

Summary
This article aims to provide the methodology for lentiviral transgenesis in rat embryos using multiple injections of a virus suspension into the zygote perivitelline space. Female rats that are mated with a fertile male strain with a different dominant fur color is used to generate pseudopregnant foster mothers.
Abstract
Transgenic animal models are fundamentally important for modern biomedical research. The incorporation of foreign genes into early mouse or rat embryos is an invaluable tool for gene function analysis in living organisms. The standard transgenesis method is based on microinjecting foreign DNA fragments into a pronucleus of a fertilized oocyte. This technique is widely used in mice but remains relatively inefficient and technically demanding in other animal species. The transgene can also be introduced into one-cell-stage embryos via lentiviral infection, providing an effective alternative to standard pronuclear injections, especially in species or strains with a more challenging embryo structure. In this approach, a suspension that contains lentiviral vectors is injected into the perivitelline space of a fertilized rat embryo, which is technically less demanding and has a higher success rate. Lentiviral vectors were shown to efficiently incorporate the transgene into the genome to determine the generation of stable transgenic lines. Despite some limitations (e.g., Biosafety Level 2 requirements, DNA fragment size limits), lentiviral transgenesis is a rapid and efficient transgenesis method. Additionally, using female rats that are mated with a fertile male strain with a different dominant fur color is presented as an alternative to generate pseudopregnant foster mothers.
Introduction
For many years, laboratory rodents, such as rats and mice, have been used to model human physiological and pathological conditions. Animal research has led to discoveries that were unattainable by any other means. Initially, genetic studies focused on the analysis of spontaneously occurring disorders and phenotypes that are considered to closely mimic the human condition1. The development of genetic engineering methods allowed the introduction or deletion of specific genes to obtain a desired phenotype. Therefore, the generation of transgenic animals is recognized as a fundamental technique in modern research that allows studies of gene function in living organisms.
Transgenic animal technology has become possible through a combination of achievements in experimental embryology and molecular biology. In the 1960s, the Polish embryologist A. K. Tarkowski published the first work on mouse embryo manipulation during the early stages of development2. Additionally, molecular biologists developed techniques to generate DNA vectors (i.e., carriers) for the inter alia introduction of foreign DNA into the animal’s genome. These vectors allow the propagation of selected genes and their appropriate modification, depending on the type of research that is conducted. The term “transgenic animal” was introduced by Gordon and Ruddle3.
The first widely accepted species that was used in neurobiology, physiology, pharmacology, toxicology, and many other fields of biological and medical sciences was the Norway rat, Rattus norvegicus4. However, because of the difficulty in manipulating rat embryos, the house mouse Mus musculus has become the dominant animal species in genetic research5. Another reason for the primacy of the mouse in such research was the availability of embryonic stem cell technology to generate knockout animals for this species. The most commonly used technique of transgenesis (2–10% of transgenic offspring relative to all born animals) is the microinjection of DNA fragments into a pronucleus of a fertilized oocyte. In 1990, this approach, which was first introduced in mice, was adapted for rats6,7. Rat transgenesis by pronuclear injection is characterized by lower efficiency8 compared with mice, which is strictly related to the presence of elastic plasma and pronuclear membranes9. Although the survival of embryos after manipulation is 40–50% lower than in mice, this technique is considered a standard in the generation of genetically modified rats10. Alternative approaches that can guarantee efficient transgene incorporation and higher survival rates of injected zygotes have been investigated.
