

Summary
Le cellule batteriche sono spazialmente altamente organizzate. Per seguire questa organizzazione nel corso del tempo in lenta crescita xanthus di Myxococcus cellule, è stato sviluppato un set-up per l'imaging di cellule vive di fluorescenza ad alta risoluzione spazio-temporale sopra parecchie generazioni. Utilizzando questo metodo, potrebbe essere determinato spatiotemporal dinamica di proteine importanti per la segregazione del cromosoma e la divisione cellulare.
Abstract
Imaging di cellule vive di fluorescenza delle cellule batteriche è un metodo a chiave nell'analisi della dinamica spaziale e temporale delle proteine e dei cromosomi alla base di eventi del ciclo cellulare centrale. Tuttavia, imaging di queste molecole in lenta crescita batteri rappresenta una sfida a causa di photobleaching di fluorofori e fototossicità durante l'acquisizione di immagini. Qui, descriviamo un protocollo semplice per aggirare queste limitazioni nel caso di xanthus di Myxococcus (che ha un tempo di generazione di 4-6 h). A tal fine, M. xanthus le cellule sono coltivate su un blocco di agar nutriente-contenente spessore in un ambiente umido a temperatura controllata. In queste condizioni, determiniamo il tempo di raddoppiamento delle cellule individuali seguendo la crescita di cellule singole. Inoltre, cellular chiave processi come segregazione cromosomica e la divisione cellulare può essere imaged da formazione immagine di cellule vive di fluorescenza delle cellule che contengono rilevanti proteine marker fluorescente contrassegnati come ParB-YFP, FtsZ-GFP e mCherry-PomX in più cicli cellulari. Successivamente, le immagini acquisite vengono elaborate per generare fotomontaggi e/o filmati.
Introduction
Le cellule batteriche sono spazialmente altamente organizzate con molte proteine localizzazione asimmetrica all'interno di compartimenti cellulari1,2,3,4. Questa localizzazione è spesso altamente dinamica e cambia nel tempo in risposta a segnali di ciclo cellulare o segnali esterni. Ugualmente, il cromosoma batterico è spazialmente altamente organizzato con singoli loci essendo posizionati a posizioni specifiche subcellulare prima e durante il processo di segregazione5. Questa organizzazione spaziale dinamica è importante per la crescita, divisione, regolazione del ciclo cellulare, differenziazione, motilità, trasduzione del segnale, nonché organizzazione di cromosoma e segregazione; così, essa colpisce essenzialmente tutti gli aspetti della funzione batterica.
La dinamica spazio-temporale di questi processi cellulari vengono analizzata in una varietà di specie batteriche diverse con Escherichia coli, Bacillus subtilis, Vibrio choleraee Crescentus di Caulobacter che serve come importante organismi di modello. Tuttavia, queste quattro specie coprono solo una piccola gamma dell'enorme diversità batterica e, forse non sorprende data la grande distanza filogenetica tra queste specie, meccanismi cellulari di organizzazione e di polarizzazione sono diversi in questi batteri. Ciò solleva la necessità di studiare altre specie batteriche per poter eventualmente estrarre principi generali la dinamica spazio-temporale delle cellule batteriche.
Il delta-proteobacterium gram-negativi M. xanthus è un organismo modello nello studio dei comportamenti sociali e la cooperazione in batteri6. M. xanthus è un aerobio rigoroso e in presenza di sostanze nutritive, forma colonie in cui cellule diffondono verso l'esterno in un'altamente coordinati, brulicante di moda e prede su altri microrganismi7. In risposta a nutrienti, cellule di avviare un programma di sviluppo che provoca la formazione di corpi fruttiferi che consiste di migliaia di cellule e all'interno del quale, le cellule motili asta-a forma di differenziano a sferica diploide spore8. Entrambi i tipi di comportamenti, cioè, brulicante e formazione del corpo fruttifero, vengono eseguiti solo da cellule che sono posti su una superficie solida. Inoltre, in entrambe le circostanze dei nutrienti, cellule impegnano nei processi che implicano contatti diretto cellula-cellula, compreso lo scambio di lipoproteine di membrana esterna che può stimolare la motilità o funzionare come tossine nel destinatario9,10 , lo scambio di LPS11, stimolazione della motilità di esopolisaccaridi su vicine cellule12e intercellulare segnalazione da una cella di superficie-ancorata segnalazione proteina13,14.
Recentemente, M. xanthus è diventato anche un organismo di modello per studiare i meccanismi alla base della motilità e suo regolamento15, divisione cellulare16,17,18e organizzazione del cromosoma19 ,20,21. Critica i passaggi M. xanthus ciclo cellulare sono stati analizzati in dettaglio da microscopia di fluorescenza utilizzando lo snap-shot immagini o registrazioni time-lapse breve su ceppi che trasportano proteine fluorescente identificati pertinenti16, 17,18,19,20. In teoria, molte cellule dovrebbero essere seguite con cella singola risoluzione da cellule vive di fluorescenza imaging per almeno un ciclo completo di cella ottenere dati quantitativi robusti sui parametri del ciclo cellulare. Tuttavia, questa è una sfida nel caso di M. xanthus dovuto al suo tempo di generazione relativamente lungo di 4-6 h in condizioni standard di laboratorio e al photobleaching di fluorofori e fototossicità durante l'acquisizione di immagini.
Qui, descriviamo un protocollo da seguire M. xanthus cellule con risoluzione di singola cellula di fluorescenza vivere-cella di imaging per almeno 24 h e coprendo diversi cicli cellulari. Importante, durante l'intero protocollo, le cellule sono mantenute su un pad di agar e a stretto contatto consentendo i contatto-dipendente di attività essenziali per lo stile di vita sociale di M. xanthus. Il protocollo inoltre permette agli utenti di monitor forma, dimensione, divisioni e sonde fluorescenti ad alta risoluzione temporale e con risoluzione di singola cellula e quindi, permette la quantificazione della cellula--cellula variabilità e la correlazione degli eventi del ciclo cellulare.
Protocol
1. preparazione e crescita di M. xanthus ceppi
Nota: Vedi tabella 1 e tabella 2.
- Preparare 1% Gengou brodo (CTT) crescita media 1% (p/v) digerito pancreatico di caseina (ad es., Bacto Gengou), 10 mM Tris-HCl pH 8.0, 1mm KH2PO4 pH 7,6, 8mm MgSO422, completati con kanamicina (50 µ g/mL) o ossitetraciclina (10 µ g/mL). Aggiungere la gentamicina (10 µ g/mL) a tutti i mezzi per ridurre il rischio di contaminazione con altri batteri, poiché M. xanthus cellule sono naturalmente resistenti ad esso.
