

Summary
As células bacterianas são espacialmente altamente organizadas. Para seguir esta organização ao longo do tempo em crescimento lento Myxococcus xanthus células, foi desenvolvido um set-up para a imagem latente de viver-pilha de fluorescência com alta resolução spatiotemporal ao longo de várias gerações. Usando esse método, pôde ser determinada spatiotemporal dinâmica de proteínas importantes para a segregação de cromossomos e divisão celular.
Abstract
Imagem latente da viver-pilha de fluorescência das células bacterianas é um método de chave na análise da dinâmica espacial e temporal de proteínas e cromossomos subjacentes eventos central ciclo celular. No entanto, imagens destas moléculas no crescimento lento bactérias representa um desafio devido fotobranqueamento fluorophores e fototoxicidade durante a aquisição de imagens. Aqui, descrevemos um protocolo simples para contornar essas limitações no caso de Myxococcus xanthus (que tem um tempo de geração de 4-6 h). Para este fim, M. xanthus células são cultivadas em um bloco de ágar nutriente contendo grosso em um ambiente úmido com temperatura controlada. Sob estas condições, nós determinamos o tempo de duplicação das células individuais, seguindo o crescimento de células únicas. Além disso, o celular chave processos tais como segregação de cromossomos e divisão celular podem ser fotografadas pela imagem latente da viver-pilha de fluorescência das células que contêm relevantes proteínas marcador fluorescente etiquetadas como ParB-YFP, FtsZ-GFP e mCherry-PomX sobre múltiplo ciclos de célula. Posteriormente, as imagens adquiridas são processadas para gerar montagens e/ou filmes.
Introduction
As células bacterianas são espacialmente altamente organizadas com muitas proteínas localizando assimetricamente dentro de compartimentos celulares1,2,3,4. Esta localização é muitas vezes altamente dinâmica e muda ao longo do tempo em resposta a sinais do ciclo celular ou sinais externos. Igualmente, o cromossomo bacteriano é espacialmente altamente organizado com loci individuais sendo posicionado para locais específicos subcellular antes e durante o processo de segregação5. Esta organização espacial dinâmica é importante para o crescimento, divisão, regulação do ciclo celular, diferenciação, motilidade, transdução de sinal, bem como organização de cromossomo e segregação; assim, ela afeta essencialmente todos os aspectos da função bacteriana.
A spatiotemporal dinâmica destes processos celulares está sendo analisada em uma variedade de diferentes espécies bacterianas com Escherichia coli, Bacillus subtilis, Vibrio choleraee Caulobacter crescentus servindo como importante organismos-modelo. No entanto, estas quatro espécies cobrem apenas um pequeno espectro da enorme diversidade bacteriana e, talvez sem surpresa dada a grande distância filogenética entre estas espécies, mecanismos de polarização e de organização celulares são diferentes em estas bactérias. Isto gera a necessidade de estudar as espécies bacterianas adicionais para poder eventualmente extrair princípios gerais subjacentes a spatiotemporal dinâmica de células bacterianas.
O delta-proteobactérias Gram-negativas M. xanthus é um organismo modelo no estudo de comportamentos sociais e cooperação em bactérias6. M. xanthus é um aeróbio estrito, e na presença de nutrientes, forma colônias em que as células se espalhou para fora em um altamente coordenados, enxame moda e ataca outros microorganismos7. Em resposta à fome de nutrientes, células de iniciar um programa de desenvolvimento que resulta na formação de corpos frutíferos que consiste de milhares de células e dentro da qual, as células motile bacilares diferenciam a esférica diploides esporos8. Ambos os tipos de comportamentos, ou seja, pululam e formação do corpo frutífero, somente são executados por células que são colocadas sobre uma superfície sólida. Além disso, ambas as condições de nutrientes, células se envolver em processos que envolvem contatos direto célula-célula, incluindo a troca de lipoproteínas membrana exterior que pode estimular a motilidade ou funcionam como toxinas do destinatário9,10 , a troca de LPS11, estimulação da motilidade por exopolissacarídeo na vizinha células12e ancorada intercelular sinalização por uma célula superfície de sinalização da proteína13,14.
Recentemente, M. xanthus também se tornou um organismo modelo para estudar os mecanismos subjacentes a motilidade e seu regulamento15, divisão celular16,17,18e organização do cromossoma19 ,20,21. Crítica entra o M. xanthus ciclo celular foram analisadas em pormenor por microscopia de fluorescência usando snap shot imagens ou gravações de lapso de tempo curtas em estirpes carregando relevantes proteínas fluorescente etiquetadas16, 17,18,19,20. Idealmente, muitas células devem ser seguidas com resolução de célula única célula viva de fluorescência da imagem latente pelo menos um ciclo completo do celular obter dados quantitativos robustos sobre parâmetros do ciclo celular. No entanto, este é um desafio no caso de M. xanthus devido a seu tempo relativamente longo de geração de 4-6 h sob condições padrão de laboratório e fotobranqueamento fluorophores e fototoxicidade durante a aquisição de imagens.
Aqui, descrevemos um protocolo a seguir M. xanthus células com resolução única célula por fluorescência viver-pilha de imagem pelo menos 24 h e cobrindo vários ciclos de célula. Importante, durante o protocolo de todo, as células são mantidas em um bloco de ágar e perto contato permitindo para as atividades de contato-dependente, essencial para o estilo de vida social de M. xanthus. O protocolo também permite que usuários de monitor de forma, tamanho, divisões e sondas fluorescentes em uma alta resolução temporal e com resolução de célula única e assim, permite a quantificação da variabilidade de célula para célula e correlações dos eventos do ciclo celular.
Protocol
1. preparação e crescimento de M. xanthus cepas
Nota: Consulte a tabela 1 e tabela 2.
- Prepare-se 1% casitone caldo (CTT) crescimento médio 1% (p/v) do pâncreas Resumo de caseína (por exemplo, Bacto casitone), 10 mM Tris-HCl pH 8.0, pH 7,6,4 2PO do KH 1 mM 8 mM MgSO422, suplementado com canamicina (50 µ g/mL) ou oxitetraciclina (10 µ g/mL). Adicionar a gentamicina (10 µ g/mL) a todos os meios para reduzir o risco de contaminação com outras bactérias, desde que M. xanthus células são naturalmente resistentes a ela.
