Atteindre des pressions modérées dans des récipients fermés hermétiquement à l’aide de neige carbonique comme Source solide CO2
Summary
Nous présentons ici un protocole permettant d’effectuer des réactions dans les cuves de réaction simple sous des pressions de faible à modéré de CO2. Les réactions peuvent être effectuées dans une variété de navires simplement en administrant le dioxyde de carbone sous forme de glace sèche, sans besoin de matériel coûteux ou élaboré ou Set-up.
Abstract
Ici est présenté une stratégie générale pour effectuer des réactions sous légère à modérée CO2 pressions à la glace sèche. Cette technique élimine le besoin d’équipement spécialisé atteindre des pressions modestes et peut même être utilisée pour atteindre des pressions plus élevées dans l’équipement plus spécialisé et cuves de réaction plus robustes. À la fin de la réaction, les flacons peuvent facilement être dépressurisés par ouverture à température ambiante. Dans l’exemple présent CO2 est aussi bien un groupe dirigeant présumé comme un moyen pour passiver les substrats amine, empêchant ainsi l’oxydation au cours de la réaction organométallique. En plus d’être facilement ajoutés, le groupe dirigeant est également supprimé sous vide, qui éviterait la purification Poussée à supprimer le groupe dirigeant. Cette stratégie permet l’arylation γ-C(sp3)-H facile des amines aliphatiques et a le potentiel pour être appliqué à une variété d’autres réactions à base d’amines.
Introduction
L’utilisation de composés gazeux dans les réactions chimiques nécessite généralement un équipement spécialisé et procédures1,2. À l’échelle de banc, certains gaz peut être ajoutés directement d’un réservoir à l’aide d’un régulateur de haute pression3. Une autre méthode consiste à condenser le gaz sous conditions cryogéniques4,5. Bien qu’utiles, ces stratégies nécessitent l’utilisation de réacteurs de pression spécialisés avec des soupapes, ce qui peuvent représenter un coût prohibitif pour l’exécution en parallèle de nombreuses réactions. Cela peut donc considérablement ralentir la vitesse à laquelle réaction dépistage peut procéder. En conséquence, les chimistes ont trouvé souhaitable d’introduire ces composés en utilisant des méthodes alternatives. L’ammoniac peut être ajouté aux réactions à l’aide de sels de carboxylate de différentes d’ammonium, en profitant de la faible équilibre entre ces sels et l’ammoniac libre6. Hydrogénation de transfert est une stratégie importante pour des réactions de réduction des oléfines, carbonyle et groupes nitro qui contourne l’utilisation du gaz d’hydrogène inflammable avec des composés tels que le formiate d’ammonium ou l’hydrazine comme transporteurs de H27. Un autre gaz d’intérêt dans ce domaine est le monoxyde de carbone8 – CO peut être généré in situ par libération de carbonyles métalliques complexes9,10, ou alternativement, il peut être généré par décarbonylation de sources telles que les formates et formamides11,12,13 ou chloroforme14,15.
Un gaz qui ne connaît pas de développement significatif à cet égard est le dioxyde de carbone,16. Une raison à cela est que les nombreuses transformations qui impliquent de CO2 exigent également des pressions et des températures élevées et donc sont automatiquement reléguées aux réacteurs spécialisés17,18. Efforts récents pour élaborer des catalyseurs plus réactifs, cependant, ont facilité l’exécution de bon nombre de ces réactions sous pressions atmosphériques de CO219,20,21,22. Nous avons récemment découvert une réaction dans laquelle le dioxyde de carbone pourraient servir d’arbitrer la γ-C (sp3) – H arylation des amines aliphatiques23. Cette stratégie devait combiner les avantages d’une approche statique groupe dirigeant dont amide24,25,26,27,28, sulfonamide 29 , 30 , 31 , 32,33,de thiocarbonyle34ou hydrazone35-base de diriger des groupes (robusticity chimique), avec la facilité d’un groupe dirigeant transitoire (étape une diminution économie)36, 37,38,,39.
