

Summary
Bakterielle celler er romlig svært organisert. For å følge denne organisasjonen over tid i langsom voksende en cellemembran celler, utviklet et oppsett for fluorescens live-celle bildebehandling med høy spatiotemporal oppløsning over flere generasjoner. Bruker denne metoden, kan spatiotemporal dynamikken i viktige proteiner for kromosom segregering og celledeling bestemmes.
Abstract
Fluorescens live-celle avbilding av bakterieceller er en viktig metode i analysen av romlige og tidsmessige dynamikken i proteiner og kromosomene underliggende sentrale celle syklus hendelser. Men imaging av disse molekylene i langsom voksende bakterier representerer en utfordring på grunn av photobleaching av fluorophores og Phototoksisitet under bildeopptak. Her beskriver vi en enkel protokoll for å omgå disse begrensningene ved en cellemembran (som har en generasjonstid med 4-6 h). Dette M. cellemembran celler er dyrket på en tykk næringsstoff inneholder agar pute i et temperaturkontrollert fuktig miljø. Under disse forholdene, kan vi finne ut dobling tid individuelle celler ved å følge veksten i enkeltceller. Videre behandler viktigste mobilnettet som kromosom segregering og celledeling kan avbildes av fluorescens live-celle tenkelig celler som inneholder relevante fluorescently merket markør proteiner som ParB-YFP, FtsZ-GFP og mCherry-PomX over flere cellen sykluser. Deretter behandles ervervet bildene for å generere montasjer og/eller filmer.
Introduction
Bakterielle celler er romlig svært organisert med mange proteiner lokalisere asymmetrisk i mobilnettet rom1,2,3,4. Denne lokalisering er ofte svært dynamisk og endringer over tid i cellen syklus signaler eller eksterne signaler. Bakteriell kromosomet er like, romlig svært organisert med personlige loci plassert til bestemte subcellular steder før og under segregering prosessen5. Denne dynamiske romlig organisering er viktig for vekst, divisjon, celle syklus regulering, differensiering, motilitet, signaltransduksjon samt kromosom organisasjon og segregering; Således, det påvirker i hovedsak alle aspekter av bakteriell funksjonen.
Spatiotemporal dynamikken i prosessene mobilnettet blir analysert i en rekke ulike bakterie-art med Escherichia coli, Bacillus subtilis, Vibrio choleraeog Caulobacter crescentus serverer viktig modell organismer. Men disse fire arter dekker bare en liten spekteret den enorme bakterieflora og, kanskje ikke overraskende gitt store Fylogenetiske avstanden mellom disse artene, mobilnettet organisasjon og polarisering mekanismer er forskjellige i disse bakterier. Dette øker behovet for å studere mer bakterie-art kunne til slutt trekke generelle prinsipper spatiotemporal dynamikken i bakterielle celler.
Den Gram-negative delta-proteobacterium M. cellemembran er en modell organisme i studiet av atferd og samarbeid i bakterier6. M. cellemembran er en streng aerob og i nærvær av næringsstoffer, danner kolonier som celler spredt utover i en svært koordinert, krydde mote og preys på andre mikroorganismer7. Svar på næringsstoffer sult, celler starte et utviklingsmessige program som resulterer i dannelsen av frukt organer som består av tusenvis av celler, og inne som stav-formet motile cellene skille til sfærisk diploide sporer8. Begge typer atferd, dvs, swarming og frukt kroppen formasjon, utføres bare av celler som er plassert på en solid overflate. Videre under begge næringsstoffer forhold deltar celler i prosesser som omfatter direkte celle-celle kontakter herunder utveksling av ytre membran lipoproteiner som kan stimulere motilitet eller fungere som giftstoffer i mottakerens9,10 , utveksling av LPS11, stimulering av motilitet av exopolysaccharides på omkringliggende celler12og intercellulære signaliserer en celle overflaten-forankret signalering protein13,14.
Nylig M. cellemembran har også blitt en modell organisme for å studere mekanismer underliggende motilitet og regulering15, celledeling16,17,18, og kromosom organisasjon19 ,20,21. Kritisk trinn i den M. cellemembran celle syklus er analysert i detalj av fluorescens mikroskopi bruke bilde bilder eller kort time-lapse opptak på stammer bære relevante fluorescently merket proteiner16, 17,18,19,20. Ideelt sett etterfølges mange celler med encellede oppløsning fluorescens levende celle imaging for minst én hele cellen syklus å skaffe robust kvantitative data på cellen syklus parametere. Men dette er en utfordring i tilfelle av M. cellemembran det relativt lang generasjonstid for 4-6 h under standard laboratorieforhold og photobleaching av fluorophores og Phototoksisitet under bildeopptak.
Her beskriver vi en protokoll for å følge M. cellemembran celler med én celle løsning etter fluorescens live-celle imaging for minst 24 timer og dekker flere cellen sykluser. Viktigst, under hele protokollen, celler vedlikeholdes på en agar pad og i nær kontakt tillater for kontakt-avhengige aktiviteter avgjørende for den sosiale livsstil av M. cellemembran. Protokollen likeledes innrømmer brukernes å dataskjerm form, størrelse, divisjoner og fluorescerende sonder høy timelige oppløsning og enkelt celle oppløsning, og dermed muliggjør kvantifisering av celle-til-celle variasjon og sammenhenger hendelsesforløpet celle syklus.
Protocol
1. forberedelse og vekst av M. cellemembran stammer
Merk: Se tabell 1 og tabell 2.
- Forberede 1% casitone kjøttkraft (CTT) vekst medium 1% (w/v) bukspyttkjertelen Sammendrag av kasein (f.eks, Bacto casitone), 10 mM Tris-HCl pH 8.0, 1 mM KH2PO4 pH 7.6, 8 mM MgSO422, supplert med kanamycin (50 µg/mL) eller Oxytetracycline (10 µg/mL). Legge til gentamycin (10 µg/mL) i alle medier å redusere risikoen for forurensning med andre bakterier, siden M. cellemembran celler er naturlig motstandsdyktig mot den.
- Vaksinere 5 mL 1% CTT som inneholder den aktuelle antibiotic(s) med en enkelt frisk dyrket koloni av ville skrive (WT) DK1622 23, SA4420 (ΔmglA)24, SA4797 (ΔmglA, ΔpomX/PpomZ mCherry-pomX )16, SA8241 (ΔmglA, ftsZ+/PnatftsZ-gfp), eller SA4749 (ΔmglA, parB+/PnatparB-yfp) i den morgenen dagen 1.