- Inocular 5ml de 1% CTT contendo o relevantes antibiotic(s) com uma única colônia recém crescida de wild digite (WT) DK1622 23, SA4420 (ΔmglA)24, SA4797 (ΔmglA, ΔpomX/ppomZ mCherry-pomX )16, SA8241 (ΔmglA, ftsZ+/pnatftsZ-gfp), ou SA4749 (ΔmglA, parB+/pnatparB-yfp) na manhã do dia 1.
- Resuspenda um único M. xanthus colônia em 500 µ l de 1% CTT suplementadas com antibióticos em um tubo estéril e transferir a suspensão inteira para um Erlenmeyer contendo 5 mL de 1% de 50 mL CTT.
Nota: Use um frasco de Erlenmeyer com 10 vezes o volume da cultura a garantia suficiente aeriation e crescimento ideal.
- Resuspenda um único M. xanthus colônia em 500 µ l de 1% CTT suplementadas com antibióticos em um tubo estéril e transferir a suspensão inteira para um Erlenmeyer contendo 5 mL de 1% de 50 mL CTT.
- Crescem as células há oito gerações (cerca de 40-48 h, com um tempo de geração de 4-6 h) a 32 ° C, agitando a 220 rpm, no escuro. Manter as células em fase de crescimento exponencial (OD550 < 1.2) e evitar que cheguem a fase estacionária. Se necessário, diluir as células em meio CTT fresco 1% contendo o antibiotic(s) relevantes para uma OD550 de 0.1 - 0.2.
Nota: Um ideal de OD550 para microscopia uma única célula é 0.5 - 0.7. Neste OD550, um número suficiente de células está presente por imagem para permitir a quantificação, bem como a análise estatística dos parâmetros celulares.
2. preparação das amostras de microscopia
Nota: As células para ser visto por microscopia são colocadas sobre uma lamela de microscópio e então cobertas por uma almofada de agarose contendo nutrientes. A lamela é colada sobre um plástico ou metal frame para fornecer mecânica suporte. Em preparação para a microscopia, um grande bloco de 1% agarose/TPM/0.2% CTT deve ser preparado com antecedência como descrito nos passos 2.1-2.3. Consulte também a Tabela de materiais para produtos específicos utilizados aqui.
- Preparar 500 mL de tampão TPM (10 mM Tris-HCl pH 7,6, pH de4 de PO para2de KH 1 mM 7.6, 8 mM de MgSO4) e filtro ou autoclave esterilizar usando um filtro superior da garrafa.
Nota: O tampão estéril pode ser armazenado por vários meses à temperatura ambiente. - Preparar a solução de microscopia de agarose 1% contendo 0,2% CTT (1 g de agarose com 80 mL de tampão TPM e 20 mL de meio CTT de 1% de mistura). Aquecer em um forno de microondas, até que a agarose é derretido.
Nota: A 0,2% CTT é suficiente para permitir que as células a crescer e evitar starvation. Concentrações mais elevadas dos CTT, no meio de microscopia resultará em fluorescência de fundo elevado. - Encha um prato de Petri com o agarose fundido a uma espessura de 0,5 cm (para um quadrado de 11,5 x 11,5 cm placa de Petri, cerca de 60 mL de agarose derretido é necessário) e deixe-o esfriar até a temperatura de quarto.
Nota: A almofada de agarose pode ser armazenada a 4 ° C, em um ambiente úmido, por até 2 dias.- Pré-aquecer a almofada CTT 1% agarose/TPM/0.2% a 32 ° C, durante pelo menos 15 min antes de usar.
Nota: Para preparar as células para microscopia, siga os passos 2.4-2.8.
- Pré-aquecer a almofada CTT 1% agarose/TPM/0.2% a 32 ° C, durante pelo menos 15 min antes de usar.
- Coloque uma lamela de vidro estéril (60 mm x 22 mm, espessura: 0,7 mm) em um quadro de plástico ou de metal que tem um buraco no meio (Figura 1A); Este quadro serve como um suporte mecânico para a lamela fina e ajuda a reduzir a deriva durante a microscopia. Corrigi a lamela para o quadro com fita adesiva.
- Para preparar o quadro, cortar um quadro de 75 mm × 25 mm de uma placa de metal de espessura de 1 mm e, em seguida, cortar um buraco de tamanho apropriado (20 mm × 30mm neste experimento) no meio.
- Adicionar 10 a 20 µ l de crescido exponencialmente M. xanthus células na lamela.
- Adicione fluorescentes 0,5 µm microesferas como marcadores fiduciais às células para simplificar o controle de células ou proteínas em gravações de lapso de tempo.
- Diluir a 1: 100 de microesferas no buffer TPM e armazenar a 4 ° C por até diversos meses. Agite cuidadosamente antes de usar e adicionar 5-10 µ l das microesferas diluídas para as células.
Nota: Aqui microesferas que são fluorescentes em todos comum azul, verde, amarelo e vermelho fluorescentes canais foram usados.
- Diluir a 1: 100 de microesferas no buffer TPM e armazenar a 4 ° C por até diversos meses. Agite cuidadosamente antes de usar e adicionar 5-10 µ l das microesferas diluídas para as células.
- Cortar uma pequena almofada, aproximadamente o tamanho da lamela da grande pré-aquecido 1% agarose/TPM/0.2% CTT almofada e coloque-o na parte superior das células (Figura 1B). Coloque uma lamela em cima da 1% agarose/TPM/0.2% CTT agarose almofada para evitar a evaporação e manter as células em um ambiente úmido.
Nota: A lamela sozinha evitará evaporação significativa pelo menos 2 h. Para mais gravações de lapso de tempo, o sanduíche de almofada e lamela CTT agarose/TPM/0.2% 1% deve ser selada com uma película de parafina para evitar a evaporação. - Incube a amostra de microscopia a 32 ° C, durante 15-20 min permitir que as células anexar a parte inferior da almofada do agarose. Em seguida, inicie as gravações de microscopia de lapso de tempo.
3. microscópio instalação e aquisição de lapso de tempo
Nota: O protocolo descrito aqui foi desenvolvido por um microscópio invertido widefield com autofocus, uma 100 X 1.30 at óleo PH3 objectivo, um X, Y motorizado estágio, uma câmera sCMOS, uma fonte de luz, filtros para verde-fluorescente, vermelho fluorescente ou amarelo-fluorescente proteínas e uma câmara de incubação controlada de temperatura. Esta câmara mantém as células protegidas da luz e a temperatura constante.
- Pré-aquece a câmara de incubação e o microscópio a 32 ° C para ~ 1-2 h antes de iniciar a microscopia.