Bien que la réaction peut se produire sous une pression atmosphérique de CO2, la nécessité d’une mise en place de Schlenk aux réactions d’écran s’est avérée beaucoup trop lent. En outre, augmentant la pression légèrement conduite à améliore les rendements des réactions, mais ne pouvait être facilement réalisé à l’aide d’une ligne de Schlenk. Nous avons donc cherché une autre stratégie, et par la suite identifié cette glace carbonique pourrait être facilement utilisé comme une source solide de CO2 qui pourraient être ajoutées à une variété de récipients d’introduire la quantité nécessaire de dioxyde de carbone pour atteindre modérée pressions (Figure 1). Bien que sous-utilisé dans la synthèse, une stratégie similaire est assez courante comme une méthode permettant de générer des liquides de CO2 d’extraction et chromatographie demandes40,41,42,43, 44. Utilisant cette stratégie a permis que notre groupe rapidement écran un grand nombre de réactions en parallèle, tandis que la capacité d’accès CO2 pressions modérées d’entre 2 à 20 atmosphères était essentiel pour améliorer les rendements des réactions. Dans ces conditions, les primaires (1°) et les amines secondaires (2°) peuvent être accès avec riche en électrons et halogénures d’aryles pauvre électron.
Protocol
Representative Results
Discussion
À l’aide de van der Waals équation d’État, la pression approximative de ces systèmes peut être calculée45
Équation 1 :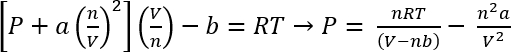
Dans les conditions dans le protocole 1, nous pouvons supposer 26,3 mg de CO2 donne n = 5,98 x 10-4 mols
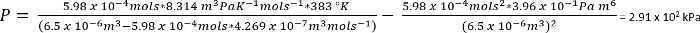
Disclosures
The authors have nothing to disclose.
Acknowledgements
Les auteurs souhaitent reconnaître des fonds de démarrage de l’Université de Toledo, ainsi que des fonds de la Fondation de l’American Chemical Society Herman Frasch dans la prise en charge partielle de cet ouvrage. M. Thomas Kina est reconnu pour son aide à l’élaboration d’un manomètre adapté pour mesurer les pressions de la réaction. M. Steve Modar est remerciée pour des discussions fructueuses.
Materials
| 7.5 mL Sample Vial with Screw Cap (Thermoset) | Qorpak | GLC-00984 | Can be reused. |
| 40 mL Sample Vial with Screw Cap (Thermoset) | Qorpak | GLC-01039 | Can be reused. |
| Pressure Tube, #15 Thread, 7" Long, 25.4 mm O.D. | Ace Glass | 8648-06 | Can be reused. |
| Pie-Block for 2 Dram Vials | ChemGlass | CG-1991-P14 | Can be reused. |
| Pie-Block for 10 Dram Vials | ChemGlass | CG-1991-P12 | Can be reused. |
| 3.2 mm PTFE Disposable Stir Bars | Fisher | 14-513-93 | Can be reused. |
| C-MAG HS 7 Control Hotplate | IKA | 20002695 | |
| Analytical Weighing Balance | Sartorius | QUINTIX2241S | |
| Double-Ended Micro-Tapered Spatula | Fisher Scientific | 21-401-10 | |
| Hei-VAP Advantage – Hand Lift Model with G5 Dry Ice Condenser Rotary Evaporator | Heidolph | 561-01500-00 | |
| Bump Trap 14/20 Joint | ChemGlass | CG-1322-01 | |
| tert-Amyl amine | Alfa Aesar | B24639-14 | Used as received. |
| 2-Methyl-N-(3-methylbenzyl)butan-2-amine | N/A | N/A | Prepared from reductive amination of tert-amyl amine and 3-tolualdehyde in the presence of sodium borohydride in methanol. |