- Inoculare 5 mL di 1% CTT contenente il relativo antibiotic(s) con una singola colonia recente sviluppata di wild digitare (WT) DK1622 23, SA4420 (ΔmglA)24, SA4797 (ΔmglA, ΔpomX/ppomZ mCherry-pomX )16, SA8241 (ΔmglA, ftsZ+/pnatftsZ-gfp), o SA4749 (ΔmglA, parB+/pnatparB-yfp) nella mattina del giorno 1.
- Risospendere un singolo M. xanthus Colonia in 500 µ l di 1% CTT completati con antibiotici in una provetta sterile e trasferire la sospensione intera a un 50 mL flacone erlenmeyer contenente 5 mL di 1% CTT.
Nota: Usate un matraccio di Erlenmeyer con 10 volte il volume della cultura a garantire sufficiente aeriation e una crescita ottimale.
- Risospendere un singolo M. xanthus Colonia in 500 µ l di 1% CTT completati con antibiotici in una provetta sterile e trasferire la sospensione intera a un 50 mL flacone erlenmeyer contenente 5 mL di 1% CTT.
- Crescere le cellule per otto generazioni (circa 40-48 h con un tempo di generazione di 4-6 h) a 32 ° C, agitando a 220 giri/min, al buio. Mantenere le cellule in fase di crescita esponenziale (OD550 < 1.2) e impedire loro di raggiungere la fase stazionaria. Se necessario, diluire le cellule nel mezzo CTT fresco 1% contenente il antibiotic(s) pertinenti a un OD550 di 0,1 - 0,2.
Nota: Un ottimale OD550 per microscopia una singola cella è 0.5 - 0.7. Presso questo OD550, un numero sufficiente di cellule è presente per ogni immagine per consentire la quantificazione nonché analisi statistica dei parametri cellulari.
2. preparazione dei campioni per microscopia
Nota: Le cellule per essere visualizzato da microscopia sono collocate su un vetrino coprioggetti microscopio e poi ricoperto da un cuscinetto di agarosio contenente sostanze nutritive. Il coprioggetto è incollato a una plastica o telaio metallico per fornire meccanici di supporto. In preparazione per la microscopia, un grande blocco di 1% agarose/TPM/0.2% CTT dovrebbe essere preparato in anticipo come descritto ai punti 2.1-2.3. Consultare anche la Tabella materiali per prodotti specifici utilizzati qui.
- Preparare 500 mL di tampone TPM (10 mM Tris-HCl pH 7,6, 1mm KH2PO4 pH 7,6, 8mm MgSO4) e autoclave o filtro sterilizzare usando un filtro superiore bottiglia.
Nota: Il tampone sterile può essere conservato per diversi mesi a temperatura ambiente. - Preparare la soluzione di microscopia di agarosio 1% contenente 0,2% CTT (mix 1 g di agarosio con 80 mL di tampone TPM e 20 mL di terreno di 1% CTT). Riscaldare nel forno a microonde fino a quando l'agarosio è fusa.
Nota: 0,2% CTT è sufficiente per consentire alle cellule di crescere e di evitare l'esaurimento. Concentrazioni più elevate di CTT nel mezzo di microscopia si tradurrà in fluorescenza di fondo elevato. - Riempire una capsula di Petri con l'agarosio fuso a uno spessore di 0,5 cm (per una piazza di 11,5 x 11,5 cm di Petri, circa 60 mL di agarosio fuso è richiesta) e lasciarlo raffreddare fino a temperatura ambiente.
Nota: Il pad di agarosio possa essere memorizzato a 4 ° C in un ambiente umido per fino a 2 giorni.- Pre-riscaldare il cuscinetto CTT 1% agarose/TPM/0.2% a 32 ° C per almeno 15 minuti prima dell'uso.
Nota: Per preparare le celle per microscopia, seguire passaggi 2.4-2.8.
- Pre-riscaldare il cuscinetto CTT 1% agarose/TPM/0.2% a 32 ° C per almeno 15 minuti prima dell'uso.
- Posizionare un coprioggetto in vetro sterile (60 mm x 22 mm, spessore: 0,7 mm) su un telaio in plastica o metallo che ha un buco in mezzo (Figura 1A); Questo telaio funge da supporto meccanico per il vetrino coprioggetti sottili e aiuta a ridurre la deriva durante la microscopia. Fissare il vetrino coprioggetto al telaio con nastro adesivo.
- Per preparare il telaio, ritagliate una cornice di 75 mm x 25 mm da una placca di metallo spessa 1 mm, quindi tagliare un foro di dimensione appropriate (20 × 30mm in questo esperimento) nel mezzo.
- Aggiungere 10-20 µ l di esponenzialmente cresciuto M. xanthus cellule sul vetrino coprioggetti.
- Aggiungere 0,5 µm fluorescente microsfere come marker fiduciali alle celle per semplificare il monitoraggio delle cellule o delle proteine nelle registrazioni time-lapse.
- Diluire 1: 100 microsfere nel buffer di TPM e conservare a 4 ° C per fino a parecchi mesi. Agitare bene prima dell'uso e aggiungere 5-10 µ l delle microsfere diluite alle celle.
Nota: Qui microsfere che sono fluorescenti in tutti i comuni blu, verde, giallo e rosso fluorescente canali sono stati utilizzati.
- Diluire 1: 100 microsfere nel buffer di TPM e conservare a 4 ° C per fino a parecchi mesi. Agitare bene prima dell'uso e aggiungere 5-10 µ l delle microsfere diluite alle celle.
- Tagliare un piccolo pad circa le dimensioni del coprivetrino dell'elettrodo grande pre-riscaldato 1% agarose/TPM/0.2% CTT e posizionarlo sopra le cellule (Figura 1B). Porre un coprivetrino in cima il pad di agarosi di 1% agarose/TPM/0.2% CTT per evitare l'evaporazione e mantenere le cellule in un ambiente umido.
Nota: Il vetrino coprioggetto da solo impedirà evaporazione significativo per almeno 2 h. Per più registrazioni time-lapse, il panino CTT 1% agarose/TPM/0.2% pad e coprioggetto deve essere sigillato con pellicola di paraffina per evitare l'evaporazione. - Incubare il campione di microscopia a 32 ° C per 15-20 minuti per lasciare che le celle fissare alla parte inferiore del riquadro dell'agarosi. Quindi avviare le registrazioni di microscopia time-lapse.
3. microscopio set-up e acquisizione di time-lapse
Nota: Il protocollo descritto qui è stato sviluppato per un microscopio invertito widefield con autofocus, un 100x / 1.30 NA olio PH3 obiettivo, una X, Y motorizzato fase, una fotocamera sCMOS, una fonte di luce, filtri per verde-fluorescente, fluorescente rosso o giallo fluorescente proteine e una camera a temperatura controllata di incubazione. Quest'Aula mantiene le cellule protette dalla luce e a temperatura costante.