- Resuspend en enkelt M. cellemembran koloni i 500 µL av 1% CTT supplert med antibiotika i et sterilt rør og overføre hele suspensjon til en 50 mL Erlenmeyer kolbe som inneholder 5 mL 1% CTT.
Merk: Bruk en Erlenmeyer kolbe med 10 ganger volumet av kulturen garantere tilstrekkelig aeriation og optimal vekst.
- Resuspend en enkelt M. cellemembran koloni i 500 µL av 1% CTT supplert med antibiotika i et sterilt rør og overføre hele suspensjon til en 50 mL Erlenmeyer kolbe som inneholder 5 mL 1% CTT.
- Vokse cellene for åtte generasjoner (ca 40 – 48 h med en generasjon av 4-6 h) på 32 ° C, rister på 220 rpm, i mørket. Vedlikeholde celler i eksponensiell vekstfase (OD550 < 1.2) og hindre dem fra å nå den stasjonære fasen. Eventuelt fortynne cellene i frisk 1% CTT medium som inneholder den aktuelle antibiotic(s) til en OD550 0,1 - 0.2.
Merk: En optimal OD550 for en enkeltcelle mikroskopi er 0,5 - 0,7. På denne OD550finnes et tilstrekkelig antall celler per bilde å tillate kvantifisering samt statistisk analyse av mobilnettet parametere.
2. forberedelse av mikroskopi prøver
Merk: Cellene skal kunne vises mikroskopi er plassert på et mikroskop dekkglassvæske og deretter dekket av en agarose pad som inneholder næringsstoffer. Dekkglassvæske er festet til en plast eller metall ramme å gi mekanisk støtte. I forberedelsene til mikroskopi, en stor blokk av 1% agarose/TPM/0.2% CTT bør være forberedt på forhånd som beskrevet i trinnene 2.1-2.3. Vennligst også se Tabellen for materiale for bestemte produkter her.
- Forberede 500 mL TPM buffer (10 mM Tris-HCl pH 7.6, 1 mM KH2PO4 pH 7.6, 8 mM MgSO4) og autoklav eller filter sterilisere bruke filtere flaske topp.
Merk: Sterile bufferen kan lagres i flere måneder ved romtemperatur. - Forberede 1% agarose mikroskopi løsning som inneholder 0,2% CTT (mix 1 g agarose med 80 mL TPM bufferen og 20 mL 1% CTT medium). Varme i en mikrobølgeovn til agarose er smeltet.
Merk: 0,2% CTT er tilstrekkelig til at cellene å vokse og hindre sult. Høyere konsentrasjoner av CTT i mikroskopi medium fører til høye fluorescens. - Fylle en Petriskål med den smeltet agarose til en tykkelse på 0,5 cm (for en 11,5 cm x 11,5 cm i firkant Petriskål, ca 60 mL av smeltet agarose er nødvendig) og la den avkjøles til romtemperatur.
Merk: Agarose puten kan lagres på 4 ° C i et fuktig miljø for opptil 2 dager.- Forvarm 1% agarose/TPM/0.2% CTT puten ved 32 ° C i minst 15 minutter før å bruke.
Merk: For å forberede cellene mikroskopi, følg trinnene 2.4-2.8.
- Forvarm 1% agarose/TPM/0.2% CTT puten ved 32 ° C i minst 15 minutter før å bruke.
- Plassere et sterilt glass dekkglassvæske (60 mm x 22 mm, tykkelse: 0,7 mm) på en plast eller metall ramme med et hull i midten (figur 1A); denne rammen fungerer som en mekanisk støtte for tynne dekkglassvæske og bidrar til å redusere drift under mikroskopi. Fikse dekkglassvæske til rammen med tape.
- Å forberede rammen, klippe ut en 75 mm × 25 mm ramme fra en 1 mm tykk metallplate og kutte ut en passende stor hull (20 mm × 30 mm i dette eksperimentet) i midten.
- Legge 10-20 µL av eksponentielt voksen M. cellemembran celler i dekkglassvæske.
- Legge til fluorescerende 0,5 µm mikrosfærer som fiducial markører i cellene å forenkle sporing av celler eller proteiner i time-lapse innspillinger.
- Fortynne 1: 100 mikrosfærer TPM buffer og lagre ved 4 ° C i opptil flere måneder. Ristes godt før bruk og legge 5-10 µL av utvannet mikrosfærer til cellene.
Merk: Her mikrosfærer som er fluorescerende i alle vanlige blå, grønn, gul og rød fluorescerende kanaler ble brukt.
- Fortynne 1: 100 mikrosfærer TPM buffer og lagre ved 4 ° C i opptil flere måneder. Ristes godt før bruk og legge 5-10 µL av utvannet mikrosfærer til cellene.
- Kutte ut en liten pad omtrent på størrelse med dekkglassvæske av store forvarmes 1% agarose/TPM/0.2% CTT puten og plasser det på cellene (figur 1B). Plass en dekkglassvæske på 1% agarose/TPM/0.2% CTT agarose pad å hindre fordampning og å vedlikeholde celler i et fuktig miljø.
Merk: Dekkglassvæske alene vil forhindre at betydelige fordampning minst 2 h. Lenger time-lapse opptak, skal 1% agarose/TPM/0.2% CTT pad og dekkglassvæske sandwich tettes med parafin film å hindre fordampning. - Inkuber mikroskopi prøven ved 32 ° C i 15-20 min la cellene feste til bunnen av agarose puten. Deretter start time-lapse mikroskopi opptakene.
3. mikroskop oppsett og Time-lapse oppkjøp
Merk: Protokollen beskrevet her ble utviklet for en invertert widefield mikroskop med autofokus, en 100 X / 1,30 NA olje PH3 mål, X, Y motoriserte scenen, et sCMOS kamera, en lyskilde, filtre for grønn-fluorescerende, rød-fluorescerende eller gul-lysstoffrør proteiner og en temperatur kontrollert inkubasjon kammer. Denne kammer holder celler beskyttet fra lys og konstant temperatur.
- Forvarm inkubasjon kammeret og mikroskopet til 32 ° C ~ 1-2 h før mikroskopi.