| Palladium Acetate | Chem-Impex International, Inc. | 4898 | Used as received. |
| Silver Trifluoroacetate | Oakwood Chemicals | 007271 | Used as received. |
| Phenyl Iodide | Oakwood Chemicals | 003461 | Used as received. |
| Acetic Acid | Fisher Chemical | A38 | Used as received. |
| 1,1,1,3,3,3-Hexafluoroisopropanol | Oakwood Chemicals | 003409 | Used as received. |
| Deionized Water | Obtained from in-house deionized water system. | ||
| Dry Ice | Carbonic Enterprises Dry Ice Inc. | Non-food grade dry ice. | |
| Concentrated Hydrochloric Acid | Fisher Chemical | A144SI | Diluted to a 1.2 M solution prior to use. |
| Diethyl Ether, Certified | Fisher Chemical | E138 | Used as received. |
| Hexanes, Certified ACS | Fisher Chemical | H292 | Used as received. |
| Saturated Ammonium Hydroxide | Fisher Chemical | A669 | Used as received. |
| Dichloromethane | Fisher Chemical | D37 | Used as received. |
| Sodium Sulfate, Anhydrous | Oakwood Chemicals | 044702 | Used as received. |
| 250 mL Separatory Funnel | Prepared in-house by staff glassblower. | ||
| 100 mL Round Bottom Flask | Prepared in-house by staff glassblower. | ||
| Scientific Disposable Funnel | Caplugs | 2085136030 | |
| Borosilicate Glass Scintillation Vials, 20 mL | Fisher Scientific | 03-337-15 | |
| 5 mm O.D. Thin Walled Precision NMR Tubes | Wilmad | 666000575 | |
| Chloroform-d | Cambridge Isotope Laboratories, Inc. | DLM-7 | Used as received. |
References
- Verboom, W. Selected Examples of High-Pressure Reactions in Glass Microreactors. Chemical Engineering and Technology. 32 (11), 1695-1701 (2009).
- Schettino, V., Bini, R. Constraining Molecules at the Closest Approach: Chemistry at High Pressure. Chemical Society Reviews. 36, 869-880 (2007).
- Hemminger, O., Marteel, A., Mason, M. R., Davies, J. A., Tadd, A. R., Abraham, M. A. Hydroformylation of 1-Hexene in Supercritical Carbon Dioxide Using a Heterogeneous Rhodium Catalyst. 3. Evaluation of Solvent Effects. Green Chemistry. 4, 507-512 (2002).
- Mo, F., Dong, G. Regioselective Ketone α-Alkylation with Simple Olefins via Dual Activation. Science. 345 (6192), 68-72 (2014).
- Schultz, A. G., Kirincich, S. J., Rahm, R. Asymmetric Organic Synthesis. Preparation and Birch Reduction-Alkylation of 2-Methyl-3,4-Dihydroisoquinolin-1-ones. Tetrahedron Letters. 36 (26), 4551-4554 (1995).
- Dong, L., Aleem, S., Fink, C. A. Microwave-Accelerated Reductive Amination Between Ketones and Ammonium Acetate. Tetrahedron Letters. 51 (39), 5210-5212 (2010).
- Wang, D., Astruc, D. The Golden Age of Transfer Hydrogenation. Chemical Reviews. 115 (13), 6621-6686 (2015).
- Morimoto, T., Kakiuchi, K. Evolution of Carbonylation Catalysis: No Need for Carbon Monoxide. Angewandte Chemie International Edition in English. 43 (42), 5580-5588 (2004).
- Iranpoor, N., Firouzabadi, H., Motevalli, S., Talebi, M. Palladium-Free Aminocarbonylation of Aryl, Benzyl, and Styryl Iodides and Bromides by Amines Using Mo(CO)6 and Norbornadiene. Tetrahedron. 69 (1), 418-426 (2013).
- Ren, W., Yamane, M. Mo(CO)6-Mediated Carbamoylation of Aryl Halides. Journal of Organic Chemistry. 75 (24), 8410-8415 (2010).
