'и 3' фланкирующие области вставляется в хромосому. После разделения заметное мутант генерируется. Выделенные мутантные клетки затем смешивали с плазмидой В , который может содержать либо (C) 1: 5 'и 3' фланкирующие области; 2: 5 'и 3' фланкирующие области с экспрессионной кассетой, содержащей интерес гены, вставленной между этими последовательностями; 3: 5 'и 3' фланкирующие области с последовательностью дикого типа с требуемыми нуклеотидных изменений, вставленных между этими последовательностями. Второй гомологичной рекомбинации происходит между 5 'и 3' фланкирующие области и гомологичных областей в хромосоме, что приводит к удалению NPT1 / SACB кассете и либо немаркированной нокаута или мутанта с прошивкой или измененном дикого станде область вводится в хромосому. Пожалуйста , нажмите здесь , чтобы посмотреть увеличенную версию этой фигуры.
| Рецепты маточного раствора | |
| химическая | Количество (г) |
| 100x BG11 (на л) | |
| NaNO 3 | 149,6 |
| MgSO 4 · 7H 2 O | 7,49 |
| CaCl 2 · 2H 2 O | 3.6 |
| Лимонная кислота | 0,6 |
| Добавить 1,12 мл 0,25 М Na 2 ЭДТА, рН 8,0 | |
| 0,25 М Na 2 ЭДТА, рН 8,0 (на 100 мл) | |
| Na 2 | 9.3 |
| Микроэлементы (на 100 мл) | |
| H 3 BO 3 | 0,286 |
| MnCl 2 · 4H 2 O | 0,181 |
| ZnSO 4 7H 2 O | 0.022 |
| Na 2 MoO 4 · 2H 2 O | 0.039 |
| CuSO 4 · 5H 2 O | 0.008 |
| Co (NO 3) 2 · 6H 2 O | 0.005 |
| Железный запас (на 100 мл) | |
| Железа цитрат аммония | 1.11 |
| Фосфаты запас (на 100 мл) | |
| K 2 HPO 4 | 3,05 |
| Na 2 CO 3 запас (на 100 мл) | |
| Na 2 CO 3 | 2 |
| TES - буфера, рН 8,2 (на 100 мл) | |
| TES | 22,9 |
| NaHCO 3 запас (на 100 мл) | |
| NaHCO 3 | 8.4 |
| HEPES, рН 8,2 (на 500 мл) | |
| HEPES | 119.15 |
| Витамин B 12 (на 50 мл) | |
| цианокобаламин | 0,02 |
| Лурия Бертани СМИ (на 500 мл) | |
| Лурия Бертани бульон | 12,5 |
| 1 М MgCl 2 (на 100 мл) | |
| MgCl 2 · 6H 2 O | 20.33 |
| MnCl 2 · 4H 2 O | 0.395 |
| CaCl 2 · 2H 2 O | 1,47 |
| 2- (N -Morpholino) гидрат этансульфоновая кислота, 4-Morpholineethanesulfonic кислоты (MES) | 0,4265 |
| Раствор А + глицерин | |
| 10 мл раствор А | |
| 1,5 мл глицерина |
Таблица 1: Решения , используемые в данном исследовании.
| грунтовка | Последовательность |
| cpcC1C2leftfor | GTAC TCTAGA GCGGCTAAATGCTACGAC |
| CPCC1C2leftrev | GATC GGATCC GCGGTAATTGTTCCCTTTGA |
| cpcC1C2rightfor | GATC GAGCTC TGCACTGGTCAGTCGTTC |
| cpcC1C2rightrev | GACT GAATTC ATCGTTGCTTGAACGGTCTC |
| M13 вперед | TGTAAAACGACGGCCAGT |
| M13 обратный | CAGGAAACAGCTATGAC |
| cpcC1C2for | GTTTTCATTGGCATCGGTCT |
| cpcC1C2rev | ATGTCCCAGGAACGACTGAC |
| A1173for | AGCAAACCGTTTTTGTGACC |
| A1173rev | TGCAAGGTGGCGAACTGTAT |
Таблица 2:. Праймеры , использованные в данном исследовании сайтами эндонуклеазы рестрикции подчеркнуты.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Наиболее важные шаги в формировании немаркированных мутантов являются: 1) тщательная разработка плазмида для обеспечения только изменен целевой области; 2) обеспечение того, чтобы образцы остаются аксенический, особенно при культивировании на сахароза; 3) металлизация трансформированные клетки помечено мутантного поколения первоначально на BG11 чашках с агаром, не имеющих антибиотики, с последующим добавлением агара плюс антибиотики 24 часа спустя; 4) культивирование отмечены мутанты в течение 4 полных дней до высева на BG11 плюс агара сахарозы пластин: 5) обеспечение того, чтобы выделенные мутанты полностью разделены и 6) тщательно подтверждая генотип мутантных штаммов. Для этого последнего шага, дополнительные праймеры, предназначенные для амплификации части удаленных символов, может быть использован для того, чтобы оно было удалено. Саузерн-блоттинг, в то время как трудоемкий, могут также быть использованы. Тем не менее, наш опыт показывает, что процедура, описанная в данном документе, является достаточным для надлежащей проверки мутантов. Эта процедура также используется для генерации отмеченных мутантов в SynechococКас elongatus PCC7942. Однако, повторим преобразование этого цианобактерии оказалось сложной задачей.
