1. Field Site Description
- Describe experimental field sites during collection periods. Determine the location of field (latitude, longitude, and altitude) using a GPS.
- Describe the sampling depth, sampling time, and soil texture.
- Environmental factors play important roles in shaping microbial communities. Record climate information such as annual mean temperature, annual precipitation, prior years’ crop rotation, tillage practices, method of fertilization, and history of the field site. Automated weather stations or other devices are useful to record the daily rainfall and temperature over a growing season.
2. Collection and Processing of Soil, Rhizosphere, and Root Field Samples
- Excavation of plants.
- Label a wash pan and bucket with a sticky note containing information about the plant material to be sampled. Include information such as plot number, plant genotype, and plant species. Carry the labeled bucket to the plot and leave the wash pan behind at the established workstation in the field.
- Pierce soil with a shovel to a depth of 30 cm to cut any of the lateral roots holding the plant in the soil. The approximate volume is 18 cm3. Randomly choose and collect two plants per plot from different areas within the plot.
- Excavate the plant roots by leveraging the shovel and place the root ball in the labeled bucket. Bring the labeled bucket with the excavated root ball back to the workstation in the field. Cut off and discard the aboveground plant biomass.
- Removal of soil from roots and collection of bulk soil.
- Shake roots to manually remove soil or use a spade or a hand-held tiller to remove soil from the roots. Shaking the roots is sufficient to remove soil in very sandy soils. Wear gloves and place roots near the processing station.
- After shaking the roots, the bulk of the soil will be in the wash pan. Mix the soil in the wash pan and break up any soil clods with a hand-held tiller. Place a sample of soil that is free of debris into a labeled, 17.7 x 19.5 cm zipper storage bag and place in a cool place or on ice.
- Collection of roots and rhizosphere.
- Using pruning scissors sterilized in 70% EtOH, excise a variety of roots, approximately 4 to 6 roots per plant and each root around 9 – 12 cm in length. Place the excised roots (cutting as needed to fit) in a labeled 50 mL tube containing 35 mL of autoclaved, phosphate buffer (6.33 g/L NaH2PO4, 8.5 g/L Na2HPO4 anhydrous, pH = 6.5, 200 µl/L surfactant).
Note: The surfactant (see Table of Materials) was added after autoclaving the phosphate buffer. The volume of the root ball and root lengths vary depending on plant age and plant species. - Shake tubes for 2 min to release the rhizosphere from the surface of the roots. With forceps sterilized in 70% EtOH, remove the roots from the tube, blot briefly on paper towels, and place in a new, labeled 50 mL tube. Place both the tube containing the rhizosphere and the one containing roots on ice.
- Using pruning scissors sterilized in 70% EtOH, excise a variety of roots, approximately 4 to 6 roots per plant and each root around 9 – 12 cm in length. Place the excised roots (cutting as needed to fit) in a labeled 50 mL tube containing 35 mL of autoclaved, phosphate buffer (6.33 g/L NaH2PO4, 8.5 g/L Na2HPO4 anhydrous, pH = 6.5, 200 µl/L surfactant).
3. Processing of Field Samples in the Laboratory
- Surface sterilization of roots after field collection.
Note: Perform surface sterilization as soon as possible after returning from the field. If it is not possible to surface sterilize the roots in the same day, store roots at 4 °C until processed.- Add approximately 35 mL of 50% bleach + 0.01% Tween 20 to 50 mL root tubes collected in the field. Shake 50 mL tubes for 30 to 60 seconds. Pour off 50% bleach and add 35 mL of 70% EtOH. Shake for another 30 to 60 seconds.
- Pour off 70% EtOH and add 35 mL of sterile, ultrapure water. Shake for 1 minute. Repeat the water wash two more times.
Note: To ensure our surface sterilization treatment is sufficient, we plated samples of water from the last rinse and observed no growth of bacteria (P. Wang, unpublished data, 2014). Other investigators have tested for root sterilization efficiency using similar methods9,10. - Blot roots dry on clean paper towels. Use a clean paper towel for each sample.