The key determinant of stable transgene expression and transmission to progeny is its integration into the host cell genome. Lentiviruses (LVs) have the distinctive feature of being able to infect both dividing and non-dividing cells. Their use as a tool for the incorporation of heterologous genes into embryos proved to be highly efficient11, and transgenic individuals are characterized by stable expression of the incorporated DNA fragment. The efficacy of lentiviral vectors has been confirmed for the genetic modification of mice12,13, rats12,14, and other species11. In this method, the LV suspension is injected under the zona pellucida of the embryo at the stage of two pronuclei. This technique essentially guarantees 100% survival of the embryos because the oolemma remains unaffected. The production of high-quality and relatively highly concentrated LV suspensions are crucial factors. However, lower concentrations of LV suspensions can be overcome by repeated injections11, which increases the amount of viral particles at the egg surface while not affecting membrane integration. Embryos that are subjected to repeated injections into the perivitelline space develop further, and transgenic offspring can transmit the transgene through the germline. The efficiency of transgenic rat generation by lentiviral transgenesis can be as high as 80%12.
Here, we describe the production of HIV-1-derived recombinant lentivirus that was pseudotyped with vesicular stomatitis virus (VSV) G envelope protein. The use of the second-generation packaging system VSV pseudotype determines the wide infectivity of viral particles and allows the production of highly stable vectors that can be concentrated by ultracentrifugation and cryopreserved. After titer verification, the vectors are ready to be used as a vehicle for transgene delivery into albino Wistar rat zygotes. After a series of injections, the embryos can be cultured overnight and transferred at the two-cell stage to foster mothers. At this point, one of two alternative approaches can be considered. The standard procedure utilizes pseudopregnant females as embryo recipients. However, when the pregnancy rate is low after mating with vasectomized males, the embryos can be implanted into pregnant Wistar/Sprague-Dawley (SD) females that are mated with fertile male rats with a dark fur color (e.g., Brown Norway [BN] rats). The color of the fur allows the distinction of offspring from natural pregnancy from offspring that originate from the transferred manipulated embryos.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
The production and application of viral vectors was in accordance with Biosafety Level 2 guidelines and was approved by the Polish Ministry of Environment. All experimental animal procedures that are described below were approved by the Local Ethical Committee. The animals were housed in individually ventilated cages at a stable temperature (21–23 °C) and humidity (50–60%) with ad libitum access to water and food under a 12 h/12 h light/dark cycle.
1. Lentiviral vector production
- Transfection of HEK 293T cells
NOTE: The protocol that is presented herein is designed for the transfection of twenty Ø10 cm culture dishes that produces approximately 200 mL of crude vector supernatant.- Culture HEK 293T cells in DMEM medium that is supplemented with fetal bovine serum (10%, v/v) in a humidified CO2 incubator at 37 °C. For transfection, prepare twenty 10 cm diameter plates, and seed 1.5–2 x 106 HEK 293T cells per dish.
- When confluence reaches ~70%, transfect the cells using polyethylenimine (PEI) reagent, pH 7.0, at a ratio of 3 μg of PEI per 1 μg of DNA.
- Prepare the transfection mixture for five plates (prepare the number of repetitions according to the total number of dishes). To 1 mL of Dulbecco’s Modified Eagle Medium (DMEM; without serum), add the mixture of three plasmids so they reach a final amount of 25 µg of VSVg plasmid, 50 µg of delta R8.2, and 50 µg of coding plasmid.
- Pipette up and down, and add 125 µL of PEI at a concentration of 3 µg/µL. Incubate at room temperature for 15 min, inverting the tube three times during incubation. Add 200 µL of the transfection mixture per plate. Next, incubate the plates in a humidified CO2 incubator at 37 °C.
- Concentration of lentiviral vectors
- Forty-eight hours after transfection, harvest the medium that contains LV particles. Use 50 mL conical tubes.
NOTE: When using a plasmid with a fluorescent tag, cells can be visualized at this point to verify transfection efficiency. A new portion of DMEM medium can be added, and cells may be incubated for an additional 24 h. The LV yield is comparable when collected at the 48 and 72 h time points after transfection. - Centrifuge the medium at 3,000 x g for 5 min and room temperature to remove detached cells.
- Filter the supernatant (0.45 μm) and pour it into new tubes.