- Pre-riscaldare la camera di incubazione e il microscopio a 32 ° C per ~ 1-2 h prima di iniziare la microscopia.
Nota: A seconda della configurazione del microscopio, riscaldamento potrebbe richiedere più tempo. Pre-riscaldamento è essenziale per ridurre la deriva e stabilizza il sistema di controllo di messa a fuoco automatica. - Accendere il microscopio e avviare il software di controllo del microscopio. Selezionare l'obiettivo corretto e la corrette specchi e filtri di acquisire fase immagini di contrasto, nonché immagini di proteine verde fluorescente, fluorescente rosso o giallo fluorescente.
Nota: Un microscopio in genere viene fornito con un comodo software per l'acquisizione di controllo e immagine del microscopio. Qui un software disponibile in commercio (Vedi la Tabella materiali) è stato usato per controllare l'acquisizione di microscopio e immagine. - Aggiungere una goccia di olio di immersione di alta qualità sulla lente dell'obiettivo e la parte inferiore del campione pre-incubato a 32 ° C. Posizionare l'obiettivo sulla Z-posizione possibile più bassa per evitare di danneggiare la lente dell'obiettivo quando il campione è posto sulla fase di microscopio. Posizionare il telaio di metallo con il campione sul palco microscopio e con il "buco-lato" verso l'obiettivo. Fissare saldamente il campione sul supporto di fase.
- Concentrarsi sulle cellule spostando il palco nella direzione Z più vicino all'obiettivo. Spostare il palco più lento quando l'olio scende sul lato inferiore del campione e la lente dell'obiettivo fare contatto. Spostare il palco in X / Y direzione fino a più celle singole sono visibili nella regione di vista, quando le cellule sono nel piano focale. Assicurarsi che almeno un microsfere fluorescenti sono nella regione di vista al fine di allineare più tardi le immagini acquisite.
Nota: In condizioni ottimali, si dovrebbe raggiungere una densità delle cellule delle cellule di 15-30 per ogni area di visualizzazione (2.048 x 2.048 pixel o 133,1 x 133,1 µm). - Aprire la procedura guidata di Acquisizione multi-dimensionale del software di controllo microscopio per impostare un esperimento di lasso di tempo che consente il microscopio per l'acquisizione immagini a più lunghezze d'onda e posizioni di fase se necessario.
- Nella scheda principale attivare Timelapse e Lunghezze d'onda Multiple. Schede aggiuntive apparirà sul lato sinistro della finestra.
- Fare clic sulla scheda risparmio e Seleziona cartella per selezionare una cartella vuota sul disco rigido del computer per salvare le immagini acquisite. Attivare il nome di base di incremento se esiste file per assicurarsi che i set di dati consecutivi non sovrascrivere quelle precedenti. Poi dare l'esperimento un nome con la data e il nome della razza o il titolo dell'esperimento.
- Fare clic sulla scheda per regolare i parametri di time-lapse Timelapse . Impostare la durata per 24 h e Intervallo di tempo a 20 min. Il Numero di intervalli di tempo cambierà automaticamente.
Nota: L'intervallo di tempo ottima dipende dall'esperimento e la funzione cellulare per essere analizzati. Acquisizioni di immagine frequente possono causare photobleaching. Così, un compromesso tra la risoluzione temporale e photobleaching deve essere trovato empiricamente. In un tempo di raddoppiamento di 4-6 h, immagini possono essere facilmente acquisite ad intervalli di 5 min (o intervalli ancora più piccoli, se lo si desidera) per microscopia di contrasto di fase. Se si desidera il microscopia di fluorescenza sopra un corso di tempo di 24 h le immagini dovrebbero essere registrate ad intervalli di circa 15-30 min. - Scegliere la scheda di lunghezze d'onda selezionate il numero di lunghezze d'onda di acquisire per ogni immagine in ogni momento modificando il numero.
Nota: per ogni lunghezza d'onda, una nuova scheda apparirà sul lato sinistro della acquisizione multi-dimensionale " guidata e lunghezze d'onda sarà acquisite nell'ordine dall'alto verso il basso. Per ogni lunghezza d'onda, le impostazioni di acquisizione possono essere modificate separatamente. - Fare clic sulla scheda prima di lunghezza d'onda dall'alto. Selezionare contrasto di fase nell'elenco a discesa illuminazione . Selezionare 100 ms per l'esposizione e selezionare Ogni volta punto nell'elenco a discesa Acquisisci . Disattivare Auto esporre selezionando mai nell'elenco a discesa.
- Ripetere il passaggio 3.5.5 per ogni lunghezza d'onda che deve essere acquisita in ogni momento. Per il set-up sperimentale e proteine fluorescente identificati descritti qui, utilizzare i seguenti parametri per esposizione: 250 ms per proteine di fusione mCherry, 200 ms per proteine di fusione di YFP e 1.000 ms per proteine di fusione di GFP.
Nota: Le impostazioni di illuminazione ottimale per ogni ceppo e la proteina fluorescente dovrebbero essere determinate in anticipo modificando l'intensità della lampada e il tempo di acquisizione immagine per ogni lunghezza d'onda. Tempi di acquisizione immagine troppo lunga aumenterà l'effetto fototossico e in ultima analisi, portare alla morte di arresto e delle cellule di crescita. Di conseguenza, dovrebbe essere raggiunto un compromesso tra l'attuabilità delle cellule e di qualità di immagine. - Acquisire immagini da posizioni multiple di fase per aumentare il numero delle cellule registrato nell'esperimento stesso.
- Per acquisire immagini da posizioni multiple di fase, è possibile attivare Più posizioni di fase nella scheda principale . Quindi fare clic sulla scheda fase e fare clic sul pulsante Live a guardare il campo di vista.
- Spostamento del tavolino in direzione X/Y fino a quando una regione di interesse (ROI) è dentro il campo di vista. Salva le coordinate X e Y-facendo clic su "+" nella scheda fase spostare il palco nuovamente in direzione X/Y finché non viene trovato un nuovo ROI e salva le coordinate nuovamente cliccando sul "+". Andare fino a quando non viene salvato il numero desiderato di regioni.
Nota: In caso di acquisizione di immagini di fluorescenza, assicurarsi che le regioni di interesse (ROI) non sono troppo vicino a vicenda per minimizzare la fototossicità.
- Controllare ancora una volta che le cellule sono a fuoco cliccando sui diverse X - e Y-posizioni salvate e avviare l'autofocus hardware facendo tenere AFC per mantenere costante la posizione Z salvata nel corso dell'esperimento.
- Avviare le registrazioni time-lapse del software di controllo del microscopio facendo acquisire nella procedura guidata di Acquisizione multi-dimensionale .