Note: Paper towels can be sterilized before use. We do not sterilize our paper towels. However, we use 1 paper towel per sample and keep towels wrapped until used. - Using sterile forceps and pruning scissors, cut the roots into approximately 5 mm pieces and place cut roots in a clean, labeled 15 mL conical tube. Store samples at -80 °C until further processed.
- Processing rhizosphere samples.
- Shake 50 mL tubes containing rhizosphere samples from the field to resuspend the entire sample. Using a sterile, 100-µm-mesh cell strainer (see Table of Materials), filter the resuspended sample into a new 50 mL tube.
- Centrifuge the tubes at 3000 x g for 5 min at room temperature. Immediately pour off and discard the supernatant.
- Place rhizosphere pellets in the 50 mL tubes on ice. Add 1.5 mL of sterile phosphate buffer (without surfactant) to rhizosphere pellets and vortex to suspend.
- Pipet the suspended liquid into a clean, labeled, 2 mL microfuge tube. Spin tubes at 15,871 x g for 2 min at room temperature. Immediately pour off the supernatant and drain the tubes on clean paper towels.
- Store the pellets at -20 °C until further processed.
Note: Steps 3.2.2 – 3.2.4 are done to reduce the sample tube size for storage. It is more space efficient to store the smaller 2 mL tubes compared to the 50 mL tubes.
- Processing soil samples for DNA extraction and soil analysis.
- Using a sterile metal spatula, fill a clean, labeled 2 mL tube with approximately 3 g of soil for DNA extraction. Avoid any small root pieces and debris. Store this soil sample at -20 °C. Rinse metal spatula in 70% EtOH between each sample.
- In a clean wash pan, empty the bag of soil into stacked sieves (see Table of Materials), larger sieve on top of the smaller sieve, and manually sieve the soil through both sieves. Use a brush to carefully clean the sieves between samples.
- Set aside 100 to 125 g of sieved soil in a 17.7 x 19.5 cm zippered bag for future soil physicochemical and texture analysis. Place the bags of soil at 4 °C for short-term storage.
- Processing soil samples for soil moisture.
- Tare a labeled brown paper bag on a scale. Measure 40 to 45 g of sieved soil into the brown paper bag. Record the weight of the brown paper bag and soil on a data sheet and place bags in a drying oven set to 55 – 60 °C.
- After 72 hours, remove the bags from the drying oven. Allow the soil bags to cool for at least 30 min and then weigh.
- To record the weight, tare the scale to zero and place the brown paper bags on the scale. Calculate the percentage of soil moisture for each sample using the formula:

Note: The soil moisture method is from Kellogg Biological Station Long Term Ecological Research (KBS LTER). KBS LTER provides a wide range of established protocols for researchers on their website (https://Lter.kbs.msu.edu/).
4. Preparation of Processed Root Samples for DNA Extractions.
- Grind frozen root material so that the sample is homogenous.
- Pour liquid nitrogen in a plastic beaker with clean spatulas and in a clean mortar and pestle to keep samples frozen throughout the grinding process.
- Place the frozen tissue in the mortar and grind with the pestle into a fine powder. Continually add liquid nitrogen throughout grinding to keep samples frozen.
- Use a spatula to place ground tissue in a clean, labeled 2 mL tube. Store at -80 °C.
5. Extraction of DNA from Soil and Rhizosphere Samples in 96-well Format
- Loading soil samples into 96-well plates.
- Wipe down the work area with 70% EtOH and 1% bleach. Wear lab gloves during these steps. Remove the soil samples from -20 °C storage and allow to thaw in an ice bucket.
- Remove the sealing mat cover from a 96-well extraction plate that is provided with the DNA extraction kit. Place the sealing mat cover between 2 paper wipes to keep clean while not in use. To avoid contamination, use adhesive 8-well PCR strips to cover the 12 columns of the 96-well extraction plate.