NOTE: This step can be omitted. - Add DNase I (RNase-free, 1 μg/mL) and MgCl2 (1 mM), and incubate in a water bath at 37 °C for 15 min.
- Transfer the medium to disposable polyethylene tubes, and ultracentrifuge in a swinging rotor at 115,000 x g and 4 °C for 1.5 h.
- After centrifuging, gently drain the walls of the tubes from the medium residues.
- Soak the pellet with sterile phosphate-buffered saline (PBS; 70–80 µL per tube).
- Incubate for 2 h at 4–8 °C.
- Resuspend the viral vectors in PBS by gentle pipetting.
CAUTION: Avoid foaming. - Transfer to a 1.5 mL centrifuge tube and centrifuge at 7,000 x g and 4 °C for 30 s. Transfer the supernatant to a new tube. Repeat this step until no cellular debris pellet is visible.
- Aliquot and freeze at -80 °C. Avoid refreezing the LV aliquot.
- Forty-eight hours after transfection, harvest the medium that contains LV particles. Use 50 mL conical tubes.
- Determination of virus titer using quantitative polymerase chain reaction
NOTE: The titration of viral vectors is performed using quantitative PCR (qPCR). This method is based on amplifying a double-stranded 84 bp long DNA fragment within the long terminal repeat region of the viral genome15.- Prepare the standard curve by making serial dilutions of the LV-coding plasmid: 1:500, 1:1,000, 1:5,000, 1:10,000, 1:100,000, and 1:1,000,000. Determine the number of copies of the plasmid that is used for the standard curve. Use the following formula: number of copies/µL = (concentration [g/µL] x 6.02 x 1023 [number/mol]) / (660 [g/mol] x plasmid size [bp]), where 6.02 x 1023 number/mol is Avogadro’s number, and 660 g/mol is the bp weight.
NOTE: Online copy number calculators may be used. - Prepare dilutions of the lentiviral suspension: 1:100, 1:500, and 1:1,000.
- Prepare the reaction mixture (volumes per well): 10 µL of qPCR Mastermix, 1 µL of 10 μM Forward primer, 1 µL of 10 μM reverse primer, and 7 µL of H2O. Pipette the mixture into the wells of 96-well plates.
NOTE: Forward primer: 5’-AGCTTGCCTTGAGTGCTTCA. Reverse primer: 5’-TGACTAAAAGGGTCTGAGGGA. - Add 1 µL of each standard dilution and lentiviral suspension in triplicate.
- Run the qPCR according to the following parameters: 50 °C for 2 min, 96 °C for 5 min, and 35 cycles of 96 °C for 20 s, 60 °C for 40 s, and 70 °C for 1 min, followed by melt curve stage: 95 °C for 1 min and 60 °C at 30 s.
- Analyze the results by comparing the number of molecules that are received for each dilution to the standard curve. Determine the concentration of vector molecules as the average of three replicates for each dilution.
NOTE: The presented quantification gives the physical concentration of the viral particles. It should not be treated as a functional titer.
- Prepare the standard curve by making serial dilutions of the LV-coding plasmid: 1:500, 1:1,000, 1:5,000, 1:10,000, 1:100,000, and 1:1,000,000. Determine the number of copies of the plasmid that is used for the standard curve. Use the following formula: number of copies/µL = (concentration [g/µL] x 6.02 x 1023 [number/mol]) / (660 [g/mol] x plasmid size [bp]), where 6.02 x 1023 number/mol is Avogadro’s number, and 660 g/mol is the bp weight.
2. Generation of transgenic rats
- Superovulation and collection of fertilized embryos
- Administer gonadotropins.
NOTE: To increase the number of collected embryos (approximately 30 per female), use immature 5-week-old Wistar females for hormonal stimulation.- On Day 1 (12 PM–1 PM), intraperitoneally inject pregnant mare’s serum gonadotropin (PMSG; 25 IU per female). Prepare 1 mL aliquots of working solution at a concentration of 125 IU/mL by dissolving hormone powder in 0.9% NaCl. Store at -20 °C for up to 1 month or -80 °C for up to 6 months.