Nota: Dependendo da configuração do microscópio, aquecimento pode levar mais tempo. Pré-aquecimento é essencial para reduzir o arrasto e estabiliza o sistema de controle de foco automático. - Ligue o microscópio e iniciar o software de controle de microscópio. Selecione o objetivo correto e os espelhos corretos e filtros para adquirir fase imagens de contraste, bem como imagens de proteínas verde-fluorescente, vermelho fluorescente ou amarelo-fluorescente.
Nota: Um microscópio normalmente é fornecido com um software preferencial para aquisição de controle e a imagem do microscópio. Aqui um software comercialmente disponível (veja a Tabela de materiais) era usado para controlar a aquisição de microscópio e imagem. - Adicionar uma gota de óleo de imersão de alta qualidade para a lente do objectivo e a parte inferior da amostra previamente incubada a 32 ° C. Coloque o objetivo a posição mais baixa possível Z-para não danificar a lente objetiva, quando a amostra é colocada na fase de microscópio. Coloque a estrutura de metal com a amostra para o palco do microscópio e com o "lado do buraco" no sentido do objectivo. Aperte firmemente a amostra no suporte do palco.
- Concentre-se nas células movendo o palco na direção-Z mais perto do objectivo. Mover o palco mais lento quando o óleo cai no lado inferior de amostra e a lente objetiva fazer contato. Mover o palco no X / Y direção até várias células únicas são visíveis na região do ponto de vista, quando as células são no plano focal. Certifique-se que pelo menos uma microesfera fluorescente é na região de vista para depois alinhar as imagens adquiridas.
Nota: Sob condições ideais, uma densidade de células de 15-30 células por região da vista (2.048 x 2.048 pixel ou 133.1 x 133.1 µm) deve ser alcançada. - Abra o assistente de Multi-dimensional de aquisição de software de controle o microscópio para configurar um experimento de lapso de tempo que permite que o microscópio adquirir imagens em vários comprimentos de onda e posições de estágio, se necessário.
- Na aba principal ative Timelapse e Múltiplos comprimentos de onda. Guias adicionais aparecerá no lado esquerdo da janela.
- Clique na guia salvar e Selecione o diretório para selecionar uma pasta vazia no disco rígido do computador para salvar as imagens adquiridas. Ative o nome base do incremento se arquivo existe para certificar-se de que os conjuntos de dados consecutivos não substituir os anteriores. Então dê o experimento um nome com a data e o nome de estirpe ou título do experimento.
- Clique na guia Timelapse para ajustar os parâmetros de lapso de tempo. Definir a duração de 24h e definir o Intervalo de tempo de 20 min. O Número de pontos de tempo mudará automaticamente.
Nota: O intervalo de tempo ideal varia de acordo com a experiência e a função celular a ser analisadas. Aquisições de imagem frequente podem causar fotobranqueamento. Assim, um trade-off entre a resolução temporal e fotobranqueamento deve ser encontrado empiricamente. Hora de uma duplicação de 4-6 h, imagens podem ser facilmente adquiridas em um intervalo de 5 min (ou intervalos ainda menores, se desejado) para a microscopia de contraste de fase. Se a microscopia de fluorescência um tempo longo de 24h é desejada imagens devem ser gravadas em um intervalo de cerca de 15-30 min. - Clique na guia de comprimentos de onda selecione o número de comprimentos de onda para adquirir para cada imagem em cada ponto do tempo, alterando o número.
Nota: para cada comprimento de onda, uma nova aba aparecerá no lado esquerdo da multi-dimensional aquisição " assistente e comprimentos de onda vão ser adquiridos na ordem de cima para baixo. Para cada comprimento de onda, as configurações de aquisição podem ser modificadas separadamente. - Clique na primeira guia de comprimento de onda do topo. Selecione o contraste de fase na lista suspensa de iluminação . Selecione a 100 ms para exposição e Ponto de cada vez na lista suspensa de adquirir . Desative o Auto expor selecionando Never na lista drop-down.
- Repita a etapa 3.5.5 para cada comprimento de onda que precisa ser adquirida em cada ponto de tempo. Para a montagem experimental e proteínas fluorescente etiquetadas descritas aqui, use os seguintes parâmetros para exposição: 250 ms para proteínas da fusão mCherry, 200 ms para proteínas da fusão YFP e 1.000 ms para proteínas da fusão de GFP.
Nota: As configurações de iluminação ideal para cada estirpe e proteína fluorescente devem ser determinadas com antecedência, alterando a intensidade da lâmpada e o tempo de aquisição de imagem para cada comprimento de onda. Tempos de aquisição de imagem muito tempo aumentará o efeito fototóxico e, finalmente, levar à morte de detenção e célula de crescimento. Portanto, um trade-off entre a viabilidade de qualidade e célula de imagem deve ser alcançado. - Adquira imagens de múltiplas posições de estágio para aumentar o número de células gravadas no mesmo experimento.
- Para adquirir imagens de múltiplas posições de estágio, ative Múltiplas posições de estágio na guia principal . Em seguida clique na guia de palco e clique no botão Live de olhar para o campo de visão.
- Mova o palco na direção X/Y, até uma região de interesse (ROI) no campo de visão. Salvar as coordenadas X e Y-clicando o "+" no guia de palco mover o palco novamente na direção X/Y até que seja encontrado um novo ROI e salvar as coordenadas novamente clicando no "+". Vá até o número desejado de regiões é salvo.
Nota: No caso de aquisição de imagens de fluorescência, certifique-se que as regiões de interesse (ROIs) não estão muito perto de cada um ao outro para minimizar a fototoxicidade.
- Verifique mais uma vez que as células estão em foco, clicando sobre a salva X - e Y-posições diferentes e iniciar o autofocus de hardware clicando AFC segura para manter a salva Z-posição constante ao longo do experimento.
- Começam as gravações de lapso de tempo no software de controle do microscópio clicando em adquirir no assistente de Aquisição de multi-dimensional .
Nota: Aparecerá uma janela para cada comprimento de onda que é adquirido e vai aparecer uma janela adicional que mostra o número de pontos adquiridos de tempo e o tempo até a próxima aquisição de imagens. - Verifique se as células estão ainda em foco após o primeiros poucos pontos de tempo-nas gravações do tempo-lapso para maximizar a qualidade das imagens e recentrar se necessário.
4. geração de filmes de lapso de tempo e alinhamento de imagem
Nota: Vários pacotes de software livre e comerciais estão disponíveis para aquisição de imagens e análise de imagem. Nós usamos um software comercialmente disponível (veja a Tabela de materiais) com vários plugins pré-instalados e ferramentas adicionais.