Nota: Verrà visualizzata una finestra per ogni lunghezza d'onda che viene acquisito e verrà visualizzata una finestra aggiuntiva che indica il numero di punti di tempo acquistato e il tempo fino a quando la prossima acquisizione di foto. - Verifica che le cellule sono ancora a fuoco dopo i primi pochi tempo-punti nelle registrazioni time-lapse a massimizzare la qualità delle immagini e riorientare se necessario.
4. generazione di filmati time-lapse e allineamento delle immagini
Nota: Alcuni pacchetti software commerciali e gratuiti sono disponibili per acquisizione di immagini e analisi delle immagini. Utilizziamo un software disponibile in commercio (Vedi la Tabella materiali) con più pre-installato plugin e strumenti aggiuntivi.
- Salvare le singole immagini da Time-lapse registrazioni su un computer che ha installato il software di elaborazione e analisi di immagine.
- Avviare il software e aprire immagini come una pila facendo Revisione multi-dimensionale dei dati | Selezionare il File di Base | Selezionare la Directory. Aprire la cartella con i dati multi-dimensionali. Controllare il dataset e fare clic su Visualizza; il dataset verrà mostrato come singole immagini da tempo punto di uno fino alla fine. Attivare la lunghezza d'onda (per la creazione di uno stack), selezionare tutte le immagini che dovrebbero essere nello stack e fare clic su Immagine (i) di carico. Ripetere questo passaggio per tutte le lunghezze d'onda e salvare pile completati.
- (Opzionale) Aprire tutte le immagini necessarie per il film utilizzando File | Aperto.
Nota: È consigliabile per aprire le immagini di una lunghezza d'onda acquisita alla volta per non rallentare il computer se la potenza di calcolo è limitata. Se alcune parti delle registrazioni time-lapse, ad esempio, inizio, fine o parecchi punti di tempo devono essere ignorate, quindi questo può essere regolato in definitiva del film. - Attivare lo stack di immagini che deve essere corretto per drift. Aprire lo strumento di allineamento di Apps | Auto Align... Check Stack come origine per le immagini e il primo aereo/tempo punto come piano di riferimento. Selezionare lo stack con pulsante stack di origine e fare clic su applica.
Nota: L'allineamento automatico ci vorrà qualche tempo e potenza di calcolo, ma è un buon modo per correggere big stack per drift dell'assetto microscopio. Questo allineamento automatico funziona bene se microsfere sono incluse, ma potrebbero anche funzionare senza di loro. - Salvare lo stack allineato.
- Utilizzare ROIs.
Nota: Microscopia fluorescente time-lapse consente di creare facilmente grandi insiemi di file di dati che occupano un sacco di potenza di calcolo e rallentano l'elaborazione a valle di questi film. Pertanto è consigliabile identificare ROIs e isolare le cellule per lavorare con file più piccoli.- Selezionate lo strumento Area rettangolare . Creare un ROI attorno alle celle di interesse disegnando manualmente un ROI sull'immagine di contrasto di fase. Assicurarsi che le celle di interesse siano visibili e messo a fuoco per tutto il film intero time-lapse.
- Apri il filmato time-lapse della lunghezza d'onda secondo dello stesso dataset. Trasferire il ROI dalle immagini di contrasto di fase per la fluorescenza immagini della lunghezza d'onda seconda utilizzano lo strumento di Trasferimento regioni con regioni | Trasferire le regioni. Selezionare il dataset di contrasto di fase come Immagine sorgente e il secondo dataset di lunghezza d'onda come Immagine di destinazione. Selezionare Tutte le regioni e premere OK.
- Ripetere il passaggio 4.6.2 per ogni lunghezza d'onda acquisita per lo stesso dataset.
- Selezionare il ROI e indicarla come una pila con modifica | Duplicare | Pila... o premere i tasti Maiusc + Ctrl + D . Quindi salvare lo stack di duplicati con File | Salvare nella stessa cartella come i dati originali.
- Ripetere il passaggio 4.6.4 per ogni ROI di ogni lunghezza d'onda acquisita per lo stesso dataset
- Per generare un filmato in formato MOV o AVI, aprire la funzione Make Movie via Stack | Fare film. Selezionare le registrazioni time-lapse con il tasto Source Stack . Selezionare il formato di output, il frame rate, il numero di fotogrammi e fare clic su Salva.
Representative Results
M. xanthus è un batterio di crescita lento che si muove su superfici solide. Per testare il nostro set-up sperimentale, abbiamo effettuato un esperimento di time-lapse con cellule motili DK1622 WT. Immagini di contrasto di fase sono state acquisite a intervalli di 5 min per 24 h (Figura 2A, B). La maggioranza delle cellule allineati in gruppi. Come previsto, le cellule visualizzato motilità e spostato prevalentemente in gruppi. Abbiamo inoltre osservato che cellule occasionalmente invertito la direzione del movimento. Questi risultati suggeriscono che cellule WT sotto le condizioni testate si comportano normalmente in termini di motilità cellulare. Tuttavia, anche quando le cellule vengono registrate ogni 5 min, l'identificazione delle singole celle è difficile. Inoltre, poiché le cellule sono motili, molte cellule fuggire o immettere il campo di vista che lo rende difficile seguire le cellule per lunghi periodi.
Al fine di tracciare lo stesso M. xanthus cellule per diversi giri del ciclo cellulare da formazione immagine di cellule vive, singoli ceppi possono essere eliminati per il gene mglA , che è essenziale per motilità25. Questo impedisce alle cellule di muoversi fuori del campo di vista durante il protocollo di formazione immagine. Delezioni in-frame vengono generati come descritto da Shi et al. 26
Come previsto, nella formazione immagine di cellule vive contrasto di fase con cellule Δ non motilimglA (Figura 3), le cellule non hanno visualizzato il movimento attivo. Siamo stati in grado di seguire la crescita e la divisione delle cellule individuali durante la formazione del microcolony. Basato sulle registrazioni time-lapse in cui immagini sono state acquisite a intervalli di 5 min per 24 h, è stato possibile quantificare il tempo di interdivision (il tempo tra due eventi di divisione cellulare) con risoluzione di singola cellula. Le cellule del mutantemglA Δ avevano un tempo Inter-divisione di 235 ± 50 min (n = 97 cellule). Con circa 4 h, il tempo interdivision è simile al tempo di raddoppiamento misurato in colture in sospensione per cellule WT. Ciò fornisce la prova che M. xanthus le cellule crescono in modo ottimale in queste condizioni sperimentali.
