Typical yields for nsRNA recovered using this ers4tU protocol are displayed in Figure 1b, this has been produced by a bioanalyzer and the trace shows yield of RNA versus size (nucleotides [nt]). Note, in both the bioanalyzer trace and the inset graph, that RNA recovery from time point 0 is a very small portion of that recovered from longer time points – approximately 0.3 µg of RNA recovered from approximately 109 cells compared with over twice as much after just 30 s of labelling (0.8 µg of nsRNA) from the same number of cells. RNA recovery at 15 s is more variable as small differences in performing the sampling have a proportionately larger effect on RNA recovery. In the bioanalyzer trace, rRNA precursors can be seen as a peak near 1000 nt and a doublet of peaks at 1700−1800 nt. The abundance of these intermediates increases as thiolation continues.
Thio-labelling was used to quantify splicing of the ACT1 transcript (Figure 3). Thiolation was performed and samples taken at 15 s intervals from the start of thio-labelling and the processing of ACT1 RNA monitored (Figure 3a,b). As can be seen, pre-mRNA is generated (by transcription), and lariats (by the first step of splicing from pre-mRNA), even after just 15 s of labelling. After about 45 s to 1 min, the amounts of lariats and pre-mRNA reach equilibrium with as much of these RNA species being created by transcription as are processed away by splicing.
To produce the data shown in Figure 3c the strain was pulsed with 4tU for 25 s and then chased with uridine. The generation of pre-mRNA and lariats reaches a maximum at 1 minute. This compares well with Figure 3b; the maximum being achieved after 45 s to reach equilibrium plus the 25 s of the labelling. After the peak, the levels decline as the thio-labelled RNAs are chased through the splicing process.
Figure 3d shows depletion of a protein splicing factor and its effect on RNA metabolism, using the β-est AID 4U system6,7. Here, Prp16p is reduced from near physiological levels to 5% of this level after 25 min of depletion. Prp16p is an essential splicing factor for the second step of splicing15. Lariats are removed during the second step of splicing (Figure 3a), but here they increase above the level of pre-mRNA as Prp16 becomes limiting. At later depletion times, other factors become limiting due to secondary effects, so that levels of lariat decrease, and pre-mRNA levels rise. The level of spliced mRNA declines.
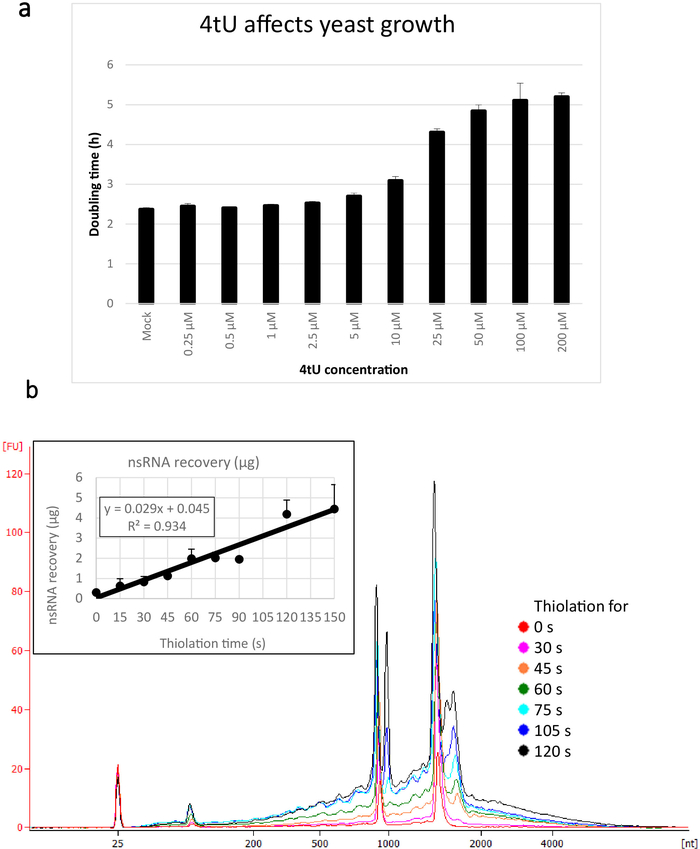