- Tare a sterile weigh-funnel (size SM) on a scale and weigh out 200 to 250 mg of soil.
Note: These sterile funnels are flattened on one side, so the funnel lays flat on a scale. The funnel is filled with soil and placed directly into a well. This technique avoids the loss of samples, minimizes spillage, and prevents cross-contamination. - To uncover the first well of the extraction plate, carefully lift the adhesive strip up, place the neck of the filled weigh-funnel into the appropriate well, and gently guide the soil sample into the appropriate well. Replace the adhesive strip to cover the well.
- Repeat this process for every well of the plate, using a new, sterile funnel for each sample until the plate is filled. Leave one well empty as an extraction blank control.
Note: One well is left blank on every extraction plate to serve as a negative (blank) control. This controls for contaminants that may be present in kit reagents11. - Replace the sealing mat cover on the extraction plate and store the plate at -20 °C until ready for DNA extraction.
- Loading rhizosphere samples into 96-well plates.
- Remove the rhizosphere samples from -20 °C storage and allow to thaw in an ice bucket.
- Follow step 5.1.2 above to prepare the DNA extraction plate.
- Place a clean paper wipe on a scale, then tare a sterile metal spatula on the scale. Use the spatula to carefully scoop out some of the rhizosphere pellet from a sample tube. Return the spatula to the scale and weigh between 200 and 250 mg of the rhizosphere sample.
- Carefully lift the adhesive strip to uncover the first well of the extraction plate, angle the filled spatula into the well, and scrape off the rhizosphere material into the appropriate well with a sterile toothpick.
- Rinse the metal spatula in water, followed by 70% EtOH between samples. Repeat this process for every well of the plate until the plate is filled, leaving one well empty as an extraction blank control.
- Extraction of DNA from rhizosphere and soil samples in 96-well format.
- Extract soil and rhizosphere DNA using a kit optimized for soil (see Table of Materials), following manufacturer’s protocol.
Note: We use this specific kit to isolate soil and rhizosphere DNA because of the ability of the proprietary reagents to remove humic acid and other powerful PCR inhibitors found in soil.
- Extract soil and rhizosphere DNA using a kit optimized for soil (see Table of Materials), following manufacturer’s protocol.
- DNA Quantification.
- Quantify 92 samples and 4 concentrations of standards using a kit (see Table of Materials; standards are included), according to manufacturer’s protocol.
- Quantify the remaining 4 samples that were removed from the plate to accommodate wells for the four standards using a kit (see Table of Materials), according to manufacturer’s protocol.
6. Extraction of DNA from Root Samples in 96-well Format.
- Loading root samples into 96-well plates.
- Wipe down the work area with 70% EtOH and 1% bleach. Wear gloves during these steps.
- Keep ground root samples frozen at all times by placing samples in a bucket with dry ice.
- Fill a plastic beaker with liquid nitrogen and place antistatic microspatulas and sterile weigh-funnels (size XSM) in the plastic beaker to cool down.
- Remove the sealing mat cover from the 96-well extraction bead plate that is provided with kit and place it between 2 paper wipes to keep clean while not in use.
Note: The newer version of this kit, released subsequent to the time we performed these DNA extractions, requires the user to supply the extraction bead plates. In the Table of Materials, we have listed the vendor catalog information needed to order these items. - To avoid contamination, use adhesive 8-well PCR strips to cover the 12 columns of the 96-well extraction plate.
- Place the extraction bead plate on dry ice to keep samples in the wells frozen.
- Carefully lift the adhesive strip up to uncover the first well of the extraction plate, place the neck of the filled weigh-funnel into the appropriate well, and add 3 spatula scoops of ground root tissue. Replace the adhesive strip to cover the well.