Merk: Avhengig av mikroskopet satt opp, oppvarming kan ta lengre tid. Forvarming er avgjørende for å redusere drift og stabiliserer autofokus kontrollsystemet. - Slå på mikroskopet og start mikroskop kontroll programvare. Velg riktig målsettingen og riktig speilene og filtre til å erverve fase kontrast bilder samt bilder av grønn-fluorescerende, rød-fluorescerende eller gul-fluorescerende proteiner.
Merk: Et mikroskop vanligvis leveres med en foretrukne programvare for mikroskop kontroll og image oppkjøpet. Her ble en kommersielt tilgjengelig programvare (se Tabell for materiale) brukt til å kontrollere mikroskop og image oppkjøpet. - Legge en dråpe høykvalitets neddyppingsolje på linsen målet og bunnen av prøven pre ruges på 32 ° C. Plass målet på lavest mulig Z-posisjon til å unngå skade på linsen når prøven er plassert på mikroskopet scenen. Plass metall-rammen med prøven på mikroskopet scenen og "hull-side" mot målet. Fest prøven ordentlig i scenen holderen.
- Fokus på cellene ved å flytte scenen i Z-retningen nærmere til målet. Flytte scenen tregere når olje dråper på undersiden eksempel og linsen gjør kontakt. Flytte scenen i X / Y-retningen til flere enkeltceller er synlige i regionen i visningen når cellene er i fokalplanet. Kontroller at minst én fluorescerende microsphere er i regionen i visningen for å justere senere ervervet bildene.
Merk: Under optimale forhold, en celle tetthet av 15-30 celler per region (2048 x 2048 piksler eller 133.1 x 133.1 µm) skal nås. - Åpne veiviseren Multi-Dimensional oppkjøpet av mikroskopet kontroll programvare å sette opp en tid forfalle eksperiment der mikroskopet hente bilder på flere bølgelengder og scenen stillinger hvis nødvendig.
- I kategorien hoved aktivere Timelapse og Flere bølgelengder. Tilleggskategoriene vises på venstre side av vinduet.
- Klikk på Lagre -kategorien og Velg mappe til å velge en tom mappe på harddisken å lagre ervervet bildene. Aktivere tilvekst Basisnavnet hvis filen finnes å sørge for at påfølgende datasett ikke overskriver tidligere de. Deretter gi eksperimentet et navn med dato og belastning navn eller tittel av eksperimentet.
- Klikk kategorien Timelapse å justere time-lapse parametere. Angi varigheten til 24 h og angi Tidsintervallet til 20 min. Tid poengsum endres automatisk.
Merk: Det optimale tidsintervallet avhenger av eksperimentet og den cellulære funksjonen som skal analyseres. Hyppige bilde oppkjøp kan forårsake photobleaching. Dermed må en avveining mellom midlertidig løsning og photobleaching være empirisk finnes. I en dobling tid med 4-6 h, kan bilder være lett kjøpt på et intervall på 5 min (eller enda mindre intervaller hvis ønskelig) for fase kontrast mikroskopi. Hvis fluorescens mikroskopi i tid-løpet av 24t ønskes bilder skal registreres på et intervall på ca 15-30 min. - Klikk kategorien bølgelengder Velg antall bølgelengder å erverve for hvert bilde på hvert tidspunkt ved å endre tallet.
Merk: For hver bølgelengde, vises en ny fane på venstre side av den Multi-Dimensional oppkjøpet " veiviseren og bølgelengder vil bli kjøpt opp i rekkefølge fra topp til bunn. For hver bølgelengde, kan oppkjøp innstillingene endres separat. - Klikk kategorien for første bølgelengde ovenfra. Velg kontrast i rullegardinlisten belysning . Velg 100 ms for eksponering og velg Hvert punkt i rullegardinlisten Hent . Deaktivere Automatisk vise ved å velge aldri i rullegardinlisten.
- Gjenta trinn 3.5.5 for hver bølgelengde som må kjøpes på hvert tidspunkt. For eksperimentelle oppsett og fluorescently merket proteiner som beskrevet her, bruker du følgende parametere for eksponering: 250 ms for mCherry fusion proteiner, 200 ms for YFP fusion proteiner og 1000 ms for GFP fusion proteiner.
Merk: Optimal belysning innstillingene for hver stamme og fluorescerende protein bør fastsettes på forhånd ved å endre lampe intensiteten og bilde oppkjøpet tid for hver bølgelengde. Lenge bilde oppkjøpet ganger vil øke fototoksisk effekten og til slutt føre til vekst arrestasjon og celle død. Derfor skal en avveining mellom bilde kvalitet og celle levedyktighet oppnås. - Hente bilder fra flere scenen posisjoner for å øke antall celler i samme eksperiment.
- Hvis du vil hente bilder fra flere scenen posisjoner, aktivere Flere scenen stillinger i kategorien hoved . Deretter klikker du kategorien scene og klikk Live for å se på synsfelt.
- Flytt scenen X/Y-retningen til et område av interesse (ROI) i synsfeltet. Lagre X - og Y-koordinatene ved å klikke flytte på "+" i kategorien scene scenen igjen i X/Y-retningen til en ny avkastning er funnet og lagre koordinatene igjen ved å klikke på "+". Gå til ønsket antall områder er lagret.
Merk: Ved fluorescens bildeopptak, kontroller at regioner av interesse (ROIs) ikke for nært til å minimere Phototoksisitet.
- Sjekk igjen at cellene er i fokus ved å klikke på ulike lagret X - og Y-stillinger og starte maskinvare autofokus ved å klikke AFC holder for å holde den lagrede Z-posisjonen konstant i løpet av eksperimentet.
- Start time-lapse opptakene i mikroskopet Kontrollprogramvaren ved å klikke Hent i veiviseren Multi-Dimensional oppkjøpet .
Merk: Ett vindu vises for hver bølgelengde som er kjøpt, og et ekstra vindu vises som viser antall ervervet tidspunkt og tid før neste bilde oppkjøpet. - Kontroller at cellene er fortsatt i fokus etter det første noen gang-poeng i time-lapse opptakene å maksimere kvaliteten på bildene og refokusere hvis nødvendig.
4. generasjon Time-lapse og justering av bilde
Merk: Flere kommersielle og fri programvare-pakker er tilgjengelige for bildeopptak og bildeanalyse. Vi bruker en kommersielt tilgjengelig programvare (se Tabell for materiale) med flere forhåndsinstallerte plugins og ekstra verktøy.