Figure 1: Growth in 4tU and RNA recovery. (a) 4-thiouracil affects growth. Increasing the concentration of 4tU in YMM drop-out growth medium without uracil increases the doubling time of S. cerevisiae (BY4741) carrying the p4FuiΔPEST plasmid. Growth of four replicate cultures was monitored at 30 °C in a Tecan Infinite Pro 200. All cultures were in log phase throughout, with OD600 between 0.1 and 0.6. Mock is a control culture with an equivalent amount of NaOH added, which does not by itself change the growth rate. This graph demonstrates that thio-labelling is compromise between rapid labelling and damage to the cell. Error bars are standard error of 4 replicates. (b) nsRNA yield increases linearly from about 15 s of labelling. The main figure shows the bioanalyzer traces of purified, nsRNA from 0 (not thiolated) to 2 min after addition of 4tU at 15 s intervals. Note that the 15 s sample is not shown, as it was indistinguishable from the unlabelled sample. The two large peaks correspond to ribosomal RNAs (rRNAs). The rRNA precursors and intermediates are visible as several peaks at greater molecular weight than mature rRNAs. The recovery of these precursors and intermediates increases with time. Results from one representative experiment are shown. The inset graph shows the recovery of nsRNA with increasing incubation with 4tU. The yield of nsRNA increases with increasing time of growth with 4tU. The recovery is remarkably linear (R2 = 0.934) throughout the timescale of this experiment and shows a slight increase over background even at 15 s labelling with 4tU even though not distinguishable from the unlabelled sample by eye from the bioanalyzer trace. Error bars show standard error for three biological replicates. Please click here to view a larger version of this figure.
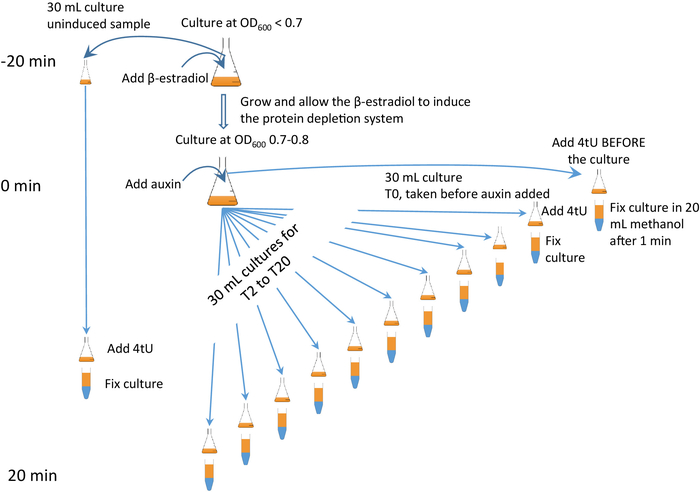
Figure 2: β-est AID 4U β-est AID 4U graphical protocol. A graphical summary of the protocol of the β-est AID 4U protocol. β-estradiol (β-est) promotes the expression of the auxin inducible degron (AID) system which in turn depletes an AID* tagged target protein, refer to Barrass et al.7 for a detailed protocol. In this case, degron system expression is initiated 25 min before protein degradation commences and thiolation at each time point is for 1 min. Samples are taken before induction and every 2 minutes during depletion. An animated version appears in the Supplementary Figure 2. Please click here to view a larger version of this figure.