Per studiare se la messa a punto permette alle cellule di crescere normalmente tenendo traccia delle proteine identificate YFP per lunghi periodi, abbiamo eseguito fluorescenza time-lapse di imaging con M. xanthus cellule che esprimono una proteina YFP-etichetta. A tal fine, abbiamo seguito ParB-YFP come indicatore per l'origine di replicazione (ori). ParB è come componente del sistema pinnaS in M. xanthus e si lega ai siti parS prossimali per gli ori; Pertanto, la segregazione di duplicazione e cromosoma di origine può essere seguita19,20,21. Con immagine acquisizione (contrasto di fase e fluorescenza, 200 ms tempo di acquisizione nel canale di YFP) ogni 20 min, cellule crebbe, dividono e visualizzata la crescita anche a 24 h (Figura 4A). All'inizio delle registrazioni, ParB-YFP formata due cluster nelle regioni subpolari nella maggior parte delle cellule (Figura 4A). Poco prima o dopo la divisione cellulare, il subpolare ParB-YFP cluster presso il vecchio palo di cella duplicato. Uno dei due cluster è rimasto presso il vecchio palo di cella mentre la seconda copia traslocata al nuovo Polo cellulare, raggiungendo la posizione subpolare finale dopo circa 40-60 min (Figura 4A, B). Queste osservazioni sono in accordo con precedenti dati generati da registrazioni di time-lapse breve utilizzando agar sottile Pad19. Concludiamo che questo set-up sperimentale permette di time-lapse microscopia di fluorescenza tenere traccia di segregazione del cromosoma nel corso di diversi cicli cellulari a crescita lenta M. xanthus cellule, senza perturbanti la crescita delle cellule o il macchinario di segregazione del cromosoma.
In un esperimento simile, abbiamo cercato di seguire gli indicatori per la divisione cellulare mediante microscopia a fluorescenza time-lapse. Simile a quasi tutti gli altri batteri, M. xanthus richiede FtsZ, un GTPase batterica di tubulina-come, per divisione cellulare16,17,18. FtsZ forma una struttura anulare al midcell, il cosiddetto Z-ring, che aiuta a reclutare tutte le altre proteine necessarie per la divisione cellulare27,28. In M. xanthus, la formazione di Z-ring e del suo posizionamento presso midcell è stimolata dalle tre PomXYZ proteine16,17. Queste tre proteine formano un complesso cromosoma-collegato che trasferisce attraverso il nucleoide dal sito di divisione delle cellule nella cellula "madre" al centro del nucleoide in due cellule della figlia. La metà del nucleoide coincide con midcell, prima di segregazione del cromosoma e qui le reclute di complessi PomXYZ FtsZ e stimola la formazione di Z-ring.
Qui, abbiamo seguito in primo luogo non motili cellule esprimenti ftsZ-gfp. Perché FtsZ-GFP generale spettacoli una fluorescenza più debole segnale di ParB-YFP, abbiamo aumentato il tempo di esposizione 5 volte per 1 s nel canale di GFP. Come previsto, forte accumulo di FtsZ-GFP è stato osservato solo a midcell e questa localizzazione dettata la posizione di costrizione di divisione delle cellule (Figura 5A). FtsZ-GFP formato prevalentemente un cluster presso midcell nella cella più a lungo. Inoltre era evidente che questo cluster aumentato d'intensità nel tempo. Dopo la divisione cellulare, abbiamo osservato che FtsZ-GFP ri-accumulato presso midcell nella figlia due cellule circa 2 ore più tardi (Figura 5B). Ciò è coerente con la constatazione che circa il 50% delle cellule in una popolazione visualizzare FtsZ localizzazione presso midcell basato su analisi snap-shot16,17.
In un secondo esperimento, abbiamo seguito le cellule dimglA Δ non motili per 24 h che esprimono mCherry-pomX. Come parte del sistema PomXYZ, PomX aiuta a guidare la formazione di Z-anello e posizionamento, stimolando in tal modo la divisione cellulare midcell16. Il segnale di fluorescenza di mCherry-PomX è forte e permette un tempo di esposizione nel canale di fluorescenza di 250 ms. d'importanza, tutte le cellule è cresciuto in dimensioni e visualizzato un evento di divisione cellulare nel corso dell'esperimento, formando microcolonies dopo 24 h ( Figura 6A). Come precedentemente segnalato16, quasi tutte le cellule hanno contenuto un cluster mCherry-PomX. La maggior parte di questi localizzato presso midcell e cluster dal midcell traslocata a midcell nel corso dell'esperimento. Durante le divisioni cellulari, mCherry-PomX cluster furono divise, con ogni cellula figlia riceve un cluster. Al contrario di FtsZ-GFP, mCherry-PomX localizzato alle midcell 80-90% del ciclo cellulare e raggiunto questa posizione presto dopo la divisione cellulare (Figura 6B).
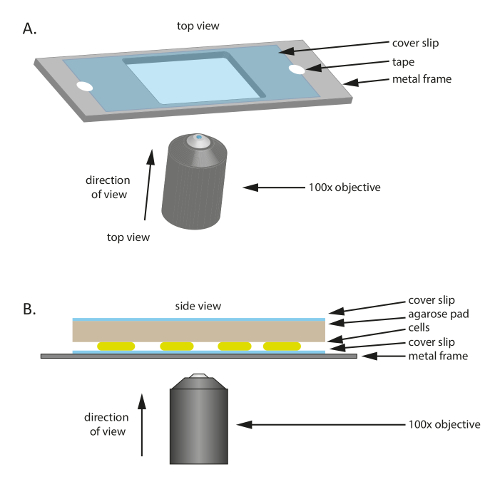
Figura 1 : Schematica della disposizione sperimentale utilizzato nel corso di questo studio. (A), A metallo o plastica telaio funge da supporto per il campione. Un vetrino coprioggetto è fissato alla struttura in metallo con nastro per ridurre il movimento del campione. Lato (B) Mostra dell'assetto campione sperimentale. Le cellule vengono montate sul coprioggetto mostrato in (A). Il pad di agarosio che fornisce le sostanze nutrienti e l'umidità alle cellule è posizionato sulla parte superiore delle celle. Il pad dell'agarosi è coperto da un vetrino coprioggetti supplementari per ridurre l'evaporazione. Per immagini di alta qualità, viene utilizzato un olio immersione fase contrasto obiettivo 100x. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 2 : Contrasto di fase microscopia time-lapse di WT M. xanthus cellule. Le cellule sono state seguite per 24 h e immagini sono state acquisite ogni 5 min (A) sono mostrati immagini rappresentative dello stesso campo di vista ogni 5 min. Le frecce colorate indicano direzionalità del movimento delle singole celle. Lo stesso colore contrassegna la cella stessa nel tempo. I numeri indicano il tempo in minuti. Barra della scala: 5 µm. (B) immagini dello stesso campo di vista dopo ogni ora sono indicate. Si noti che viene visualizzato lo stesso campo di vista, ma perché le cellule sono in movimento, le cellule sono costantemente entrano ed escono il campo visivo. I numeri indicano il tempo in ore. Barra della scala: 5 µm. PH: contrasto di fase. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 3 : Contrasto di fase microscopia time-lapse di non motili M. xanthus cellule. Le cellule ΔmglA sono state seguite per 24 h. immagini sono state acquisite ogni 5 min e immagini rappresentative dopo ogni ora vengono visualizzati. Divisione cellulare selezionato costrizioni sono contrassegnati con frecce arancioni. I numeri indicano il tempo in ore. PH: contrasto di fase. Clicca qui per visualizzare una versione più grande di questa figura.