Если выделенные мутанты не могут быть отделены , то различных условиях окружающей среды высокой CO 2, низкий свет (<20 мкмоль фотонов м -2 с -1) или дополнительные питательные вещества (т.е. глюкоза) может быть проверена. Например, добавление глюкозы имеет важное значение для того , чтобы генерировать мутантов 21 фотосистемы II. Если отмечены мутанты никогда полностью отделить то ген, вероятно, важное значение для жизнеспособности. Тем не менее, существуют примеры из литературы , где некоторые исследовательские группы не смогли нокаут гена (например, VIPP в Synechocystis) 22, только для других групп , чтобы позже показать , что ген не является существенным 23. Это может быть связано с различиями в штаммов дикого типа или неправильной конструкции плазмиды, в результате чего в полярных эффектов на соседних, важных генов. Если мутант не полностьюразделяться мы рекомендуем плазмиду , содержащую NPT1 кассету из pUC18K 20 между левой и правой фрагментов можно использовать для трансформации. Легче проверить наличие полос , соответствующих дикого типа и мутанта с помощью ПЦР, так как этот фрагмент приблизительно 1,2 т.п.н., по сравнению с 3,8 кб NPT1 / SACB кассете. Этот результат является важной частью доказательств, свидетельствующих о том, что ген имеет важное значение.
Генерация немаркированных мутантов с вставленными кассет экспрессии, как правило, более сложной задачей, чем развитие штаммов нокаутом. Как правило , мы экспрессируют гены под контролем сильного промотора cpcBAC1C2D 13. В некоторых случаях это может уменьшить вероятность успешного включения гена кассеты, если избыточная экспрессия белка, является безвредным для клетки. Более слабые промоторы должны затем быть проверены. В целом мы обнаружили, что чем больше ген кассета, тем труднее вSERT его в геном. Мы не смогли вставить генные кассеты размером более 5 кб. Кроме того, следует принимать в выборе сайтов для вставки кассеты экспрессии в геном. Нейтральные сайты, которые не влияют на жизнеспособность клеток или рост должен быть использован. Примеры в Synechocystis включают phaAB и Phace, которые кодируют белки , кодирующие полигидрооксибутирате путь биосинтеза 24,25. Совсем недавно обширный список нейтральных сайтов в Synechocystis было определено 26.
Генерация немаркированных мутантов в цианобактерий является медленным процессом, принимая около 5-7 недель, если все шаги проводят должным образом. Это медленнее, чем стандартный метод генерации отмеченных нокауты, которые используются большинством исследовательских групп, расследующих цианобактерии. Тем не менее, гибкость в возможности вводить дополнительные мутации в немаркированных мутантов частично компенсирует это, так как дополнительные плазмид продAining ряд кассет, придающие устойчивость к различным антибиотикам, не должны быть построены. Для исследовательских целей способность мутировать множественные гены иногда необходимо для того, чтобы полностью характеризовать функциональную роль белков. Например, мы определили пагубное фенотип только при удалении двух концевых оксидаз электронных моек , локализованных на тилакоидную мембране, так как потеря только один из этих комплексов может быть компенсирована за счет активности другой 14. Разработка штамма для промышленного применения также потребует нескольких модификаций штамма, а не только для введения чужеродных генов, но и повысить эффективность фотосинтеза, легкую оптимизацию уборки и удаления конкурирующих путей для желаемой подложки.
Основным фактором, ограничивающим скорость немаркированной мутантного поколения является медленным временным разделением видов модели цианобактерий, между 8-20 ч в зависимости от условий освещенности. ООНдер более высокие интенсивности света и концентрации СО 2, рост происходит быстрее. Тем не менее, существует риск того, что мутантные штаммы , которые не могут терпеть ни высокого света или CO 2 будет выбран в отношении, или что мутантные штаммы претерпит нежелательные изменения до начала фенотипической характеристики. Поэтому это не рекомендуется. Тем не менее, было бы весьма выгодно, если более быстрый протокол для генерации немаркированных мутантов был разработан. В целом, это способствовало бы развитию штаммов как для фундаментальных исследований и прикладных применений. Такие штаммы могут быть использованы для биотоплива, биомассы или химического производства или в понимании многих аспектов цианобактерий биохимии, генетики и физиологии.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Мы благодарны Environmental Services Association Education Trust, синтетической биологии в Кембриджском СинБио фонда и министерства социальной справедливости и расширения прав и возможностей, правительство Индии, за финансовую поддержку.
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. 3rd Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