- On day 3 (12 PM–1 PM), intraperitoneally inject human chorionic gonadotrophin (hCG; 30 IU per female). Prepare 1 mL aliquots of working solution (150 IU/mL) by dissolving hormone powder in 0.9% NaCl. Store at -20 °C for up to 1 month or -80 °C for up to 6 months.
- After hCG administration, mate females 1:1 with sexually fertile males (3-10 months old).
- The next morning (day 4 at 8–10 AM), check the females for the presence of a vaginal plug. Check the vaginal opening for the presence of a whitish mating plug, which for best visualization should be checked early in the morning after the mating night. For embryo collection, use only females with a visible plug.
- Collect embryos at 10 AM. Sacrifice the animals to excise the oviducts, and collect the oviducts in a dish with pre-warmed M2 medium.
- Transfer the oviducts to a 35 mm dish that contains pre-warmed M2 medium with hyaluronidase from bovine testes at a concentration of 0.5 mg/mL.
- Open the walls of the oviduct using fine forceps under a stereomicroscope and press the ampulla (i.e., the swollen part of the oviduct that contains fertilized embryos that are surrounded by cumulus cells) until the embryos are freed.
NOTE: Hyaluronidase enzymatically digests cumulus cells, releasing embryos.
CAUTION: Prolonged exposure to hyaluronidase is deleterious to embryos; therefore, this step should last no longer than 5 min. - To facilitate the release of embryos from cumulus cells, gently pipette them up and down using a glass transfer pipette that is connected to a mouth-operated aspirator tube.
- To produce the transfer pipette, pull a glass Pasteur pipette over a flame to produce a straight ~5-10 cm tip. Break the pipette leaving a ~4 cm tip.
- Wash the embryos a few times in M2 medium to remove hyaluronidase and cellular debris. Transfer the embryos to a 60 mm dish that contains (~50 µL) drops of pre-equilibrated M16 medium, covered by liquid paraffin or mineral oil, in a humidified 37 °C incubator with a 5% CO2 atmosphere.
- Administer gonadotropins.
- Microinjection of lentiviral vectors to one-cell-stage embryo under the zona pellucida
NOTE: Use one-cell-stage embryos with two visible pronuclei for microinjection (Figure 1).- Thaw the LV aliquot at room temperature and centrifuge at 10,000 x g and RT for 2 min to pellet any remaining cellular debris.
- Microinjection setup
- Prepare glass holding pipettes (borosilicate glass capillary) using a microforge. Pull the glass capillary over a flame to produce a 5–10 cm tip. Break the pipette leaving a ~4 cm tip. The outside diameter should be ~80–120 µm.
NOTE: Ensure that the pipette tip is perfectly straight and smooth. - Assemble the pulled pipette in a microforge with the tip in front of the heating filament. Heat the filament very close to the pipette tip and allow it to shrink to a diameter of ~15 µm (approximately 20% of embryo size). Position the pipette perpendicularly to the heating filament, 2–3 mm from the pipette tip, and begin to heat. The glass will soften. Heat until it reaches a 15° angle.
- Prepare microinjection borosilicate glass capillaries with a filament using a pipette puller. Insert the capillary in the pulling chamber. Run a ramp test (for the first time for new glass and every time after changing the filament). Set the Heat to the ramp value -10, Pull to 100, Velocity to 150 and Time to 100.
NOTE: Modify the parameters to obtain an optimal injection capillary. - Under a biosafety laminar flow hood, load approximately 2 µL of the viral solution into the microinjection pipette with a microloader tip.
- Prepare a microinjection dish (lid of 60 mm Petri dish) with a 100 µL drop of M2 medium (in the middle), covered by liquid paraffin or mineral oil.
- Mount the holding pipette and microinjection capillary that is loaded with viral solution to a micromanipulator and microinjection dish under an inverted microscope.