Note: The soil and rhizosphere are thawed on ice prior to weighing, whereas plant material is weighed out frozen. Frozen plant tissue, especially small amounts, is difficult to weigh on a scale without thawing. Weigh tests were done on ground root tissue to determine how many spatula scoops were sufficient. Note that the manufacturer of the kit does not require an exact amount of tissue but recommends around 50 mg. Different plant sample types will vary and the user will need to determine the appropriate amount. - Repeat this process for every well of the plate until the plate is filled.
- Store plate at -20 °C until ready for DNA extraction.
- DNA extraction of root tissue.
- Extract DNA using a kit optimized for plants (see Table of Materials) following the manufacturer's protocol.
Note: We use this kit that is designed specifically for plant tissue to achieve maximum yields from the root samples. Unlike soil and rhizosphere, the humic acid and other contaminants are less of a problem for the root tissue.
- Extract DNA using a kit optimized for plants (see Table of Materials) following the manufacturer's protocol.
- Quantify the DNA as in step 5.4.
7. Amplification and Sequencing the Isolated DNA.
- Amplify the V4 region of the 16S gene with a proof reading polymerase (see Table of Materials) as described in Gohl et al.12 Barcode samples with different indexing primers and pool before sequencing. Sequence using the methods described by Gohl et al12 using a two step PCR process with V4 primers (Supplemental File 1).
Note: Some of the methods for these steps are described in detail elsewhere12,13,14 and therefore will not be described here. For the root samples, PNA blockers were added to reduce the amount of plastid DNA amplified from the plant tissue, which has previously been fully described15. Two controls were used in sequencing, a negative control which included the extraction plate blank controls (see note after step 5.1.5), and a mock community of a known population of bacterial DNA (see Table of Materials) which served as a positive control.
Note: In most cases, the MiSeq Reagent Kits v3 are used in the 2 X 300 base paired end mode. For the samples in this manuscript, the Illumina HiSeq 2500 was used in rapid mode with 250 Paired-end (2 x 250) mode. All the sample were sequenced in the same lane. - Process the sequencing data through a pipeline for microbial community analyses (USEARCH v9.2.64, QIIME v1.9.1 and RStudio v3.4.3 16).
- Prepare the sequencing data using USEARCH17.
Note: USEARCH is available online with full instructions (https://www.drive5.com/usearch/). - Demultiplex the sequencing data using index reads or barcodes to assign Illumina reads to samples.
- Merge the paired-end reads to get consensus sequences. Use the command: usearch -fastq_mergepairs *R1*.fastq -relabel @ -fastq_maxdiffs 10 -fastq_minmergelen 230 -fastq_maxmergelen 320 -fastq_pctid 80 -fastqout merged.fq.
Note: The parameters are set through referencing the USEARCH instruction manual. - Remove the primers from the sequencing data to avoid substitutions in the primer sequences, which may be caused by the PCR reaction. Use the command: usearch -fastx_truncate merge.fq -stripleft 19 -stripright 20 -fastqout stripped.fq.
- Filter sequencing data to remove the low-quality reads and keep high quality operational taxonomic unit (OTU) sequences. Use the command: usearch -fastq_filter stripped.fq -fastq_maxee 1.0 -fastaout filtered.fa.
- Prepare the sequencing data using USEARCH17.
- Generate OTUs in USEARCH.
- Perform the dereplication to identify the set of unique OTU sequences. Use the command: usearch -fastx_uniques filtered.fa -fastaout uniques.fa -sizeout -relabel Uniq.
- Cluster OTUs with 97 – 100% sequence similarity to designate the unique OTUs. Use the command: usearch -cluster_otus uniques.fa -minsize 2 -otus otus.fa -relabel Otu.
Note: This step also incorporates the removal of singletons from the clustered OTUs and removal of chimeras from sequencing data. - Create an OTU table in USEARCH. Use the command: usearch -usearch_global stripped.fq -db otus.fa -strand plus -id 0.97 -otutabout otutable.txt.