- Salve as imagens individuais de gravações de lapso de tempo em um computador que tenha o software de análise/processamento de imagem instalado.
- Inicie o software e abrir imagens como uma pilha clicando dados de multi-dimensional de revisão | Selecione o arquivo de Base | Selecione o diretório. Abra a pasta com os dados multidimensionais. Verificar o dataset e clique em modo de exibição; o dataset será mostrado como única imagens de tempo ponto um até o fim. Ativar o comprimento de onda (para a criação de uma pilha), selecione todas as imagens que devem estar na pilha e clique em Carregar imagem (ns). Repita este passo para todos os comprimentos de onda e salvar pilhas concluídas.
- (Opcional) Abrir todas as imagens necessárias para o filme usando o arquivo | Aberto.
Nota: É aconselhável abrir imagens por um comprimento de onda adquirido ao mesmo tempo para não abrandar o computador se poder computacional é limitado. Se certas partes das gravações as lapso de tempo, por exemplo, o início, fim ou vários pontos de tempo devem ser ignorados, então isto pode ser ajustado no filme concluído. - Ative a pilha de imagens que precisa ser corrigido para drift. Abra a ferramenta de alinhamento por Apps | Auto Align... verificação de pilha como a fonte para as imagens e o primeiro avião/tempo apontar como o plano de referência. Selecione a pilha com o botão de pilha de fonte e clique em aplicar.
Nota: O alinhamento automático levará algum tempo e poder computacional, mas é uma boa maneira para corrigir grandes stacks para drift de set-up o microscópio. Este alinhamento automático funciona bem se microesferas são incluídas, mas também podem funcionar sem eles. - Salve a pilha alinhada.
- Use ROIs.
Nota: A microscopia fluorescente de lapso de tempo cria facilmente grandes conjuntos de arquivos de dados que pegam um monte de poder computacional e retardar o processamento a jusante desses filmes. Portanto recomendamos ROIs de identificar e isolar as células para trabalhar com arquivos menores.- Selecione a ferramenta Região retangular . Crie um ROI ao redor das células de interesse pelo desenho manualmente um ROI na imagem de contraste de fase. Certifique-se que as células de interesse estão visíveis e em foco durante o filme todo lapso de tempo.
- Abra o filme lapso de tempo da segunda onda do mesmo conjunto de dados. Para transferir o ROI das imagens de contraste de fase para a fluorescência imagens de comprimento de onda segundo usam a ferramenta de Transferência de regiões com regiões | Regiões de transferência. Selecione o dataset de contraste de fase como a Imagem de origem e o segundo conjunto de dados de comprimento de onda como Imagem de destino. Selecione Todas as regiões e pressione Okey.
- Repita a etapa 4.6.2 para cada comprimento de onda adquirido para o mesmo conjunto de dados.
- Selecione o ROI e duplicá-lo como uma pilha com Editar | Duplicate | Pilha... ou pressione as teclas Shift + Ctrl + D . Em seguida, salve a pilha duplicada com arquivo | Salvar na mesma pasta em que os dados originais.
- Repita a etapa 4.6.4 para cada ROI de cada comprimento de onda adquirido para o mesmo conjunto de dados
- Para gerar um filme em formatos MOV ou AVI, abrir a função de Fazer filme através de pilha | Fazer filme. Selecione as gravações de lapso de tempo com o botão de Fonte pilha . Selecione o formato de saída, a taxa de quadros, o número de quadros e clique em salvar.
Representative Results
M. xanthus é uma bactéria de crescimento lenta que se move sobre superfícies sólidas. Para testar nossa montagem experimental, foi realizado um experimento de lapso de tempo com células de DK1622 WT motile. Imagens de contraste de fase foram adquiridas em intervalos de 5 min para 24 h (Figura 2A, B). A maioria das células alinhadas em grupos. Como esperado, células exibido motilidade e mudou-se predominantemente em grupos. Ainda observamos que células ocasionalmente inverter a direção do movimento. Estes achados sugerem que as células WT nas condições testadas se comportar normalmente em termos de motilidade celular. No entanto, mesmo quando as células são registradas a cada 5 min, a identificação de células individuais é difícil. Além disso, porque as células são motile, muitas células escapar ou insira o campo de visão tornando difícil seguir células por períodos prolongados.
A fim de rastrear o mesmo M. xanthus células para várias rodadas de ciclo celular pela imagem latente da viver-pilha, cepas individuais podem ser excluídas para o gene mglA , que é essencial para motilidade25. Isso impede que as células mover-se fora do campo de visão durante o protocolo de imagem. Armação de exclusões são geradas conforme descrito por Shi et al. 26
Como esperado, na imagem latente de viver-pilha de contraste de fase com células Δ non-motilemglA (Figura 3), as células não exibir movimento ativo. Fomos capazes de acompanhar o crescimento e divisão das células individuais durante a formação de microcolônias. Com base nas gravações lapso de tempo em que as imagens foram adquiridas em intervalos de 5 min por 24 h, foi possível quantificar o tempo de interdivision (o tempo entre dois eventos de divisão celular) com resolução de célula única. Células do mutantemglA Δ tinham um tempo de Divisão Inter de 235 ± 50 min (n = 97 células). Com cerca de 4 h, o tempo de interdivision é semelhante o duplicação tempo medido em culturas de suspensão para células WT. Isto fornece provas de que M. xanthus células crescem otimamente nestas condições experimentais.
Para investigar se a nossa configuração permite que as células a crescer normalmente, enquanto o rastreamento proteínas YFP-labeled durante longos períodos, realizamos o lapso de tempo de imagem com fluorescência M. xanthus células que expressam uma proteína YFP-tag. Para este fim, nós seguimos ParB-YFP como um marcador para a origem de replicação (ori). ParB é como componente do sistema de José PereiraS em M. xanthus e vincula os sites parS proximais aos ori; Portanto, a segregação de duplicação e o cromossomo de origem pode ser seguido19,20,21. Com imagem aquisição (contraste de fase e fluorescência, tempo de aquisição 200 ms no canal YFP) a cada 20 min, células cresceram, dividido e exibido o crescimento nem a 24 h(Figura 4). No início das gravações, ParB-YFP formado dois clusters nas regiões subpolares, na maioria das células(Figura 4). Pouco antes ou depois da divisão celular, a subpolar ParB-YFP cluster no polo celular velho duplicado. Um dos dois aglomerados permaneceu no polo celular velho, enquanto a segunda cópia translocado para o novo polo da célula, atingindo posição final subpolar após cerca de 40-60 min (Figura 4A, B). Estas observações estão de acordo com dados anteriores, gerados a partir de gravações de lapso de tempo curtas usando almofadas de ágar fina19. Podemos concluir que esta montagem experimental permite a microscopia de fluorescência lapso de tempo acompanhar a segregação de cromossomos ao longo de vários ciclos de células em crescimento lento M. xanthus células, sem o crescimento de células perturbando ou a maquinaria de segregação do cromossomo.