- Lagre bilder enkeltvis fra time-lapse innspillinger på en datamaskin som har den analyse/bildebehandling installert.
- Start programvaren og åpne bilder som en stabel ved gjennomgang Multi-Dimensional Data | Velg Base fil | Velg katalog. Åpne mappen med flerdimensjonale data. Sjekk datasettet og klikk Vis. datasettet vises som enkeltbilder fra tid peker inntil enden. Aktivere bølgelengde (for å lage en stabel), velge alle bilder som skal være i stakken og klikk Last inn på nytt. Gjenta dette trinnet for alle bølgelengder og lagre fullførte stabler.
- (Valgfritt) Åpne alle bilder kreves for filmen bruker filen | Åpne.
Merk: Det anbefales å åpne bilder av en ervervet bølgelengde samtidig ikke tregere datamaskinen hvis regnekraft er begrenset. Hvis deler av time-lapse opptakene, f.eks, start, slutt eller flere tidspunkt skal overses, kan dette justeres i den ferdige filmen. - Aktivere stabelen av bilder som må rettes for drift. Åpne verktøyet for justering av Apps | Auto Juster... av stabelen som kilde for bilder og første fly/gang peker som referanseplanet. Velg bunken med knappen Source stabel og klikk Bruk.
Merk: Den automatiske justeringen vil ta litt tid og databehandlingskraft men er en god måte å korrigere store stabler for drift av mikroskopet oppsettet. Dette automatisk justering fungerer bra hvis mikrosfærer er inkludert, men kan også fungere uten. - Lagre justert stabelen.
- Bruk ROIs.
Merk: Fluorescerende time-lapse mikroskopi skaper lett store datasett datafiler som tar opp mye av databehandlingskraft og tregere nedstrøms behandlingen av disse filmene. Derfor anbefaler vi sterkt å identifisere ROIs og isolere celler å arbeide med mindre filer.- Velg Rektangel -verktøyet. Opprette en avkastning rundt cellene rundt ved å manuelt trekke en avkastning på fase kontrast bildet. Kontroller at cellene rundt er synlig og i fokus gjennom hele time-lapse filmen.
- Åpne time-lapse filmen med andre bølgelengden til samme datasettet. Overføre Avkastningen fra fase kontrast bildene til fluorescensen bilder av andre bølgelengden verktøyet Overføring områder med regioner | Overføre områder. Velg fase kontrast datasettet som Kildebildet og andre bølgelengde datasettet som Målbildet. Velg Alle regioner og trykk OK.
- Gjenta trinn 4.6.2 for hver bølgelengde ervervet for samme datasettet.
- Velg Avkastningen og kopiere denne som en bunke med Rediger | Kopiere | Stabel... eller trykker Skift + Ctrl + D . Lagrer dupliserte stabelen med fil | Lagre i samme mappe som de opprinnelige dataene.
- Gjenta trinn 4.6.4 for hver avkastning av hver bølgelengde ervervet for samme datasettet
- Vil generere en film i MOV eller AVI format, åpne funksjonen Lage film via stabel | Lage film. Velg time-lapse opptakene med knappen Source stabel . Velg utdataformat, bildefrekvens, antall rammer, og klikk Lagre.
Representative Results
M. cellemembran er en langsom voksende bakterie som beveger seg på faste flater. For å teste våre eksperimentelle set-up, utført vi en time-lapse eksperimentere med motile DK1622 WT celler. Fase kontrast bildene ble anskaffet i intervaller på 5 min 24 h (figur 2A, B). Fleste cellene justeres i grupper. Som forventet, celler vises motilitet og flyttet overveiende i grupper. Videre observerte vi at celler av og reversert bevegelsesretning. Disse funnene tyder på at WT celler under testet forhold fungerer normalt i cellen motilitet. Men selv når cellene registreres hvert 5 min, er identifikasjon av individuelle celler vanskelig. Videre, fordi cellene motile, mange celler flykte eller angi synsfelt gjør det vanskelig å følge celler i lengre perioder.
For å spore den samme M. cellemembran celler for flere runder av cellen syklus av live-celle tenkelig, individuelle stammer kan bli slettet for mglA genet, som er avgjørende for motilitet25. Dette hindrer at cellene flytte ut av synsfeltet under tenkelig protokollen. I-ramme slettinger genereres som beskrevet av Shi et al. 26
Som forventet, i fase kontrast live-celle bildebehandling med ikke-motile ΔmglA celler (Figur 3), vise celler ikke aktiv bevegelse. Vi var kjøpedyktig følge vekst og delingen av individuelle celler under microcolony formasjon. Basert på time-lapse opptakene som bildene ble anskaffet i intervaller på 5 min 24 h, var det mulig å kvantifisere interdivision tiden (tiden mellom to celledeling hendelser) med enkelt celle oppløsning. Celler med ΔmglA mutant hadde en Inter divisjon tid på 235 ± 50 min (n = 97 celler). Med ca 4 h ligner interdivision tiden dobling tiden målt i suspensjon kulturer for WT celler. Dette gir bevis for at M. cellemembran cellene vokse optimalt under disse eksperimentelle forhold.
For å undersøke om vår organisering tillater celler å vokse normalt under sporing av YFP-merket proteiner over lengre perioder, vi utført fluorescens time-lapse tenkelig med M. cellemembran celler som uttrykker et YFP-merket protein. Dette fulgte vi ParB-YFP som en markør for opprinnelsen til replikering (ori). ParB er som komponent i ParABS i M. cellemembran og bindes til webområdene parS proksimale til ori; Derfor kan opprinnelse duplisering og kromosom segregering føle etter19,20,21. Med image vinningen (kontrast og fluorescens, 200 ms anskaffet i YFP kanalen) hver 20 min, vokste, delt og vist vekst selv på 24 h (Figur 4A). I begynnelsen av opptak dannet ParB-YFP to klynger i subpolar områder i fleste cellene (Figur 4A). Kort tid før eller etter celledeling klynge av subpolar ParB-YFP på gamle celle pole dupliseres. En av to klynger forble på gamle celle pole mens den andre kopien translocated til nye cellen Polen, nå sin endelige subpolar posisjon etter ca 40-60 min (Figur 4A, B). Disse observasjonene er enig med tidligere data generert kort time-lapse opptak med tynne agar pads19. Vi konkludere med at dette eksperimentelle set-up tillater fluorescens time-lapse mikroskopi spore kromosom segregering over flere cellen sykluser i langsom voksende M. cellemembran celler, uten perturbing cellevekst eller kromosom segregering maskiner.