Note: This command generates a table with the number of reads (counts) of all OTUs for each sample. The OTU table is used for downstream steps including differential abundance analyses and microbial diversity analyses. Example of OTU table is shown in supplemental figure 2. - Conduct a rarefaction analysis in QIIME v1.9.1 18.
Note: The rarefaction curve is calculated using the OTU table to determine whether the depth of the sequencing properly samples the microbial community. Example of rarefaction curves are shown in Supplemental Figure 3. - Conduct an alpha diversity analysis18. Use alpha_rarefaction.py in QIIME v1.9.1 to calculate the diversity of the microbial community within each sample.
Note: This analysis calculates diversity indices such as Shannon19, Simpson20, and Chao121. - Conduct a beta diversity analysis18,22. Use the Python script: beta_diversity_through_plots.py in QIIME v1.9.1 Bray–Curtis dissimilarity matrix.
Note: This analysis compares the microbial community composition between samples. - Conduct statistical analyses between groups. Use the distance matrices calculated for PERMONOVA using the adonis and anova function in the vegan package23 v2.4.5 in RStudio16. Conduct canonical analysis of principal coordinates (CAP) analysis using the capscale function in the vegan package. Visualize data using ggplot2 package24 v2.2.1 in RStudio.
The representative results presented in this manuscript come from a field site established in 2012 at the University of Nebraska-Lincoln Agriculture Research Division Farm near Mead, NE. Prior to the experiment, the site had been managed as a corn-soybean rotation. The study site was located on three different types of soils, but the data was analyzed as if all changes in measured soil properties were due to the treatments imposed.
The field site contained two, pure, stands of switchgrass (P. virgatum cv Liberty) and big bluestem (A. gerardii) as well as a low-diversity grass mixture containing big bluestem, indiangrass (S. nutans), and 'Butte' sideoats grama (B. curtipendula). The three warm-season grass plots were in a randomized complete block design that was replicated three times. Nested into the three different grass plots were two nitrogen (N) fertilization treatments, which were 56 (N1) and 112 (N2) kg N ha-1 of applied urea. At the time of microbiome sampling at the end of the growing season, the soil contained 8.0 ± 1.1 (mean ± SD) ppm nitrate in the plots fertilized with 112 kg N ha-1 and 6.8 ± 0.7 (mean + SD) ppm nitrate in the plots fertilized with 56 kg N ha-1. The plots had been fertilized once a year. The warm-season grass plots were designated as the main plots (8000 m2) and N treatments were the split plots (4000 m2). Big bluestem was seeded as a 50:50 blend of 'Bonanza' and 'Goldmine' and Indiangrass was seeded as a 50:50 blend of 'Scout' and 'Warrior'. The plots were planted in 2012, and the first N application occurred in the spring of 2013.
The soil and root sampling was conducted on September 15, 2014. The work described below was conducted on a field that was set up as a split-plot randomized design with three replicates (Figure 1). The average sequencing depth of all the samples for endosphere were as follows: 4871 ± 5711 (mean ± SD), rhizosphere: 40726 ± 14684, soil: 38184 ± 9043. One of the largest sources of variation in these experiments, using the methods described, is the difference in microbial communities found between sample types (Figure 2). In this representative data set, the rhizosphere and soil appear to be more similar in composition to each other than the endosphere (Figure 2A). However, there were also highly significant (p = 0.001) differences in microbial community composition between rhizosphere and soil (Figure 2B). The total variation accounted for in these experiments analyzed by sample type was 26%.
Alpha diversity analysis showed that the microbial communities in the endosphere were lower in sample diversity as compared to soil and rhizosphere (Figure 3). The only significant differences in diversity between the grass species in any compartment were between the endosphere samples of big bluestem and switchgrass, with switchgrass having significantly higher microbial species diversity (Figure 3). The relative abundance analysis (Figure 4) highlights the dominance of Proteobacteria followed by Actinobacteria in all sample types. Soil and rhizosphere are also dominated by Acidobacteria and Chloroflexi whereas the endosphere had a larger relative abundance of Bacteriodetes.