Em um experimento similar, procuramos seguir marcadores para a divisão celular por microscopia de fluorescência de lapso de tempo. Semelhante à quase todas as outras bactérias, M. xanthus requer FtsZ, uma GTPase de tubulina, como bacteriana, por divisão celular16,17,18. FtsZ forma uma estrutura em anel no midcell, o chamado Z-ring, que ajuda a recrutar todas as outras proteínas necessárias para a divisão celular27,28. Em M. xanthus, a formação de o Z-ring e seu posicionamento no midcell é estimulada por três PomXYZ proteínas16,17. Estes três proteínas formam um complexo de cromossomo associada que transfere através de nucleoide, do site da divisão celular na célula "mãe" para o meio do nucleoide em duas células-filhas. No meio do nucleoide coincide com midcell, antes de segregação de cromossomas e aqui os recrutas de complexo de PomXYZ FtsZ e estimula a formação de Z-ring.
Aqui, seguimos primeiro non-motile células expressando ftsZ-gfp. Porque FtsZ-GFP geral mostra uma fluorescência mais fraca do sinal do que ParB-YFP, aumentamos o tempo de exposição de 5-fold para 1 s no canal da GFP. Como esperado, forte acumulação de FtsZ-GFP foi observada apenas em midcell e esta localização ditou a posição da constrição de divisão celular (Figura 5A). FtsZ-GFP predominantemente formado um cluster no midcell na célula mais tempo. Também ficou evidente que este aglomerado aumentou em intensidade ao longo do tempo. Após a divisão celular, observamos que a FtsZ-GFP re-acumulado no midcell na filha duas células aproximadamente 2h mais tarde (Figura 5B). Isto é consistente com a constatação de que cerca de 50% das células em uma população apresentar FtsZ localização na midcell com base na análise de snap shot16,17.
Em um segundo experimento, seguimos Δ non-motilemglA células para 24h expressam mCherry-pomX. Como parte do sistema de PomXYZ, PomX ajuda a orientar a formação de Z-ring e posicionamento, assim, estimulando a divisão celular em midcell16. O sinal de fluorescência de mCherry-PomX é forte e permite um tempo de exposição no canal da fluorescência de 250 MS. importante, todas as células cresceram em tamanho e exibido um evento de divisão celular ao longo do experimento, formando microcolonies após 24 h ( Figura 6A). Como relatado anteriormente16, quase todas as células continham um cluster mCherry-PomX. A maioria destes localizados na midcell e clusters longe midcell translocados para midcell durante o curso do experimento. Durante as divisões celulares, clusters de mCherry-PomX foram divididos, com cada célula filha recebe um cluster. Em oposição a FtsZ-GFP, mCherry-PomX localizadas em midcell 80-90% do ciclo celular e alcançou esta posição logo após a divisão celular (Figura 6B).
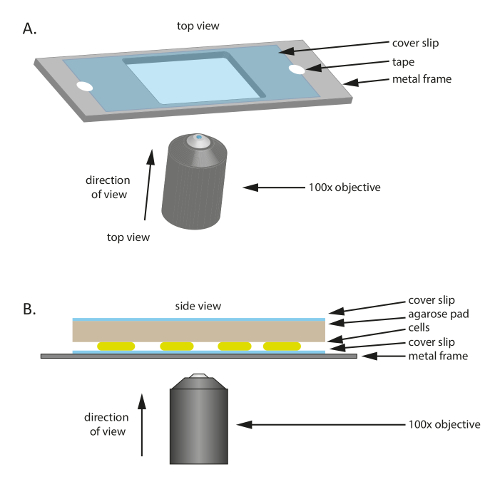
Figura 1 : Esquemático da montagem experimental utilizada ao longo deste estudo. (A), A metal ou plástico frame serve como um suporte para a amostra. Uma lamela é fixada para o frame do metal com fita adesiva para reduzir o movimento da amostra. Lado (B) vista de set-up a amostra experimental. As células são montadas no sentido do lamela mostrado em (A). A almofada de agarose que fornece nutrientes e umidade para as células é colocada em cima das células. A almofada de agarose é coberta por uma lamela adicional para reduzir a evaporação. Para imagens de alta qualidade, é usado um 100 X óleo imersão fase contraste objetivo. Clique aqui para ver uma versão maior desta figura.

Figura 2 : Microscopia de lapso de tempo da WT de contraste de fase M. xanthus células. As células foram seguidas por 24 h e imagens foram adquiridas a cada 5 min. (A) são mostradas imagens representativas do mesmo campo de visão cada 5 min. Setas coloridas indicam a direção do movimento de células individuais. A mesma cor marca a mesma célula ao longo do tempo. Os números indicam o tempo em minutos. Barra de escala: 5 µm. (B) imagens do mesmo campo de visão após cada hora são mostradas. Observe que o mesmo campo de visão é mostrado, mas porque as células estão se movendo, as células são constantemente entram e saem do campo de visão. Os números indicam o tempo em horas. Barra de escala: 5 µm. PH: contraste de fase. Clique aqui para ver uma versão maior desta figura.

Figura 3 : Microscopia de lapso de tempo de non-motile de contraste de fase M. xanthus células. Células ΔmglA foram seguidas por 24 h. imagens foram adquiridas a cada 5 min e são mostradas imagens representativas após cada hora. Divisão de células selecionado constrições são marcadas com setas laranja. Os números indicam o tempo em horas. PH: contraste de fase. Clique aqui para ver uma versão maior desta figura.