- Wang, H., Dong, B., Wang, Y., Li, J., Shi, Y. A Palladium-Catalyzed Regioselective Hydroesterification of Alkenylphenols to Lactones with Phenyl Formate as CO Source. Organic Letters. 16 (1), 186-189 (2014).
- Zhang, Y., Chen, J. -. L., Chen, Z. -. B., Zhu, Y. -. M., Ji, S. -. J. Palladium-Catalyzed Carbonylative Annulation Reactions Using Aryl Formate as a CO Source: Synthesis of 2-Substituted Indene-1,3(2H)-Dione Derivatives. Journal of Organic Chemistry. 80 (21), 10643-10650 (2015).
- Wan, Y., Alterman, M., Larhed, M., Hallberg, A. Dimethylformamide as a Carbon Monoxide Source in Fast Palladium-Catalyzed Aminocarbonylations of Aryl Bromides. Journal of Organic Chemistry. 67 (17), 6232-6235 (2002).
- Gockel, S. N., Hull, K. L. Chloroform as a Carbon Monoxide Precursor: In or Ex Situ Generation of CO for Pd-Catalyzed Aminocarbonylations. Organic Letters. 17 (13), 3236-3239 (2015).
- Zhao, H., Du, H., Yuan, X., Wang, T., Han, W. Iron-Catalyzed Carbonylation of Aryl Halides with Arylborons Using Stoichiometric Chloroform as the Carbon Monoxide Source. Green Chemistry. 18, 5782-5787 (2016).
- Chen, P., Xu, C., Yin, H., Gao, X., Qu, L. Shock Induced Conversion of Carbon Dioxide to Few Layer Graphene. Carbon. , 471-476 (2017).
- Iijima, T., Yamaguchi, T. Efficient Regioselective Carboxylation of Phenol to Salicylic Acid with Supercritical CO2 in the Presence of Alumnium Bromide. Journal of Molecular Catalysis A: Chemical. 295 (1-2), 52-56 (2008).
- Jevtovikj, I., Manzini, S., Hanauer, M., Rominger, F., Schaub, T. Investigations on the Catalytic Carboxylation of Olefins with CO2 Towards α, β-Unsaturated Carboxylic Acid Salts: Characterization of Intermediates and Ligands as well as Substrate Effects. Dalton Transactions. 44, 11083-11094 (2015).
- Juliá-Hernández, F., Moragas, T., Cornella, J., Martin, R. Remote Carboxylation of Halogenated Aliphatic Hydrocarbons with Carbon Dioxide. Nature. 545, 84-88 (2017).
- North, M., Pasquale, R. Mechanism of Cyclic Carbonate Synthesis from Epoxides and CO2. Angewandte Chemie International Edition. 48 (16), 2946-2948 (2009).
- Yeung, C. S., Dong, V. M. Beyond Aresta’s Complex: Ni- and Pd-Catalyzed Organozinc Coupling to CO2. Journal of the American Chemical Society. 130 (25), 7826-7827 (2008).
- Zhu, D. -. Y., Fang, L., Han, H., Wang, Y., Xia, J. -. B. Reductive CO2 Fixation via Tandem C-C and C-N Bond Formation: Synthesis of Spiro-Indopyrrolidines. Organic Letters. 19 (16), 4259-4262 (2017).
- Kapoor, M., Liu, D., Young, M. C. Carbon Dioxide Mediated C(sp3)–H Arylation of Amine Substrates. J. Am. Chem. Soc. , (2018).
- Zhang, Y. -. F., Zhao, H. -. W., Wang, H., Wei, J. -. B., Shi, Z. -. J. Readily Removable Directing Group Assisted Chemo- and Regioselective C(sp3)-H Activation by Palladium Catalysis. Angewandte Chemie International Edition. 54 (46), 13686-13690 (2015).
- He, G., Chen, G. A Practical Strategy for the Structural Diversification of Aliphatic Scaffolds Through the Palladium-Catalyzed Picolinamide-Directed Remote Functionalization of Unactivated C(sp3)-H Bonds. Angewandte Chemie International Edition. 50 (22), 5192-5196 (2011).