I et lignende eksperiment forsøkte vi å følge markører for celledeling ved time-lapse fluorescens mikroskopi. Ligner på nesten alle andre bakterier, M. cellemembran krever FtsZ, en bakteriell tubulin-lignende GTPase, celledeling16,17,18. FtsZ danner en ring-lignende struktur på midcell, den såkalte Z-ringen, som bidrar til å rekruttere alle andre proteiner kreves for celledeling27,28. I M. cellemembran, dannelsen av Z-ringen og sin plassering på midcell er stimulert av de tre PomXYZ proteiner16,17. Disse tre proteiner danner et kromosom-assosiert kompleks som overfører over nucleoid området av cellen divisjon i "mor" celle til midten av nucleoid i to datter celler. Midten av nucleoid sammenfaller med midcell, før kromosom segregering, og her PomXYZ komplekse rekrutter FtsZ og stimulerer Z-ring formasjon.
Her, fulgte vi først ikke-motile celler som uttrykker ftsZ-gfp. Fordi FtsZ-GFP totale viser en svakere fluorescens signal enn ParB-YFP, økte vi eksponeringstid 5-fold til 1 s i GFP kanalen. Som forventet, sterk opphopning av FtsZ-GFP ble kun observert på midcell og denne lokalisering diktert plasseringen av cellen divisjon innsnevring (figur 5et). FtsZ-GFP dannet overveiende en klynge på midcell i lengre celle. Det var også tydelig at sektorgruppen økt intensitet over tid. Etter celledeling observerte vi at FtsZ-GFP re samlet på midcell i to datter celler ca 2 timer senere (figur 5B). Dette stemmer overens med å finne at ca 50% av celler i en befolkning vises FtsZ lokalisering i midcell basert på bilde analyse16,17.
I et andre forsøk fulgte vi ikke-motile ΔmglA celler for 24 h som uttrykker mCherry-pomX. Som en del av PomXYZ bidrar PomX til å veilede Z-ring dannelse og posisjonering, derved stimulere celledeling midcell16. Fluorescens signalet fra mCherry-PomX er sterk og gjør en eksponeringstid i fluorescens kanalen av 250 ms. viktigere, alle celler vokste i størrelse og vises en celledeling hendelse i løpet av eksperimentet, danner microcolonies etter 24 h ( Figur 6A). Som tidligere rapportert16inneholdt nesten alle celler en mCherry-PomX klynge. Fleste av disse lokalisert på midcell og klynger fra midcell translocated til midcell i løpet av eksperimentet. Under celledelinger, ble mCherry-PomX klynger splittet, med hver datter celle mottar en klynge. I motsetning til FtsZ-GFP, mCherry-PomX lokalisert på midcell 80-90% av cellen syklus og nådde denne stillingen etter celledeling (figur 6B).
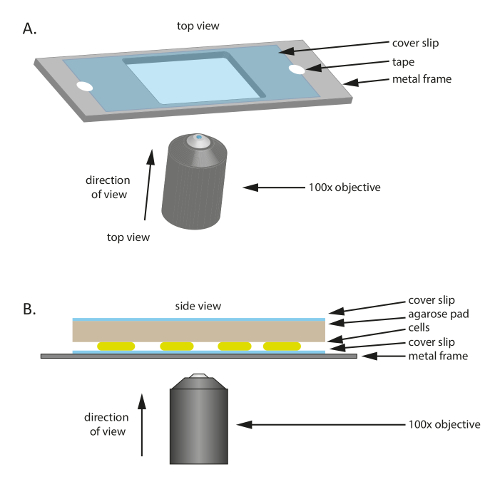
Figur 1 : Skjematisk av den eksperimentelle set-up brukes i hele denne studien. (A) A metall eller plast ramme fungerer som en støtte for prøven. En dekkglassvæske er festet til metallramme med tape for å redusere bevegelse av prøven. (B) Side visning av eksperimentelle eksempel oppsettet. Celler er montert på dekkglassvæske vises i (A). Agarose puten som leverer næringsstoffer og fuktighet til cellene er plassert øverst i celler. Agarose pad er dekket av en ekstra dekkglassvæske å redusere fordampning. For høy kvalitet bilder brukes et 100 X olje nedsenking fase kontrast mål. Klikk her for å se en større versjon av dette tallet.

Figur 2 : Fase kontrast time-lapse mikroskopi av WT M. cellemembran celler. Cellene ble fulgt i 24 timer og bildene ble anskaffet hver 5 min. (A) representant bilder av samme synsfelt hver 5 min vises. Fargede piler angir retningen av individuelle celler. Samme farge markerer den samme cellen over tid. Tall angir tiden i minutter. Skala bar: 5 µm. (B) bilder av samme synsfelt hver time vises. Merk at samme synsfelt vises men fordi celler er i bevegelse, cellene stadig inn og forlate synsfelt. Tall angir tid i timer. Skala bar: 5 µm. PH: fase kontrast. Klikk her for å se en større versjon av dette tallet.

Figur 3 : Fase kontrast time-lapse mikroskopi av ikke-motile M. cellemembran celler. ΔmglA celler ble fulgt for 24 h. bildene ble anskaffet hver 5 min og representant bilder hver time vises. Valgte celledeling constrictions er markert med oransje piler. Tall angir tid i timer. PH: kontrast. Klikk her for å se en større versjon av dette tallet.