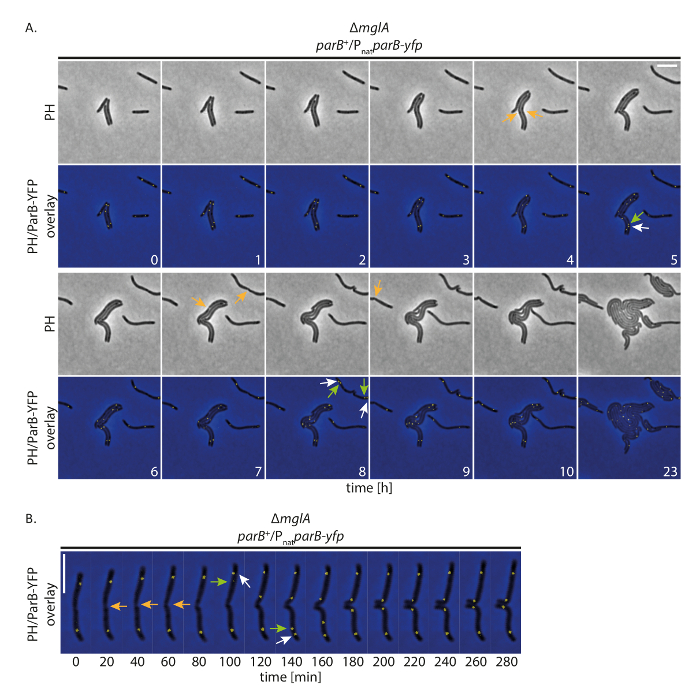
Figura 4 : Time-Lapse microscopia de fluorescência de ParB-YFP em non-motile M. xanthus células. Células de um ΔmglA mutanteparB-yfp expressando na presença de nativo parB (SA4749; ΔmglA; parB +/PnatparB-yfp) foram seguidos por 24h por microscopia de contraste e fluorescência fase. Imagens do (A) foram adquirido a cada 20 min e representante imagens a cada hora até 10 h são mostrados, juntamente com as células mesmas depois de 24 h. imagens são mostradas em contraste de fase (PH) e sobreposição de contraste de fase e a YFP sinal. Divisões de células selecionadas são marcadas com setas laranja. Brancas e verdes setas indicam eventos de duplicação de cluster ParB-YFP, com as setas verdes marcando o cluster translocating. Os números indicam o tempo em horas. Barra de escala: 5 µm. (B) imagens foram adquiridas como em (A), mas são mostradas em maior resolução temporal. Os números indicam o tempo em minutos. As setas são como em (A). Barra de escala: 5 µm. clique aqui para ver uma versão maior desta figura.

Figura 5 : Time-Lapse microscopia de fluorescência de FtsZ-GFP em non-motile M. xanthus células. Células de um mutante demglA Δ expressando gfp-ftsZ na presença de nativo ftsZ (SA8241; ΔmglA; ftsZ +/PnatftsZ-gfp) foram seguidos por 24h por microscopia de contraste e fluorescência de fase. Imagens do (A) foram adquirido a cada 20 min e cada hora até 10h de imagens representativas são mostradas, juntamente com as mesmas células após 24 h. imagens são mostradas em contraste de fase (PH) e como sobreposição de contraste de fase e sinal de GFP. Divisões de células selecionadas são marcadas com setas laranja. Setas brancas indicam clusters de FtsZ-GFP no midcell. Os números indicam o tempo em horas. Barra de escala: 5 µm. (B) imagens foram adquiridas como em (A), mas são mostradas em maior resolução temporal. Os números indicam o tempo em minutos. As setas verdes e brancas marcam clusters de FtsZ-GFP nas células esquerdas e direita, respectivamente. As setas laranja indicam divisões celulares. Barra de escala: 5 µm. clique aqui para ver uma versão maior desta figura.

Figura 6 : Time-Lapse microscopia de fluorescência de mCherry-PomX em non-motile M. xanthus células. Células depomX Δ non-motile acumulando mCherry-PomX (SA4797; ΔmglA; ΔpomX/ppomZ mCherry-pomX) foram seguidas por 24h por microscopia de contraste e fluorescência fase cada 20 min. (A) representante imagens a cada hora até 10 h são mostradas, juntamente com as mesmas células após 24 h. imagens são mostradas em contraste de fase (PH) e como sobreposição de contraste de fase e mCherry sinal. Divisões de células selecionadas são marcadas com setas laranja. Brancas e verdes setas indicam grupos de mCherry-PomX antes e após a divisão de eventos, respectivamente. Os números indicam o tempo em horas. Barra de escala: 5 µm. (B) imagens foram adquiridas como em (A) e são mostradas em maior resolução temporal. As setas são como em (A). Barra de escala: 5 µm. clique aqui para ver uma versão maior desta figura.
| Estirpe bacteriana | Relevantes do genótipo1 | Referência |
| DK1622 | Sua | 23 |
| SA4420 | ΔmglA | 24 |
| SA4749 | ΔmglA; parB+/attB:: PnatparB-yfp (pAH7) | Este estudo |
| SA4797 | ΔmglA; ΔpomX / attB::PpomZ mCherry-pomX (pAH53) | 16 |
| SA8241 | ΔmglA; ftsZ+/ mxan18-19::PnatftsZ-gfp (pDS150) | Este estudo |
| Plasmídeos entre parêntesis contêm gene indicado fusões e foram intergated nos locais indicados no genoma. Plasmídeos integrados no site attB ou região intergênica mxan18-19 foram expressos de seu nativo promotor P (nat) ou o promotor nativo do pomZ (PpomZ). |
||
Tabela 1: Lista de estirpes de bactérias utilizadas neste estudo.
| Plasmídeos | Características relevantes | Referência |
| pAH7 | PnatparB-yfp; Mx8 attP; TetR | 19 |
| pAH53 | PpomZ mCherry-pomX; Mx8 attP ; KmR | 16 |
| pDS150 1 | PnatftsZ-gfp ; mxan18-19 ; TetR | Este estudo |
| pMR3691 | Plasmídeo de expressão gênica inducible vanillate | 18 |
| pKA51 | PnatftsZ-gfp ; Mx8 attP; TetR | 17 |
| 1 pDS150: pDS150 é um derivado do pKA51 em que o site Mx8 attP foi substituído com a região intergênica mxan18-19 . Por isso a região intergênica mxan18-19 foi amplificada de pMR3691 com primers Mxan18-19 fwd BsdRI (GCGATCATTGCGCGCCAGACGATAACAGGC) e Mxan18-19 rev BlpI (GCGGCTGAGCCCGCGCCGACAACCGCAACC) e clonado em pKA51. |
||
Tabela 2: Lista de plasmídeos utilizados neste estudo.