- Nack, W. A., Wang, X., Wang, B., He, G., Cheng, G. Palladium-Catalyzed Picolinamide-Directed Iodination of Remote ortho-C-H Bonds of Arenes: Synthesis of Tetrahydroquinolines. Beilstein Journal of Organic Chemistry. 12, 1243-1249 (2016).
- Feng, P., Li, M., Ge, H. Room Temperature Palladium-Catalyzed Decarboxylative ortho-Acylation of Acetanilides with α-Oxocarboxylic Acids. Journal of the American Chemical Society. 132 (34), 11898-11899 (2010).
- Coomber, C. E., Benhamou, L., Bučar, D. -. K., Smith, P. D., Porter, M. J., Sheppard, T. D. Silver-Free Palladium-Catalyzed C(sp3)-H Arylation of Saturated Bicyclic Amine Scaffolds. Journal of Organic Chemistry. 83 (5), 2495-2503 (2018).
- Mei, T. -. S., Wang, X., Yu, J. -. Q. Pd(II)-Catalyzed Amination of C-H Bonds Using Single-Electron or Two-Electron Oxidants. Journal of the American Chemical Society. 131 (31), 10806-10807 (2009).
- Xie, W., Yang, J., Wang, B., Li, B. Regioselective Ortho Olefination of Aryl Sulfonamide via Rhodium-Catalyzed Direct C-H Bond Activation. Journal of Organic Chemistry. 79 (17), 8278-8287 (2014).
- Rodriguez, N., Romero-Revilla, J. A., Fernández-Ibáñez, M. &. #. 1. 9. 3. ;., Carretero, J. C. Palladium-Catalyzed N-(2-pyridyl)sulfonyl-Directed C(sp3)-H γ-Arylation of Amino Acid Derivatives. Chemical Science. 4, 175-179 (2013).
- Zheng, Y., Song, W., Zhu, Y., Wei, B., Xuan, L. Pd-Catalyzed Acetoxylation of γ-C(sp3)-H Bonds of Amines Directed by a Removable Bts-Protecting Group. Journal of Organic Chemistry. 83 (4), 2448-2454 (2018).
- Jain, P., Verma, P., Xia, G., Yu, J. -. Q. Enantioselective Amine α-Functionalization Via Palladium-Catalysed C-H Arylation of Thioamides. Nature Chemistry. 9, 140-144 (2017).
- Tran, A. T. Practical Alkoxythiocarbonyl Auxiliaries for Ir(I)-Catalyzed C-H Alkylation of Azacycles. Angewandte Chemie International Edition. 56 (35), 10530-10534 (2017).
- Huang, Z., Wang, C., Dong, G. A Hydrazone-Based exo-Directing Group Strategy for β-C-H Oxidation of Aliphatic Amines. Angewandte Chemie International Edition. 55 (17), 5299-5303 (2016).
- Xu, Y., Young, M. C., Wang, C., Magness, D. M., Dong, G. Catalytic C(sp3)-H Arylation of Free Primary Amines via an in situ Generated Exo-Directing Group. Chemie International Edition. 55 (31), 9084-9087 (2016).
- Liu, Y., Ge, H. Site-Selective C-H Arylation of Primary Aliphatic Amines Enabled by a Catalytic Transient Directing Group. Nature Chemistry. 9, 26-32 (2017).
- Wu, Y., Chen, Y. -. Q., Liu, T., Eastgate, M. D., Yu, J. -. Q. Pd-Catalyzed γ-C(sp3)-H Arylation of Free Amines Using a Transient Directing Group. Journal of the American Chemical Society. 138 (44), 14554-14557 (2016).
- Yada, A., Liao, W., Sato, Y., Murakami, M. Buttressing Salicylaldehydes: A Multipurpose Directing Group for C(sp3)-H Bond Activation. Angewandte Chemie International Edition. 56 (4), 1073-1076 (2017).