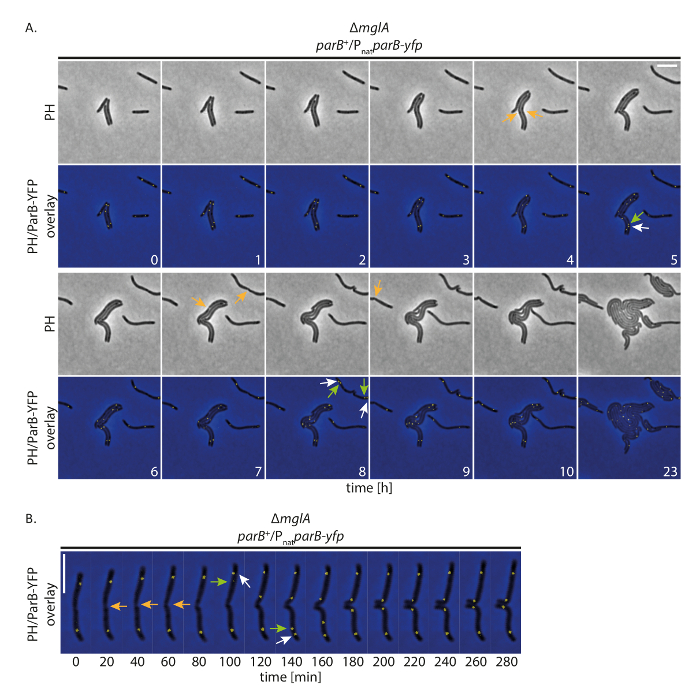
Figur 4 : Fluorescens time-lapse mikroskopi av ParB-YFP i ikke-motile M. cellemembran celler. Cellene i en ΔmglA mutantuttrykke parB-yfp i nærvær av innfødte parB (SA4749; ΔmglA; parB +/PnatparB-yfp) ble fulgt i 24 timer av fase kontrast og fluorescens mikroskopi. (A) bildene ble anskaffet hvert 20 min og representant bilder hver time til 10t vises, sammen med de samme cellene etter 24 h. bilder vises i kontrast (PH) og overlapping av kontrast og YFP signal. Valgte celledelinger er markert med oransje piler. Hvite og grønne pilene angir ParB-YFP klyngen duplisering hendelser, med de grønne pilene merking translocating klyngen. Tall angir tid i timer. Skala bar: 5 µm. (B) bilder ble anskaffet som (A), men vises i høyere midlertidig løsning. Tall angir tiden i minutter. Pilene er som (A). Skala bar: 5 µm. Klikk her for å se en større versjon av dette tallet.

Figur 5 : Fluorescens time-lapse mikroskopi av FtsZ-GFP i ikke-motile M. cellemembran celler. Cellene i en ΔmglA mutant uttrykke ftsZ-gfp i nærvær av innfødte ftsZ (SA8241; ΔmglA; ftsZ +/PnatftsZ-gfp) ble fulgt i 24 timer av fase kontrast og fluorescens mikroskopi. (A) bildene ble anskaffet hvert 20 min og representant bilder hver time til 10t vises, sammen med de samme cellene etter 24 h. bilder vises i kontrast (PH) og overlapping av kontrast og GFP signal. Valgte celledelinger er markert med oransje piler. Hvite pilene viser FtsZ-GFP klynger på midcell. Tall angir tid i timer. Skala bar: 5 µm. (B) bilder ble anskaffet som (A), men vises i høyere midlertidig løsning. Tall angir tiden i minutter. Grønne og hvite pilene merke FtsZ-GFP-klynger i venstre og høyre cellene, henholdsvis. Oransje pilene angir celledelinger. Skala bar: 5 µm. Klikk her for å se en større versjon av dette tallet.

Figur 6 : Fluorescens time-lapse mikroskopi av mCherry-PomX i ikke-motile M. cellemembran celler. Ikke-motile ΔpomX celler samler mCherry-PomX (SA4797, ΔmglA, ΔpomX/PpomZ mCherry-pomX) ble fulgt i 24 timer av fase kontrast og fluorescens mikroskopi hver 20 min. (A) representant bilder hver time til 10t vises, sammen med de samme cellene etter 24 h. bilder vises i kontrast (PH) og som overlegg kontrast og mCherry signal. Valgte celledelinger er markert med oransje piler. Hvite og grønne pilene angir mCherry-PomX klynger før og etter dele arrangementer, henholdsvis. Tall angir tid i timer. Skala bar: 5 µm. (B) bilder ble anskaffet som (A) og vises i høyere midlertidig løsning. Pilene er som (A). Skala bar: 5 µm. Klikk her for å se en større versjon av dette tallet.
| Bakterielle belastningen | Relevante genotype1 | Referanse |
| DK1622 | Wildtype | 23 |
| SA4420 | ΔmglA | 24 |
| SA4749 | ΔmglA; parB+/attB:: PnatparB-yfp (pAH7) | Denne studien |
| SA4797 | ΔmglA; ΔpomX / attB::PpomZ mCherry-pomX (pAH53) | 16 |
| SA8241 | ΔmglA; ftsZ+/ mxan18-19::PnatftsZ-gfp (pDS150) | Denne studien |
| Plasmider i parentes inneholder angitt genet fusjoner og ble intergated på angitte steder på genomet. Plasmider integrert på attB område eller mxan18-19 intergenisk regionen ble uttrykt fra deres innfødte promoter (Pnat) eller innfødt arrangøren av pomZ (PpomZ). |
||
Tabell 1: Liste over bakteriell påkjenningen brukes i denne studien.
| Plasmider | Relevante egenskaper | Referanse |
| pAH7 | PnatparB-yfp; Mx8 attP; TetR | 19 |
| pAH53 | PpomZ mCherry-pomX; Mx8 attP ; KmR | 16 |
| pDS150 1 | PnatftsZ-gfp ; mxan18-19 ; TetR | Denne studien |
| pMR3691 | Plasmider for vanillate induserbart genuttrykk | 18 |
| pKA51 | PnatftsZ-gfp ; Mx8 attP; TetR | 17 |
| 1 pDS150: pDS150 er et derivat av pKA51 der webområdet Mx8 attP ble erstattet med mxan18-19 intergenisk regionen. For denne ble mxan18-19 intergenisk regionen forsterket fra pMR3691 med primer Mxan18-19 fwd BsdRI (GCGATCATTGCGCGCCAGACGATAACAGGC) og Mxan18-19 rev BlpI (GCGGCTGAGCCCGCGCCGACAACCGCAACC) og klonet i pKA51. |
||
Tabell 2: Liste over plasmider brukt i denne studien.
Discussion
Fluorescens live-celle bildebehandling har blitt et kraftig verktøy for å studere spatiotemporal dynamikken i bakterielle celler. Time-lapse fluorescens mikroskopi motile og langsom voksende bakterier som M. cellemembran, men har vært utfordrende og var bare utført for kort tid varighet. Her presenterer vi en lett-å-bruk og robust metode for live-celle avbildning av M. cellemembran av time-lapse fluorescens mikroskopi. Denne metoden tillater brukeren å følge celler og fluorescently merket proteiner for flere runder av cellen syklus med enkeltcelle oppløsning.