In this experiment, plants were grown with two different amounts of N fertilizer and therefore we analyzed the data to determine whether there were treatment effects. Treatment effects accounted for 12% of the total variation but were not significantly different although in the ordination the two treatments look different (Figure 5). This highlights the importance of statistical analyses for these datasets rather than visual inspection or qualitative judgments.
Plant-influenced differences in the microbiome of plant tissues and soil were visualized using a constrained method of ordination. Statistical differences were determined using a PERMANOVA analysis to test whether specific variables, such as species, result in significantly different microbial community composition between samples. When all the sample types were analyzed together, a highly significant difference was found in microbial community composition due to plant species (Figure 6). In this experiment, the amount of variation accounted for by plant species was 6.7%. Finally, each sample type was analyzed individually to determine which of the sample types might be driving the significant plant species effect. Only in the endosphere was there a highly significant difference (p = 0.001) between the microbial community compositions of the different plant species (Figure 7). In the other sample types, the species effect was not significant when analyzed individually. In the endosphere, the percent variation due to species was 27%, whereas it was lower in rhizosphere (18%) and soil (15%). This further highlights the importance of analyzing each tissue type individually.

Figure 1: Example of the experimental field design. Experimental field design illustrating a randomized complete block design in triplicate of the field site located at the University of Nebraska-Lincoln Eastern Nebraska Research and Extension Center near Mead, NE. For full site description see the Results section. N1 is the low (56 kg N ha-1 urea) and N2 (112 kg N ha-1 urea) is the higher nitrogen rate that was applied. Please click here to view a larger version of this figure.
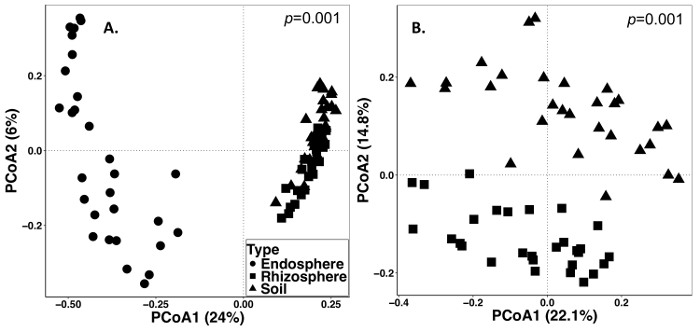
Figure 2: Beta diversity analysis comparing the microbial composition in the different sample types including endosphere, rhizosphere, and soil from the perennial grass sampling in 2014. The analysis was carried out using a Python script in QIIME1.9.1 to produce the Bray-Curtis dissimilarity matrix. Principal coordinates analysis (PCoA) based on the Bray-Curtis dissimilarity matrix was visualized in RStudio. PCoA1 and PCoA2 indicate the first and second largest variance explained by the PCoA analysis. PERMANOVA statistical analysis was performed to determine the significance between sample types, and the p value is shown on the top right corner. Each symbol in the figures represent the entire microbial community for each sample. (A) Endosphere, rhizosphere and soil sample types were analyzed together. All 87 samples were rarefied to 486 sequences per sample. (B) Rhizosphere and soil samples were analyzed together. All 59 samples were rarefied with 8231 sequences. Please click here to view a larger version of this figure.