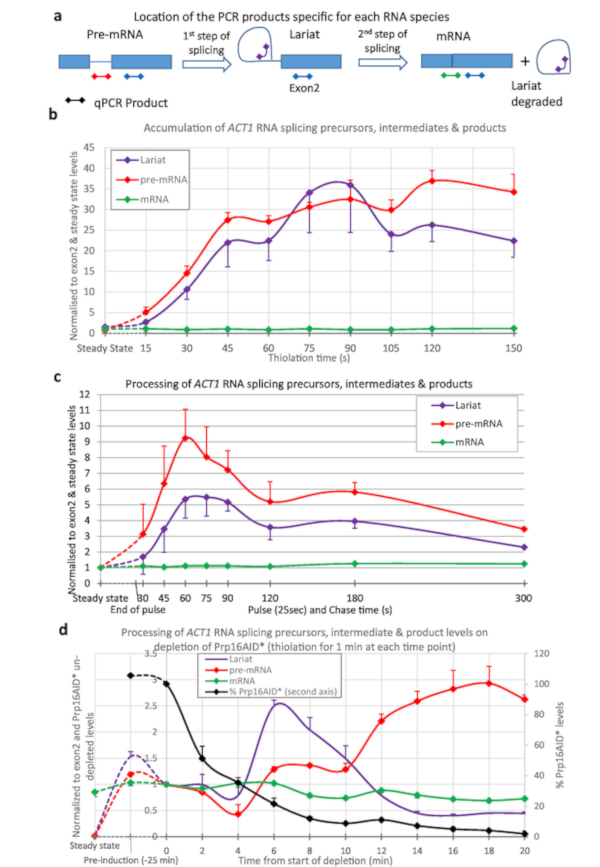
Figure 3: Precursors and intermediates of ACT1 RNA splicing. Splicing of ACT1 pre-mRNA transcripts was monitored by quantitative reverse transcription PCR16. The levels of ACT1 precursor (pre-mRNA), intermediate lariat-exon2 (Lariat) and spliced product (mRNA) are shown normalized against the level of ACT1 Exon2 and steady state levels of these RNAs. (a) Location of qPCR products on the ACT1 transcript. Schematic of the locations of the qPCR products used to assay the levels of precursors, intermediates and products of the splicing reaction of the ACT1 transcripts16. Exons are represented by boxes, intron as a line and the qPCR products as lines with diamonds at either end, the color matches those used in the graphs. The pre-mRNA PCR is specific for pre-mRNA and not any intermediates of splicing as this product crosses the branch point which is disrupted after the first step of splicing. Lariat PCR will detect the product of the first step of splicing and the excised lariat produced after the second. The mRNA PCR is specific for the product of splicing, mRNA. Results from the exon PCR (present in all precursors, intermediates and products, except the excised lariat) is not shown in the graphs as this was used to normalize the data and is therefore always equal to 1. (b) Continuous thiolabelling. The amount of pre-mRNA increases with time as 4tU is incorporated by transcription and, after a short delay, splicing converts it to lariat-exon2 intermediate and spliced products. The levels of these pre-mRNA and lariat species are detectable above background after as little as 15 s of growth with 4tU and reach a maximum after approximately 45 s of continuous labelling with 4tU, at which point their production is balanced by conversion to spliced mRNA and/or degradation. Values are normalized to their steady state (left-most point of the graph), and exon 2 levels to show their appearance and processing in comparison to transcription of exon 2. As RNA splicing of ACT1 is largely co-transcriptional4,17 spliced mRNA rapidly becomes the most abundant species, its level is similar to that of exon 2. Standard error of three biological replicates, each assayed in triplicate. (c) Pulse/chase. Thiolation pulse of 25 seconds followed by chase with uridine. Compared to the steady state levels of these RNAs (left-most point), they are initially very abundant in the newly synthesized pool. The levels gradually decline as they are processed into mRNA (or degraded), approaching levels very similar to steady state levels by 5 min. Standard error of three biological replicates, each assayed in triplicate. (d) nsRNA and protein depletion. Splicing of ACT1 pre-mRNA transcripts monitored by quantitative reverse transcription PCR as in panel (a) upon depletion of the Prp16 protein using the auxin degron system as described in Figure 2. The Prp16 protein levels are also displayed in the graph plotted against the second Y-axis as percentage of levels prior to auxin depletion. Prp16 is a vital component of the spliceosome, particularly important for the second step of splicing shown in panel (a), after which lariats are degraded. When this step becomes limiting lariats accumulate initially. At later time points splicing fails completely, lariats are no longer produced and pre-mRNA levels rise. Error bars are standard error of three biological replicates, each assayed in triplicate. Please click here to view a larger version of this figure.

Figure 4: Graphical summary of the protocol sections 1 to 3. The cells are thiolated with 4tU and allowed to grow to incorporate the modified nucleotide into the RNA. A thiolated S. pombe spike can be added to allow normalization across time points and experiments. The pulse of 4tU can be chased using un-thiolated uridine. Labelling can either be performed continuously from 4tU addition or from a change to growth conditions, the culture split and 4tU added to cultures at increasing times from the growth condition change, but each labelling only for a brief time. The cells are collected, and RNA prepared from the cells, preferably using a homogenizer and phenol-based methods. The RNA is biotinylated and then the biotinylated RNA purified from unincorporated biotin using a size exclusion column. The nsRNA is now ready for purification with streptavidin beads (section 4, Figure 5). Numbers in red correspond to the step numbers in the protocol. Please click here to view a larger version of this figure.