Det er flere forutsetninger som påvirker suksessen til live-celle avbilding av langsom voksende M. cellemembran celler inkludert: 1) en solid overflate for cellen vedlegg; 2) tilgjengeligheten av næringsstoffer og oksygen; 3) konstant fuktighet og temperatur; og 4) optimalisering av eksperimentelle forhold som eksponering og oppkjøp frekvens.
I vår eksperimentelle set-up bruker vi tykke agarose pads med næringsstoffer. Ved hjelp av tykk agarose pads i motsetning til microfluidic enheter for å følge enkeltceller har noen grunnleggende fordeler, men også noen ulemper. Først agarose pad gir ikke bare en overflate for M. cellemembran celle vedlegg og bevegelse, men også tilstrekkelig næringsstoffer for vekst for minst 24 timer. Andre snap skudd analyser brukes vanligvis til å studere intracellulær lokalisering av fluorescently merket proteiner var tidligere gjort av samme type agarose pads16,17,29. Derfor kan data fra snap shot analyser være direkte forhold til innhentet med metoden beskrevet her. For det tredje agarose pads kan lett endres og supplert med antibiotika eller andre kosttilskudd som CuSO4 og vanillate som brukes vanligvis for gene expression induksjon18,30. Til slutt, fordi cellene er tillatt å skjemaet microcolonies i løpet av et eksperiment, dette oppsettet kan også studere effekten av direkte celle-celle samhandling på en bestemt parameter blir analysert. Dette er spesielt viktig i tilfelle av M. cellemembran fordi denne bakterie viser flere kontakt-avhengige interaksjoner. Den viktigste ulempen med denne metoden er at eksperimentelle forhold er forhåndsinnstilt for varigheten av et eksperiment. I motsetning kan microfluidic enheter vanligvis endre eksperimentelle forhold i løpet av et eksperiment ved å legge for eksempel antibiotika31.
Fri programvarepakker (f.eksMicrobeJ, Oufti) er tilgjengelig å automatisk analysere veksten av enkeltceller og protein lokalisering i enkeltceller. Men disse programvare er bare egnet for analyse av enkeltceller eller små grupper av celler. Dermed er det fortsatt en utfordring å automatisk analysere dataene som genereres for 24 h innspillingene beskrevet her.
I sammendraget, vi beskrev en lett-å-bruke og reproduserbar protokoll for å utføre live-celle bildebehandling med langsom voksende M. cellemembran bakterier. Vi viser at enkel nærings-supplert agarose pads er tilstrekkelig til å opprettholde vekst for minst 24 timer og tillate observere og analysere protein lokalisering og vekst med enkeltcelle oppløsning over flere generasjoner.
Disclosures
Forfatterne erklærer at de har ingen konkurrerende økonomiske interesser.
Acknowledgments
Dette arbeidet ble støttet av tysk Research Council (DFG) innenfor rammen av Transregio 174 "Spatiotemporal dynamikken i bakterieceller" og Max Planck Society.
Materials
| Name | Company | Catalog Number | Comments |
| DMI6000B with AFC | Leica microsystems | 11888945 | Automated inverted widefield fluorescence microscope with adaptive focus control |
| Universal mounting frame | Leica microsystems | 11532338 | Stage holder for different sample sizes |
| HCX PL FLUOTAR 100x/1.30 oil PH3 | Leica microsystems | 11506197 | Phase contrast objective |
| Orca Flash 4.0 camera | Hamamatsu | 11532952 | 4.0 megapixel sCMOS camera for picture aquisition |
| Filter set TXR ET, k | Leica microsystems | 11504170 | Fluorescence filter set, Ex: 560/40 Em: 645/75 |
| Filter set L5 ET, k | Leica microsystems | 11504166 | Fluorescence filter set, Ex: 480/40 Em: 527/30 |
| Filter set YFP ET, k | Leica microsystems | 11504165 | Fluorescence filter set, Ex: 500/20 Em: 535/30 |
| ProScan III | Prior | H117N1, V31XYZEF, PS3J100 | Microscope automation controller with interactive control center |
| EL 6000 light source | Leica microsystems | 11504115 | External fluorescence light source |
| Incubator BLX Black | Pecon | 11532830 | Black incubation chamber surrounding the microscope |
| Tempcontrol 37-2 digital | Leica microsystems | 11521719 | Automated temperature control for incubation chamber |
| Gentmycin sulphate | Carl Roth | 0233.4 | Gentamycin |
| Oxytetracylin dihydrate | Sigma Aldrich | 201-212-8 | Oxytetracyclin |
| Kanamycin sulphate | Carl Roth | T832.3 | Kanamycin |
| Filtropur BT25 0.2 bottle top filter | Sarstedt | 831,822,101 | Bottle top filter for sterilization of buffers |
| Deckgläser | VWR | 630-1592 | Glass cover slip (60 x 22 mm, thickness: 0.7 mm) |
| Seakem LE agarose | Lonza | 50004 | Agarose for microscopy slides |
| Leica Metamorph AF | Leica microsystems | 11640901 | Microscope control software and software for picture analysis |
| Tetraspeck Microsperes, 0.5 µm | ThermoFisher | T7281 | Fluorescent microspheres |
| petri dish | Greiner Bio-one | 688102 | 120 mm x 120 mm x 17 mm squared petri dish for agarose pads |
| BD Bacto Casitone | Becton Dickinson | 225930 | Casitone |
| Parafilm M | VWR | 291-1213 | Parafilm |
| Tris(hydroxymethyl)-aminomethane | Carl Roth | AE15.2 | Tris |
| Magnesium sulphate heptahydrate | Carl Roth | P027.2 | Magnesium sulphate |
| Potassium dihydrogen phosphate p.a. | Carl Roth | 3904.1 | Potassium dihydrogen phosphate |
| 1% CTT medium: 1 % (w/v) BD Bacto™ casitone, 10 mM Tris-HCl ph 8.0, 1 mM potassium phosphate buffer pH 7.6, 8 mM MgSO4 | Cultivation medium for M.xanthus | ||
| TPM buffer: 10 mM Tris-HCl ph 8.0, 1 mM potassium phosphate buffer pH 7.6, 8 mM MgSO4 | Buffer for preparation of microscopy slides for M.xanthus |
References
- Shapiro, L., McAdams, H. H., Losick, R. Why and how bacteria localize proteins. Science. 326 (5957), 1225-1228 (2009).