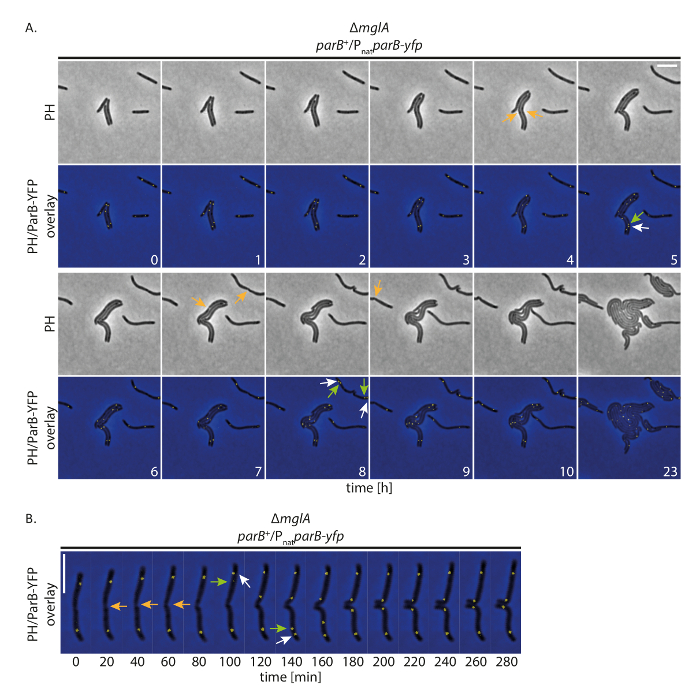
Figura 4 : Time-lapse microscopia di fluorescenza di ParB-YFP in non motili M. xanthus cellule. Cellule di un mutante dimglA esprimenti parB-yfp Δ in presenza di nativi parB (SA4749; ΔmglA; parB +/PnatparB-yfp) sono stati seguiti per 24 h di microscopia di fluorescenza e contrasto di fase. (A) le immagini sono state acquisite ogni 20 min e rappresentante immagini ogni ora fino a 10 h sono indicati, con le stesse cellule dopo 24 h. immagini vengono mostrate in contrasto di fase (PH) e come sovrapposizione di contrasto di fase e la YFP del segnale. Divisioni cellulari selezionate sono contrassegnate con frecce arancioni. Frecce bianche e verdi indicano eventi di duplicazione cluster ParB-YFP, con le frecce verdi marcatura del cluster translocating. I numeri indicano il tempo in ore. Barra della scala: 5 µm. (B) le immagini sono state acquisite come in (A), ma vengono visualizzate a una risoluzione temporale superiore. I numeri indicano il tempo in minuti. Le frecce sono come in (A). Barra della scala: 5 µm. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 5 : Time-lapse microscopia di fluorescenza di FtsZ-GFP in non motili M. xanthus cellule. Cellule di un mutante dimglA Δ esprimendo ftsZ-gfp in presenza di nativo ftsZ (SA8241; ΔmglA; ftsZ +/PnatftsZ-gfp) sono stati seguiti per 24 h di microscopia di fluorescenza e contrasto di fase. (A) le immagini sono state acquisite ogni 20 min e immagini rappresentative ogni ora fino alle 10 h vengono mostrati, insieme con le stesse cellule dopo 24 h. immagini vengono mostrate in contrasto di fase (PH) e come la sovrapposizione di contrasto di fase e segnale GFP. Divisioni cellulari selezionate sono contrassegnate con frecce arancioni. Frecce bianche indicano i cluster di FtsZ-GFP presso midcell. I numeri indicano il tempo in ore. Barra della scala: 5 µm. (B) le immagini sono state acquisite come in (A), ma vengono visualizzate a una risoluzione temporale superiore. I numeri indicano il tempo in minuti. Frecce verdi e bianche contrassegno i cluster di FtsZ-GFP nelle cellule sinistro e destro, rispettivamente. Frecce arancioni indicano divisioni cellulari. Barra della scala: 5 µm. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 6 : Time-lapse microscopia di fluorescenza di mCherry-PomX in non motili M. xanthus cellule. Cellule dipomX Δ non-motile accumulando mCherry-PomX (SA4797; ΔmglA; ΔpomX/ppomZ mCherry-pomX) sono state seguite per 24 h di microscopia di fluorescenza e contrasto di fase ogni 20 min (A) rappresentante le immagini vengono visualizzate ogni ora fino alle h 10, insieme con le stesse cellule dopo 24 h. immagini vengono mostrate in contrasto di fase (PH) e come la sovrapposizione di contrasto di fase e mCherry segnale. Divisioni cellulari selezionate sono contrassegnate con frecce arancioni. Frecce bianche e verdi indicano i cluster mCherry-PomX prima e dopo la divisione eventi, rispettivamente. I numeri indicano il tempo in ore. Barra della scala: 5 µm. (B) le immagini sono state acquisite come in (A) e vengono visualizzate a una risoluzione temporale superiore. Le frecce sono come in (A). Barra della scala: 5 µm. Clicca qui per visualizzare una versione più grande di questa figura.
| Ceppo batterico | Rilevanti di genotipo1 | Riferimento |
| DK1622 | Wildtype | 23 |
| SA4420 | ΔmglA | 24 |
| SA4749 | ΔmglA; parB+/attB:: PnatparB-yfp (pAH7) | Questo studio |
| SA4797 | ΔmglA; ΔpomX / attB::PpomZ mCherry-pomX (pAH53) | 16 |
| SA8241 | ΔmglA; ftsZ+/ mxan18-19::PnatftsZ-gfp (pDS150) | Questo studio |
| Plasmidi tra parentesi contengono fusioni del gene indicato e sono stati intergated presso i siti indicati sul genoma. Plasmidi integrato presso il sito attB o la regione intergenica di mxan18-19 sono state espresse da loro promotore nativo P (nat) o il promotore nativo di pomZ (PpomZ). |
||
Tabella 1: Elenco dei ceppi batterici utilizzati in questo studio.
| Plasmidi | Caratteristiche rilevanti | Riferimento |
| pAH7 | PnatparB-yfp; Mx8 attP; TetR | 19 |
| pAH53 | PpomZ mCherry-pomX; Mx8 attP ; KmR | 16 |
| pDS150 1 | PnatftsZ-gfp ; mxan18-19 ; TetR | Questo studio |
| pMR3691 | Plasmidi per l'espressione genica viscoelastica Vanillato | 18 |
| pKA51 | PnatftsZ-gfp ; Mx8 attP; TetR | 17 |
| 1 pDS150: pDS150 è un derivato del pKA51 in cui il sito attP Mx8 è stato sostituito con la regione intergenica di mxan18-19 . Per questo la regione intergenica di mxan18-19 è stata amplificata da pMR3691 con primer fwd Mxan18-19 BsdRI (GCGATCATTGCGCGCCAGACGATAACAGGC) e Mxan18-19 rev BlpI (GCGGCTGAGCCCGCGCCGACAACCGCAACC) e clonato in pKA51. |
||
Tabella 2: Elenco dei plasmidi utilizzati in questo studio.