- Prepare glass holding pipettes (borosilicate glass capillary) using a microforge. Pull the glass capillary over a flame to produce a 5–10 cm tip. Break the pipette leaving a ~4 cm tip. The outside diameter should be ~80–120 µm.
- Perform the microinjection.
- Transfer 15–20 one-cell-stage embryos to the M2 drop on the microinjection dish. Hold the embryo using a holding pipette.
- Using 400x magnification, inject the LV solution under the zona pellucida to the perivitelline space using the glass capillary that is connected to an automatic injector. Hold the capillary under the zona pellucida for a moment.
NOTE: Using gentle positive pressure, the viral solution will flow continuously out of the injection capillary, but the volume of the suspension that is delivered cannot be controlled. - Using a fine pipette, return the embryos to the culture dish in the incubator at 37 °C in a 5% CO2 atmosphere. The number of injections of one zygote may vary and can be adapted based on the viral vector concentration.
NOTE: The injected embryos can be transferred to foster mothers at the one-cell stage or incubated O/N in M16 medium before being transferred at the two-cell stage. Prolonged in vitro culture of rat embryos should be avoided.
- Transfer of injected embryos to foster mothers
- Prepare foster mothers by mating sexually mature SD females with fertile BN males or with vasectomized SD males (the vasectomy procedure is described in section 3 below) on day 3 (for transferring embryos at the one-cell stage) or day 4 (for transferring embryos at the two-cell stage).
NOTE: For oviduct transfer, use 0.5 days post coitum (dpc) females. - The next morning, check SD females for a vaginal plug, and use only those with a visible plug.
- Perform embryo transfer.
NOTE: Conduct the surgical procedure with sterile instruments under a stereomicroscope. Before the day of surgery, autoclave scissors, fine forceps, needle holder, and scalpel holder.- Anesthetize a female with i.p. administration of ketamine (50 mg/kg) and medetomidine (0.5 mg/kg) solution.Test for reflexes to confirm anesthesia before starting the surgical procedure.
- Inject the animal subcutaneously with tolefenamic acid (2 mg/kg), butorphanol tartrate (1 mg/kg), and enrofloksacin (5–10 mg/kg) to prevent inflammation, pain, and infection, respectively.
- Apply ophthalmic ointment lubrication to both eyes to prevent corneal drying. Shave the fur from the back, and sterilize the skin with surgical scrub followed by 70% alcohol using sterile non-adhering pads. Allow the skin to dry.
- Inject the animal subcutaneously with 100 µL of 0.25% bupivacaine (local anesthetic) at the incision site. Transfer the animal in a prone position to a clean surface on a heating pad under the objective of a surgical microscope. Cover the rat with a sterile drape with a small hole cut over the lower back.
- Perform an approximately 2 cm skin incision, parallel to the lumbar vertebral column.
- Using sharp scissors, make a cut in the abdominal wall. Grab an ovarian fat pad using forceps, and pull out the ovary and oviduct and place them on gauze that is wetted with 0.9% NaCl.
- Aspirate M2 medium, three bubbles of air, and the embryos into the transfer capillary. Recommended total number of embryos to be transferred (unilateral or bilateral): pregnant female (≤ 15–16 embryos), pseudopregnant female (≤ 30 embryos).
- Make a small incision in the oviduct (between the infundibulum and ampulla) using micro-scissors, and insert the transfer pipette in the oviduct.
- Gently expel embryos and air bubbles from the pipette to the oviduct. With blunt forceps, place the reproductive tract back in the abdominal cavity.
- Suture the abdominal wall with polyglycolic acid absorbable sutures and close the skin incision with wound clips. Depending on the number of embryos that are available, repeat this procedure for the other oviduct.
- Inject the animal intraperitoneally with atipamezole (0.5 mg/kg) to reverse the effect of anesthesia.