Discussion
Imagem latente da viver-pilha de fluorescência tornou-se uma poderosa ferramenta para estudar a dinâmica spatiotemporal de células bacterianas. Microscopia de fluorescência do tempo-lapso de motile e lentas crescimento bactérias como M. xanthus, no entanto, tem sido um desafio e só foi realizada para durações de tempo curto. Aqui, apresentamos um método fácil de usar e robusto para a imagem latente de viver-pilha de M. xanthus por microscopia de fluorescência de lapso de tempo. Esse método permite que o usuário siga as células e proteínas fluorescente etiquetadas por várias rodadas do ciclo celular com resolução de célula única.
Existem vários pré-requisitos que influenciam o sucesso da imagem latente da viver-pilha de crescimento lento M. xanthus células incluindo: 1) uma superfície sólida para fixação da célula; 2) a disponibilidade de nutrientes e oxigênio; 3) constante umidade e temperatura; e 4) a otimização das condições experimentais, tais como frequência de aquisição de imagem e tempo de exposição.
Em nossa montagem experimental, utilizamos almofadas grossas agarose suplementadas com nutrientes. Usando almofadas grossas de agarose em oposição a dispositivos microfluídicos seguir células únicas tem alguns benefícios fundamentais mas também algumas desvantagens. Primeiro, a almofada de agarose não só fornece uma superfície de M. xanthus celular acessório e movimento, mas também nutrientes suficientes para o crescimento pelo menos 24 h. Em segundo lugar, snap shot análises comumente usadas para estudar a localização intracelular de proteínas fluorescente etiquetadas anteriormente foi feito no mesmo tipo de agarose almofadas16,17,29. Portanto, dados de snap shot análises podem ser directamente comparados aos dados obtidos com o método descrito aqui. Em terceiro lugar, almofadas de agarose podem ser facilmente modificadas e complementadas com antibióticos ou outros suplementos como o CuSO4 e vanillate que são comumente usados para a expressão de gene a indução18,30. Finalmente, porque as células são permitidas de forma microcolonies no decurso de uma experiência, esta configuração também permite estudar o efeito das interações célula-célula direto no parâmetro particular sendo analisado. Este aspecto é particularmente importante no caso de M. xanthus porque esta bactéria exibe várias interações de contato-dependente. A principal desvantagem deste método é que as condições experimentais são predefinidas para a duração de um experimento. Por outro lado, dispositivos microfluídicos geralmente permitem alterar as condições experimentais no decurso de uma experiência, adicionando, por exemplo, antibióticos,31.
Pacotes de software livre (por exemplo, MicrobeJ, Oufti) estão disponíveis para analisar automaticamente o crescimento de células únicas e a localização da proteína dentro das células individuais. No entanto, estes softwares são apenas well-suited para a análise de células individuais ou pequenos grupos de células. Assim, continua a ser um desafio para automaticamente analisar os dados gerados para as gravações de 24 h descritas aqui.
Em resumo, nós descrevemos um protocolo fácil de usar e pode ser reproduzido para executar com crescimento lento, imagem latente da viver-pilha M. xanthus bactérias. Nós mostramos que almofadas simples nutriente-suplementado agarose são suficientes para sustentar o crescimento pelo menos 24 h e permitir a observando e analisando a localização da proteína e o crescimento com resolução de célula única ao longo de várias gerações.
Disclosures
Os autores declaram que eles têm não tem interesses financeiro concorrente.
Acknowledgments
Este trabalho foi apoiado pelo Conselho alemão de pesquisa (DFG) no âmbito da Transregio 174 "Spatiotemporal dinâmica de células bacterianas" e a sociedade Max Planck.
Materials
| Name | Company | Catalog Number | Comments |
| DMI6000B with AFC | Leica microsystems | 11888945 | Automated inverted widefield fluorescence microscope with adaptive focus control |
| Universal mounting frame | Leica microsystems | 11532338 | Stage holder for different sample sizes |
| HCX PL FLUOTAR 100x/1.30 oil PH3 | Leica microsystems | 11506197 | Phase contrast objective |
| Orca Flash 4.0 camera | Hamamatsu | 11532952 | 4.0 megapixel sCMOS camera for picture aquisition |
| Filter set TXR ET, k | Leica microsystems | 11504170 | Fluorescence filter set, Ex: 560/40 Em: 645/75 |
| Filter set L5 ET, k | Leica microsystems | 11504166 | Fluorescence filter set, Ex: 480/40 Em: 527/30 |
| Filter set YFP ET, k | Leica microsystems | 11504165 | Fluorescence filter set, Ex: 500/20 Em: 535/30 |
| ProScan III | Prior | H117N1, V31XYZEF, PS3J100 | Microscope automation controller with interactive control center |
| EL 6000 light source | Leica microsystems | 11504115 | External fluorescence light source |
| Incubator BLX Black | Pecon | 11532830 | Black incubation chamber surrounding the microscope |
| Tempcontrol 37-2 digital | Leica microsystems | 11521719 | Automated temperature control for incubation chamber |
| Gentmycin sulphate | Carl Roth | 0233.4 | Gentamycin |
| Oxytetracylin dihydrate | Sigma Aldrich | 201-212-8 | Oxytetracyclin |
| Kanamycin sulphate | Carl Roth | T832.3 | Kanamycin |
| Filtropur BT25 0.2 bottle top filter | Sarstedt | 831,822,101 | Bottle top filter for sterilization of buffers |
| Deckgläser | VWR | 630-1592 | Glass cover slip (60 x 22 mm, thickness: 0.7 mm) |
| Seakem LE agarose | Lonza | 50004 | Agarose for microscopy slides |
| Leica Metamorph AF | Leica microsystems | 11640901 | Microscope control software and software for picture analysis |
| Tetraspeck Microsperes, 0.5 µm | ThermoFisher | T7281 | Fluorescent microspheres |
| petri dish | Greiner Bio-one | 688102 | 120 mm x 120 mm x 17 mm squared petri dish for agarose pads |
| BD Bacto Casitone | Becton Dickinson | 225930 | Casitone |
| Parafilm M | VWR | 291-1213 | Parafilm |
| Tris(hydroxymethyl)-aminomethane | Carl Roth | AE15.2 | Tris |
| Magnesium sulphate heptahydrate | Carl Roth | P027.2 | Magnesium sulphate |
| Potassium dihydrogen phosphate p.a. | Carl Roth | 3904.1 | Potassium dihydrogen phosphate |
| 1% CTT medium: 1 % (w/v) BD Bacto™ casitone, 10 mM Tris-HCl ph 8.0, 1 mM potassium phosphate buffer pH 7.6, 8 mM MgSO4 | Cultivation medium for M.xanthus | ||
| TPM buffer: 10 mM Tris-HCl ph 8.0, 1 mM potassium phosphate buffer pH 7.6, 8 mM MgSO4 | Buffer for preparation of microscopy slides for M.xanthus |
References
- Shapiro, L., McAdams, H. H., Losick, R. Why and how bacteria localize proteins. Science. 326 (5957), 1225-1228 (2009).