Discussion
Imaging di cellule vive di fluorescenza è diventato un potente strumento per studiare la dinamica spazio-temporale delle cellule batteriche. Microscopia di fluorescenza time-lapse di batteri crescente motili e lenti come M. xanthus, tuttavia, è stato impegnativo ed è stato eseguito solo per breve tempo durate. Qui, presentiamo un metodo facile da usare e affidabile per l'imaging di cellule vive di M. xanthus da microscopia di fluorescenza time-lapse. Questo metodo consente all'utente di seguire le cellule e proteine fluorescente contrassegnati per diversi giri del ciclo cellulare con risoluzione di singola cellula.
Esistono diversi prerequisiti che influenzano il successo di formazione immagine di cellule vive di crescita lenta M. xanthus cellule tra cui: 1) una superficie solida per il collegamento delle cellule; 2) la disponibilità di nutrienti e ossigeno; 3) costante umidità e temperatura; e 4) l'ottimizzazione delle condizioni sperimentali quali frequenza di acquisizione immagine e tempo di esposizione.
Nel nostro set-up sperimentale, usiamo agarosio spessore pastiglie completate con sostanze nutritive. Utilizzando pastiglie spesse agarosio al contrario di dispositivi microfluidici per seguire celluli ha alcuni vantaggi fondamentali ma anche alcuni svantaggi. In primo luogo, il pad di agarosio non solo fornisce una superficie di M. xanthus cella allegato e movimento ma anche nutrienti sufficienti per la crescita per almeno 24 h. In secondo luogo, snap shot analisi comunemente usati per studiare la localizzazione intracellulare delle proteine fluorescente contrassegnati in precedenza è stato fatto sullo stesso tipo di agarosio pastiglie16,17,29. Pertanto, i dati da snap shot analisi possono essere direttamente confrontati ai dati ottenuti con il metodo descritto qui. In terzo luogo, agarosio pastiglie possono essere facilmente modificate e completate con antibiotici o altri integratori come CuSO4 e Vanillato che sono comunemente usati per gene espressione induzione18,30. Infine, perché le cellule sono autorizzate a forma microcolonies nel corso di un esperimento, questo set-up anche permette di studiare l'effetto delle interazioni cellula-cellula diretto sul parametro particolare analizzato. Questo aspetto è particolarmente importante nel caso di M. xanthus perché questo batterio Visualizza diverse interazioni di contatto-dipendente. Il principale svantaggio di questo metodo è che le condizioni sperimentali sono preimpostate per la durata di un esperimento. Al contrario, dispositivi microfluidici generalmente consentono cambiando le condizioni sperimentali nel corso di un esperimento aggiungendo per esempio antibiotici31.
Pacchetti di software libero (ad es., MicrobeJ, Oufti) sono disponibili per analizzare automaticamente la crescita di cellule singole e la localizzazione della proteina all'interno di singole celle. Tuttavia, questi software sono solo adatto per l'analisi di cellule singole o piccoli gruppi di cellule. Così, rimane una sfida di analizzare automaticamente i dati generati per le registrazioni di 24h descritte qui.
In sintesi, abbiamo descritto un protocollo riproducibile e facile da usare per eseguire imaging di cellule vive con crescita lenta M. xanthus batteri. Indichiamo che semplice sostanza nutriente-completati agarosio pastiglie sono sufficienti per sostenere la crescita per almeno 24 h e consente di osservare e analizzare la localizzazione della proteina e crescita con risoluzione singola cella sopra parecchie generazioni.
Disclosures
Gli autori dichiarano di non avere nessun concorrenti interessi finanziari.
Acknowledgments
Questo lavoro è stato sostenuto dal Consiglio di ricerca tedesca (DFG) nel quadro della Transregio 174 "dinamica spazio-temporale delle cellule batteriche" e la società Max Planck.
Materials
| Name | Company | Catalog Number | Comments |
| DMI6000B with AFC | Leica microsystems | 11888945 | Automated inverted widefield fluorescence microscope with adaptive focus control |
| Universal mounting frame | Leica microsystems | 11532338 | Stage holder for different sample sizes |
| HCX PL FLUOTAR 100x/1.30 oil PH3 | Leica microsystems | 11506197 | Phase contrast objective |
| Orca Flash 4.0 camera | Hamamatsu | 11532952 | 4.0 megapixel sCMOS camera for picture aquisition |
| Filter set TXR ET, k | Leica microsystems | 11504170 | Fluorescence filter set, Ex: 560/40 Em: 645/75 |
| Filter set L5 ET, k | Leica microsystems | 11504166 | Fluorescence filter set, Ex: 480/40 Em: 527/30 |
| Filter set YFP ET, k | Leica microsystems | 11504165 | Fluorescence filter set, Ex: 500/20 Em: 535/30 |
| ProScan III | Prior | H117N1, V31XYZEF, PS3J100 | Microscope automation controller with interactive control center |
| EL 6000 light source | Leica microsystems | 11504115 | External fluorescence light source |
| Incubator BLX Black | Pecon | 11532830 | Black incubation chamber surrounding the microscope |
| Tempcontrol 37-2 digital | Leica microsystems | 11521719 | Automated temperature control for incubation chamber |
| Gentmycin sulphate | Carl Roth | 0233.4 | Gentamycin |
| Oxytetracylin dihydrate | Sigma Aldrich | 201-212-8 | Oxytetracyclin |
| Kanamycin sulphate | Carl Roth | T832.3 | Kanamycin |
| Filtropur BT25 0.2 bottle top filter | Sarstedt | 831,822,101 | Bottle top filter for sterilization of buffers |
| Deckgläser | VWR | 630-1592 | Glass cover slip (60 x 22 mm, thickness: 0.7 mm) |
| Seakem LE agarose | Lonza | 50004 | Agarose for microscopy slides |
| Leica Metamorph AF | Leica microsystems | 11640901 | Microscope control software and software for picture analysis |
| Tetraspeck Microsperes, 0.5 µm | ThermoFisher | T7281 | Fluorescent microspheres |
| petri dish | Greiner Bio-one | 688102 | 120 mm x 120 mm x 17 mm squared petri dish for agarose pads |
| BD Bacto Casitone | Becton Dickinson | 225930 | Casitone |
| Parafilm M | VWR | 291-1213 | Parafilm |
| Tris(hydroxymethyl)-aminomethane | Carl Roth | AE15.2 | Tris |
| Magnesium sulphate heptahydrate | Carl Roth | P027.2 | Magnesium sulphate |
| Potassium dihydrogen phosphate p.a. | Carl Roth | 3904.1 | Potassium dihydrogen phosphate |
| 1% CTT medium: 1 % (w/v) BD Bacto™ casitone, 10 mM Tris-HCl ph 8.0, 1 mM potassium phosphate buffer pH 7.6, 8 mM MgSO4 | Cultivation medium for M.xanthus | ||
| TPM buffer: 10 mM Tris-HCl ph 8.0, 1 mM potassium phosphate buffer pH 7.6, 8 mM MgSO4 | Buffer for preparation of microscopy slides for M.xanthus |
References
- Shapiro, L., McAdams, H. H., Losick, R. Why and how bacteria localize proteins. Science. 326 (5957), 1225-1228 (2009).