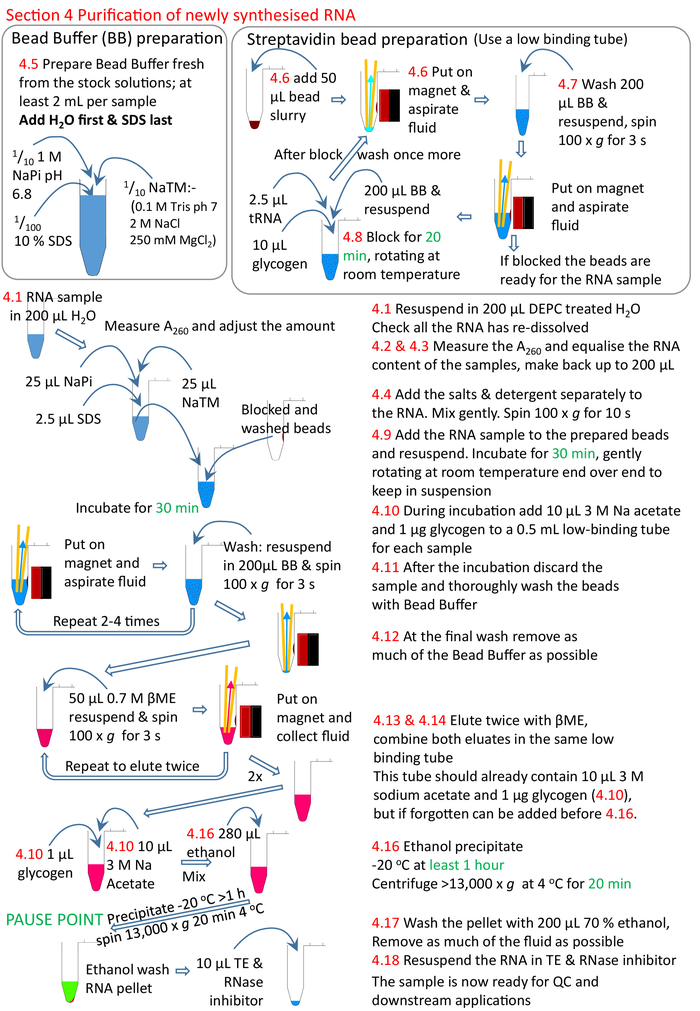
Figure 5: Graphical summary of the protocol section 4. Following on from sections 1 to 3 (Figure 4), the streptavidin beads are blocked and the biotinylated RNA sample added to the prepared beads. The biotinylated RNA binds to the streptavidin beads and the un-biotinylated RNA removed and washed. The biotinylated RNA is eluted from the beads using βME and precipitated ready for further research. Numbers in red correspond to the step numbers in the protocol. Please click here to view a larger version of this figure.
Supplementary Figure 1: Improvement of nsRNA recovery from yeast cells with and without additional copies of the importer at 1 and 3 minutes of thio-labelling. Note that Fui1 is the yeast's own promoter expressed from a 2 µm plasmid. The genomic copy of this gene is present in both of these strains. Please click here to download this file.
Supplementary Figure 2: Animated version of the β-est AID 4U β-est AID 4U graphical protocol. Please click here to download this file.
Supplementary File 1: 4tU_experiment_template.xltx. Please click here to download this file.
| Plasmid Name | Importer/permease | Marker | Comment | |
| p4Fui | S. cerevisiae Fui1 | URA3 | Fui1 imports Uracil and Uridine, making it ideal for pulse/chase experiments. | |
| pAT2 | S. cerevisiae Fui1 | LEU2 | ||
| p4Fui-ΔPEST | S. cerevisiae Fui1 | URA3 | The PEST motif of Fui1 has been deactivated, so the permease is not degraded when there is sufficient intracellular uracil for the cell’s needs. Works well in labelling experiments and improves pulse/chase performance. | |
| p4Fur | S. cerevisiae Fur4 | URA3 | Uracil permease | |
| YEpEBI311 | H. Sapiens ENT1 | LEU2 | Miller et al.11. Also contains an HSV thymidine kinase gene. | |
| (equilibrative nucleoside transporter) | ||||
| All plasmids are 2 µm based. All p4 plasmids and pAT are based on the pRS16 series of plasmids. FUI1 and FUR4 are expressed from their own, endogenous promoters. | ||||
Table 1: Plasmids used with this protocol.