- Treuner-Lange, A., Søgaard-Andersen, L. Regulation of cell polarity in bacteria. J Cell Biol. 206 (1), 7-17 (2014).
- Laloux, G., Jacobs-Wagner, C. Spatiotemporal control of PopZ localization through cell cycle-coupled multimerization. J Cell Biol. 201, 827-841 (2013).
- Rudner, D. Z., Losick, R. Protein subcellular localization in bacteria. Cold Spring Harb. Perspect. Biol. 2 (4), 000307 (2010).
- Badrinarayanan, A., Le, T. B. K., Laub, M. T. Bacterial chromosome organization and segregation. Annu Rev Cell Dev Biol. 31 (1), 171-199 (2015).
- Munoz-Dorado, J., Marcos-Torres, F. J., Garcia-Bravo, E., Moraleda-Munoz, A., Perez, J. Myxobacteria: Moving, Killing, Feeding, and Surviving Together. Front Microbiol. 7, 781 (2016).
- Berleman, J. E., Kirby, J. R. Deciphering the hunting strategy of a bacterial wolfpack. FEMS Microbiol Rev. 33 (5), 942-957 (2009).
- Konovalova, A., Petters, T., Søgaard-Andersen, L. Extracellular biology of Myxococcus xanthus. FEMS Microbiol. Rev. 34, 89-106 (2010).
- Nudleman, E., Wall, D., Kaiser, D. Cell-to-cell transfer of bacterial outer membrane lipoproteins. Science. 309, 125-127 (2005).
- Vassallo, C. N., et al. Infectious polymorphic toxins delivered by outer membrane exchange discriminate kin in myxobacteria. eLife. 6, 29397 (2017).
- Vassallo, C., et al. Cell rejuvenation and social behaviors promoted by LPS exchange in myxobacteria. Proc Natl Acad Sci USA. 112 (22), 2939-2946 (2015).
- Li, Y., et al. Extracellular polysaccharides mediate pilus retraction during social motility of Myxococcus xanthus. Proc. Natl. Acad. Sci. USA. 100, 5443-5448 (2003).
- Kim, S. K., Kaiser, D. Cell alignment required in differentiation of Myxococcus xanthus. Science. 249, 926-928 (1990).
- Lobedanz, S., Søgaard-Andersen, L. Identification of the C-signal, a contact dependent morphogen coordinating multiple developmental responses in Myxococcus xanthus. Genes Dev. 17, 2151-2161 (2003).
- Schumacher, D., Søgaard-Andersen, L. Regulation of cell polarity in motility and cell division in Myxococcus xanthus. Annu Rev Microbiol. 71 (1), 61-78 (2017).
- Schumacher, D., et al. The PomXYZ proteins self-organize on the bacterial nucleoid to stimulate cell division. Dev Cell. 41 (3), 299-314 (2017).
- Treuner-Lange, A., et al. PomZ, a ParA-like protein, regulates Z-ring formation and cell division in Myxococcus xanthus. Mol Microbiol. 87 (2), 235-253 (2013).
- Iniesta, A. A., Garcia-Heras, F., Abellon-Ruiz, J., Gallego-Garcia, A., Elias-Arnanz, M. Two systems for conditional gene expression in Myxococcus xanthus inducible by isopropyl-beta-D-thiogalactopyranoside or vanillate. J Bacteriol. 194 (21), 5875-5885 (2012).
- Harms, A., Treuner-Lange, A., Schumacher, D., Søgaard-Andersen, L. Tracking of chromosome and replisome dynamics in Myxococcus xanthus. reveals a novel chromosome arrangement. PLoS Genet. 9 (9), 1003802 (2013).
- Iniesta, A. A. ParABS system in chromosome partitioning in the bacterium Myxococcus xanthus. PLoS One. 9 (1), 86897 (2014).
- Lin, L., Osorio Valeriano, M., Harms, A., Søgaard-Andersen, L., Thanbichler, M. Bactofilin-mediated organization of the ParABS chromosome segregation system in Myxococcus xanthus. Nat Commun. 8 (1), 1817 (2017).
- Hodgkin, J., Kaiser, D. Cell-to-cell stimulation of movement in nonmotile mutants of Myxococcus. Proc Natl Acad Sci U S A. 74 (7), 2938-2942 (1977).
- Kaiser, D. Social gliding is correlated with the presence of pili in Myxococcus xanthus. Proc Natl Acad Sci USA. 76 (11), 5952-5956 (1979).
- Miertzschke, M., et al. Structural analysis of the Ras-like G protein MglA and its cognate GAP MglB and implications for bacterial polarity. EMBO J. 30 (20), 4185-4197 (2011).
- Hodgkin, J., Kaiser, D. Genetics of gliding motility in Myxococcus xanthus. (Myxobacterales): Two gene systems control movement. Mol Gen Genet. 171, 177-191 (1979).
- Shi, X., et al. Bioinformatics and experimental analysis of proteins of two-component systems in Myxococcus xanthus. J Bacteriol. 190 (2), 613-624 (2008).
- Bi, E. F., Lutkenhaus, J. FtsZ ring structure associated with division in Escherichia coli. Nature. 354 (6349), 161-164 (1991).
- Lutkenhaus, J., Pichoff, S., Du, S. Bacterial cytokinesis: From Z ring to divisome. Cytoskeleton. 69 (10), 778-790 (2012).
- McLoon, A. L., et al. MglC, a Paralog of Myxococcus xanthus GTPase-Activating Protein MglB, Plays a Divergent Role in Motility Regulation. J Bacteriol. 198 (3), 510-520 (2015).
- Gomez-Santos, N., et al. Comprehensive set of integrative plasmid vectors for copper-inducible gene expression in Myxococcus xanthus. Appl Environ Microbiol. 78 (8), 2515-2521 (2012).
- Treuner-Lange, A., et al. The small G-protein MglA connects to the MreB actin cytoskeleton at bacterial focal adhesions. J Cell Biol. 210 (2), 243-256 (2015).