- Treuner-Lange, A., Søgaard-Andersen, L. Regulation of cell polarity in bacteria. J Cell Biol. 206 (1), 7-17 (2014).
- Laloux, G., Jacobs-Wagner, C. Spatiotemporal control of PopZ localization through cell cycle-coupled multimerization. J Cell Biol. 201, 827-841 (2013).
- Rudner, D. Z., Losick, R.
- Badrinarayanan, A., Le, T. B. K., Laub, M. T. Bacterial chromosome organization and segregation. Annu Rev Cell Dev Biol. 31 (1), 171-199 (2015).
- Munoz-Dorado, J., Marcos-Torres, F. J., Garcia-Bravo, E., Moraleda-Munoz, A., Perez, J. Myxobacteria: Moving, Killing, Feeding, and Surviving Together. Front Microbiol. 7, 781 (2016).
- Berleman, J. E., Kirby, J. R. Deciphering the hunting strategy of a bacterial wolfpack. FEMS Microbiol Rev. 33 (5), 942-957 (2009).
- Konovalova, A., Petters, T., Søgaard-Andersen, L.
- Nudleman, E., Wall, D., Kaiser, D. Cell-to-cell transfer of bacterial outer membrane lipoproteins. Science. 309, 125-127 (2005).
- Vassallo, C. N., et al. Infectious polymorphic toxins delivered by outer membrane exchange discriminate kin in myxobacteria. eLife. 6, 29397 (2017).
- Vassallo, C., et al. Cell rejuvenation and social behaviors promoted by LPS exchange in myxobacteria. Proc Natl Acad Sci USA. 112 (22), 2939-2946 (2015).
- Li, Y., et al. Extracellular polysaccharides mediate pilus retraction during social motility of Myxococcus xanthus. Proc. Natl. Acad. Sci. USA. 100, 5443-5448 (2003).
- Kim, S. K., Kaiser, D. Cell alignment required in differentiation of Myxococcus xanthus. Science. 249, 926-928 (1990).
- Lobedanz, S., Søgaard-Andersen, L. Identification of the C-signal, a contact dependent morphogen coordinating multiple developmental responses in Myxococcus xanthus. Genes Dev. 17, 2151-2161 (2003).
- Schumacher, D., Søgaard-Andersen, L. Regulation of cell polarity in motility and cell division in Myxococcus xanthus. Annu Rev Microbiol. 71 (1), 61-78 (2017).
- Schumacher, D., et al. The PomXYZ proteins self-organize on the bacterial nucleoid to stimulate cell division. Dev Cell. 41 (3), 299-314 (2017).
- Treuner-Lange, A., et al. PomZ, a ParA-like protein, regulates Z-ring formation and cell division in Myxococcus xanthus. Mol Microbiol. 87 (2), 235-253 (2013).
- Iniesta, A. A., Garcia-Heras, F., Abellon-Ruiz, J., Gallego-Garcia, A., Elias-Arnanz, M. Two systems for conditional gene expression in Myxococcus xanthus inducible by isopropyl-beta-D-thiogalactopyranoside or vanillate. J Bacteriol. 194 (21), 5875-5885 (2012).
- Harms, A., Treuner-Lange, A., Schumacher, D., Søgaard-Andersen, L. Tracking of chromosome and replisome dynamics in Myxococcus xanthus. reveals a novel chromosome arrangement. PLoS Genet. 9 (9), 1003802 (2013).
- Iniesta, A. A. ParABS system in chromosome partitioning in the bacterium Myxococcus xanthus. PLoS One. 9 (1), 86897 (2014).
- Lin, L., Osorio Valeriano, M., Harms, A., Søgaard-Andersen, L., Thanbichler, M. Bactofilin-mediated organization of the ParABS chromosome segregation system in Myxococcus xanthus. Nat Commun. 8 (1), 1817 (2017).
- Hodgkin, J., Kaiser, D. Cell-to-cell stimulation of movement in nonmotile mutants of Myxococcus. Proc Natl Acad Sci U S A. 74 (7), 2938-2942 (1977).
- Kaiser, D. Social gliding is correlated with the presence of pili in Myxococcus xanthus. Proc Natl Acad Sci USA. 76 (11), 5952-5956 (1979).
- Miertzschke, M., et al. Structural analysis of the Ras-like G protein MglA and its cognate GAP MglB and implications for bacterial polarity. EMBO J. 30 (20), 4185-4197 (2011).
- Hodgkin, J., Kaiser, D. Genetics of gliding motility in Myxococcus xanthus. (Myxobacterales): Two gene systems control movement. Mol Gen Genet. 171, 177-191 (1979).
- Shi, X., et al. Bioinformatics and experimental analysis of proteins of two-component systems in Myxococcus xanthus. J Bacteriol. 190 (2), 613-624 (2008).
- Bi, E. F., Lutkenhaus, J. FtsZ ring structure associated with division in Escherichia coli. Nature. 354 (6349), 161-164 (1991).
- Lutkenhaus, J., Pichoff, S., Du, S. Bacterial cytokinesis: From Z ring to divisome. Cytoskeleton. 69 (10), 778-790 (2012).
- McLoon, A. L., et al. MglC, a Paralog of Myxococcus xanthus GTPase-Activating Protein MglB, Plays a Divergent Role in Motility Regulation. J Bacteriol. 198 (3), 510-520 (2015).
- Gomez-Santos, N., et al. Comprehensive set of integrative plasmid vectors for copper-inducible gene expression in Myxococcus xanthus. Appl Environ Microbiol. 78 (8), 2515-2521 (2012).
- Treuner-Lange, A., et al. The small G-protein MglA connects to the MreB actin cytoskeleton at bacterial focal adhesions. J Cell Biol. 210 (2), 243-256 (2015).