- Treuner-Lange, A., Søgaard-Andersen, L. Regulation of cell polarity in bacteria. J Cell Biol. 206 (1), 7-17 (2014).
- Laloux, G., Jacobs-Wagner, C. Spatiotemporal control of PopZ localization through cell cycle-coupled multimerization. J Cell Biol. 201, 827-841 (2013).
- Rudner, D. Z., Losick, R. Protein subcellular localization in bacteria. Cold Spring Harb. Perspect. Biol. 2 (4), 000307 (2010).
- Badrinarayanan, A., Le, T. B. K., Laub, M. T. Bacterial chromosome organization and segregation. Annu Rev Cell Dev Biol. 31 (1), 171-199 (2015).
- Munoz-Dorado, J., Marcos-Torres, F. J., Garcia-Bravo, E., Moraleda-Munoz, A., Perez, J. Myxobacteria: Moving, Killing, Feeding, and Surviving Together. Front Microbiol. 7, 781 (2016).
- Berleman, J. E., Kirby, J. R. Deciphering the hunting strategy of a bacterial wolfpack. FEMS Microbiol Rev. 33 (5), 942-957 (2009).
- Konovalova, A., Petters, T., Søgaard-Andersen, L. Extracellular biology of Myxococcus xanthus. FEMS Microbiol. Rev. 34, 89-106 (2010).
- Nudleman, E., Wall, D., Kaiser, D. Cell-to-cell transfer of bacterial outer membrane lipoproteins. Science. 309, 125-127 (2005).
- Vassallo, C. N., et al. Infectious polymorphic toxins delivered by outer membrane exchange discriminate kin in myxobacteria. eLife. 6, 29397 (2017).
- Vassallo, C., et al. Cell rejuvenation and social behaviors promoted by LPS exchange in myxobacteria. Proc Natl Acad Sci USA. 112 (22), 2939-2946 (2015).
- Li, Y., et al. Extracellular polysaccharides mediate pilus retraction during social motility of Myxococcus xanthus. Proc. Natl. Acad. Sci. USA. 100, 5443-5448 (2003).
- Kim, S. K., Kaiser, D. Cell alignment required in differentiation of Myxococcus xanthus. Science. 249, 926-928 (1990).
- Lobedanz, S., Søgaard-Andersen, L. Identification of the C-signal, a contact dependent morphogen coordinating multiple developmental responses in Myxococcus xanthus. Genes Dev. 17, 2151-2161 (2003).
- Schumacher, D., Søgaard-Andersen, L. Regulation of cell polarity in motility and cell division in Myxococcus xanthus. Annu Rev Microbiol. 71 (1), 61-78 (2017).
- Schumacher, D., et al. The PomXYZ proteins self-organize on the bacterial nucleoid to stimulate cell division. Dev Cell. 41 (3), 299-314 (2017).
- Treuner-Lange, A., et al. PomZ, a ParA-like protein, regulates Z-ring formation and cell division in Myxococcus xanthus. Mol Microbiol. 87 (2), 235-253 (2013).
- Iniesta, A. A., Garcia-Heras, F., Abellon-Ruiz, J., Gallego-Garcia, A., Elias-Arnanz, M. Two systems for conditional gene expression in Myxococcus xanthus inducible by isopropyl-beta-D-thiogalactopyranoside or vanillate. J Bacteriol. 194 (21), 5875-5885 (2012).
- Harms, A., Treuner-Lange, A., Schumacher, D., Søgaard-Andersen, L. Tracking of chromosome and replisome dynamics in Myxococcus xanthus. reveals a novel chromosome arrangement. PLoS Genet. 9 (9), 1003802 (2013).
- Iniesta, A. A. ParABS system in chromosome partitioning in the bacterium Myxococcus xanthus. PLoS One. 9 (1), 86897 (2014).
- Lin, L., Osorio Valeriano, M., Harms, A., Søgaard-Andersen, L., Thanbichler, M. Bactofilin-mediated organization of the ParABS chromosome segregation system in Myxococcus xanthus. Nat Commun. 8 (1), 1817 (2017).
- Hodgkin, J., Kaiser, D. Cell-to-cell stimulation of movement in nonmotile mutants of Myxococcus. Proc Natl Acad Sci U S A. 74 (7), 2938-2942 (1977).
- Kaiser, D. Social gliding is correlated with the presence of pili in Myxococcus xanthus. Proc Natl Acad Sci USA. 76 (11), 5952-5956 (1979).
- Miertzschke, M., et al. Structural analysis of the Ras-like G protein MglA and its cognate GAP MglB and implications for bacterial polarity. EMBO J. 30 (20), 4185-4197 (2011).
- Hodgkin, J., Kaiser, D. Genetics of gliding motility in Myxococcus xanthus. (Myxobacterales): Two gene systems control movement. Mol Gen Genet. 171, 177-191 (1979).
- Shi, X., et al. Bioinformatics and experimental analysis of proteins of two-component systems in Myxococcus xanthus. J Bacteriol. 190 (2), 613-624 (2008).
- Bi, E. F., Lutkenhaus, J. FtsZ ring structure associated with division in Escherichia coli. Nature. 354 (6349), 161-164 (1991).
- Lutkenhaus, J., Pichoff, S., Du, S. Bacterial cytokinesis: From Z ring to divisome. Cytoskeleton. 69 (10), 778-790 (2012).
- McLoon, A. L., et al. MglC, a Paralog of Myxococcus xanthus GTPase-Activating Protein MglB, Plays a Divergent Role in Motility Regulation. J Bacteriol. 198 (3), 510-520 (2015).
- Gomez-Santos, N., et al. Comprehensive set of integrative plasmid vectors for copper-inducible gene expression in Myxococcus xanthus. Appl Environ Microbiol. 78 (8), 2515-2521 (2012).
- Treuner-Lange, A., et al. The small G-protein MglA connects to the MreB actin cytoskeleton at bacterial focal adhesions. J Cell Biol. 210 (2), 243-256 (2015).