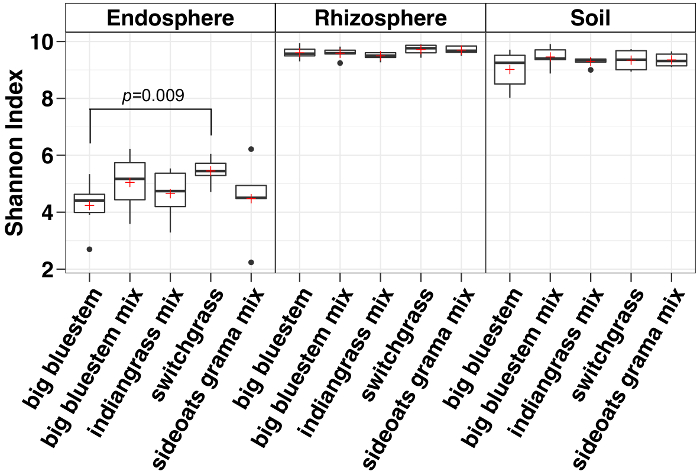
Figure 3: Alpha diversity analysis using Shannon index for each species in the endosphere, rhizosphere and soil. The analysis was carried out using a Python script in the QIIME1.9.1. Rarefaction was done for the endosphere, rhizosphere, and soil sample types respectively with 486, 17154 and 8231 sequences per sample. Boxes indicate the 25th and 75th percentiles (first and third quartiles). The horizontal line within the box denotes the median and the red plus shows the mean. Whiskers show the range of the data excluding outliers (which are shown as black dots) that fell more than 1.5 times the interquartile range (n = 6 for each sample except for sideoats grama mix where n = 5). The Shannon index of all five species in the endosphere were lower than both rhizosphere and soil. Non-parametric Wilcoxon rank sum test was used to determine the significance between the species and only significant differences between species were shown on top of the boxes. Please click here to view a larger version of this figure.

Figure 4: Relative abundance on the phylum level in the endosphere, rhizosphere, and soil. Samples were analyzed to compare the abundance of microbial phyla among different samples types (n = 29 for each sample type). The analysis was carried out using a Python script in QIIME1.9.1 from the OTU table. The different colors inside the pie chart denote the phyla. The percentage indicates the relative abundance of each phylum in each sample type. The phylum information was annotated using the Ribosomal Database Project classifier (RDP)25. Please click here to view a larger version of this figure.

Figure 5: Analysis using treatment as constraining factor between all sample types. Canonical analysis of principal coordinates (CAP) analysis was performed to determine whether there were differences in microbial community composition between treatments. For each N treatment, n = 42 for N1 (56 kg N ha-1) and n = 45 for N2 (112 kg N ha-1). The Bray-Curtis dissimilarity matrix was generated using a python script in the QIIME1.9.1. CAP analysis based on the Bray-Curtis dissimilarity matrix was done by constraining the treatment as the factor in RStudio. PERMANOVA analysis was performed to determine whether treatment differences were significant, and the p value is shown on the top right corner. Please click here to view a larger version of this figure.

Figure 6: Analysis using plant species as constraining factor between all sample types. Analysis was conducted to determine whether there were differences in the microbial community composition between plant species in all sample types. Principal coordinates ordination and CAP analysis of all sample types (endosphere, rhizosphere, and soil) were done using a Bray-Curtis dissimilarity matrix. The Bray-Curtis dissimilarity matrix was generated using the Python script in QIIME1.9.1. CAP analysis based on the Bray-Curtis dissimilarity matrix was done by constraining the plant species as the factor in RStudio. PERMANOVA statistical analysis was performed to determine the significance between plant species, and the P value is shown on the top right corner. Each symbol in the figures represents the entire microbial community for that sample. n = 18 for each species in all sample types except n = 15 for the sideoats grama mix. Please click here to view a larger version of this figure.

Figure 7: Example of CAP analysis using species as constraining factor for each sample type individually. Principal coordinate ordination and CAP analysis of each sample type (endosphere, rhizosphere, and soil) using Bray-Curtis dissimilarity matrix. Each sample type was rarefied to 486, 17154, and 8231 reads per sample respectively in endosphere, rhizosphere and soil. Species was used as the factor to constrain the ordination. PERMANOVA statistical analysis was performed to determine the significance between plant species in each sample type, and the p value is shown on the top right corner. Each symbol in the figure represents the entire microbial community for each sample. Sample size is n = 29 for each sample type, n = 6 for each plant species in each sample type except for the sideoats grama mix (n = 5). Please click here to view a larger version of this figure.