- Baldwin, B. W., Kuntzleman, T. S. Liquid CO2 in Centrifuge Tubes: Separation of Chamazulene from Blue Tansy (Tanacetum annum) Oil via Extraction and Thin-Layer Chromatography. Journal of Chemical Education. 95 (4), 620-624 (2018).
- McKenzie, L. C., Thompson, J. E., Sullivan, R., Hutchison, J. E. Green Chemical Processing in the Teaching Laboratory: A Convenient Liquid CO2 Extraction of Natural Products. Green Chemistry. 6, 355-358 (2004).
- Hudson, R., Ackerman, H. M., Gallo, L. K., Gwinner, A. S., Krauss, A., Sears, J. D., Bishop, A., Esdale, K. N., Katz, J. L. CO2 Dry Cleaning: A Benign Solvent Demonstration Accessible to K-8 Audiences. Journal of Chemical Education. 94, 480-482 (2017).
- Barcena, H., Chen, P. An Anesthetic Drug Demonstration and an Introductory Antioxidant Activity Experiment with "Eugene, the Sleepy Fish.". Journal of Chemical Education. 93, 202-205 (2016).
- Bodsgard, B. R., Lien, N. R., Waulters, Q. T. Liquid CO2 Extraction and NMR Characterization of Anethole from Fennel Seed: A General Chemistry Laboratory. Journal of Chemical Education. 93, 397-400 (2016).
- Fishbane, P. M., Gasiorowicz, S. G., Thornton, S. T. . Physics for Scientists and Engineers. , (2005).
- Rumpf, B., Xia, J., Maurer, G. Solubility of Carbon Dioxide in Aqueous Solutions Containing Acetic Acid or Sodium Hydroxide in the Temperature Range from 313 to 433 K and at Total Pressures up to 10 MPa. Industrial & Engineering Chemistry Research. 37, 2012-2019 (1998).
- Luo, J., Larrosa, I. C-H Carboxylation of Aromatic Compounds Through CO2 Fixation. ChemSusChem: Chemistry & Sustainability, Energy & Materials. 10, 3317-3332 (2017).
- Manjolinho, F., Arndt, M., Gooßen, K., Gooßen, L. J. Catalytic C-H Carboxylation of Terminal Alkynes with Carbon Dioxide. ACS Catalysis. 2, 2014-2021 (2012).
- Banerjee, A., Dick., G. R., Yoshino, T., Kanan, M. W. Carbon Dioxide Utilization via Carbonate-Promoted C-H Carboxylation. Nature. 531, 215-219 (2016).
- Fei, H., Sampson, M. D., Lee, Y., Kubiak, C. P., Cohen, S. M. Photocatalytic CO2 Reduction to Formate Using a Mn(I) Molecular Catalyst in a Robust Metal-Organic Framework. Inorganic Chemistry. 54, 6821-6828 (2015).
- Chabolla, S. A., Yang, J. Y. For CO2 Reduction, Hydrogen-Bond Donors Do the Trick. ACS Central Science. 4, 315-317 (2018).
- Kim, D., Kley, C. S., Li, Y., Yang, P. Copper Nanoparticle Ensembles for Selective Electroreduction of CO2 to C2-C3 Products. Proceedings of the National Academy of Sciences of the United States of America. , C2-C3 (2017).
- Liu, Q., Wu, L., Jackstell, R., Beller, M. Using carbon dioxide as a building block in organic synthesis. Nature Communications. 6, 5933-5945 (2015).
- Hâncu, D., Green, J., Beckman, E. J. H2O2 in CO2 Sustainable Production and Green Reactions. Accounts of Chemical Research. 35, 757-764 (2002).
- Ballivet-Tkatchenko, D., Camy, S., Condoret, J. S., Lichtofouse, E., Scwarzbauer, J., Robert, D. Carbon Dioxide, a Solvent and Synthon for Green Chemistry. Environmental Chemistry. , 541-552 (2005).
- Hyatt, J. A. Liquid and Supercritical Carbon Dioxide as Organic Solvents. Journal of Organic Chemistry. 49, 5097-5101 (1984).