- Transfer the animal to a clean cage and keep it on a warming plate to fully recover from anesthesia. Delivery in rats occurs after ~21 days.
NOTE: When male BN rats are used for mating, only white pups are potentially transgenic; brown pups are from natural pregnancy. - Collect tissue fragments (preferably from the ear) to genotype 3-week-old pups.
- Prepare foster mothers by mating sexually mature SD females with fertile BN males or with vasectomized SD males (the vasectomy procedure is described in section 3 below) on day 3 (for transferring embryos at the one-cell stage) or day 4 (for transferring embryos at the two-cell stage).
3. Vasectomy
NOTE: Before the day of surgery, autoclave scissors, fine forceps and needle holder.
- Anesthetize a 5-week-old male SD rat with i.p. administration of ketamine (50 mg/kg) and medetomidine (0.5 mg/kg) solution. Test for reflexes to confirm anesthesia before starting the surgical procedure.
- Administer tolfenamic acid (2 mg/kg), butorphanol tartrate (1 mg/kg), and enrofloksacin (5–10 mg/kg) subcutaneously to prevent inflammation, pain, and infection, respectively.
- Apply ophthalmic ointment lubrication to both eyes to prevent corneal drying. Place the rat supine on a clean surface on a heating pad, and sterilize the skin on the testes with surgical scrub followed by 70% alcohol using sterile non-adhering pads. Allow the skin to dry. Cover the rat with a sterile drape with a small hole cut over the testes. Gently press the abdomen to expose the testes in the scrotal sac.
- Using surgical scissors, make a ~0.5 cm incision in the middle of the scrotal sac. Locate the midline wall (whitish line) between the testes.
- Make a 5 mm incision in the testis membrane close to the left side of the midline wall.
- Carefully push the testis to the left and locate vas deferens (between the testis and midline) as a white duct with a single blood vessel.
- Gently pull the vas deferens out of the scrotal sac using a watchmaker’s forceps. Hold the vas deferens with one pair of forceps, and cut it with fine scissors (or cauterize with red-hot tips of a second pair of forceps). Remove a ~1 cm fragment of the duct.
NOTE: If cauterization is performed, hold the tip of the second pair of forceps in the flame. - Repeat the above procedure for the other testis. Suture the skin with polyglycolic acid absorbable sutures and inject the animal intraperitoneally with atipamezole (0.5 mg/kg).
- Place the rat in a clean cage on a warming plate until the animal recovers from anesthesia.
NOTE: Males can be used in the test matings after a ~2-week recovery period. After sterility is confirmed, they can be used for pseudopregnancy induction.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Using the protocol described herein, lentiviral vectors that carried the Syn-TDP-43-eGFP construct were produced (physical LV titer = 3.4 x 108/µL) and then could be used for one-cell-stage embryo subzonal injections. Only embryos with two visible pronuclei were subjected to the procedure. The number of injections of viral suspensions was determined experimentally. High implantation efficiency and a simultaneous lack of transgenic offspring were considered indicators of an insufficient number of viral particles for successful transduction. In this case, the number of injections was increased. The single administration of LV resulted in the birth of 20 F0-generation rats, none of which were transgenic. An increase in the number of injections by one order of magnitude did not result in the birth of rats, but 100% of the embryos developed to the two-cell stage. In subsequent experiments, the number of injections was increased by one compared with the value for which the offspring were obtained. For the variant of two injections, eight rats were born, three of which were confirmed to carry the transgene (summarized in Table 1). One of the founders did not transfer the transgene to offspring. The number of embryos that were injected and transferred in each experimental variant was 48 in variants LV x1 and LV x2 and 45 in LV x10. Three foster females were used for each experimental setup. The chosen approach allowed the generation of stable transgenic rat lines that expressed the TDP-43-eGFP fusion protein under control of the neuronal Synapsin-1 promoter throughout the entire central nervous system (Figure 2A,B)14. Lentivirus-based transgenesis resulted in a single copy insertion of the transgene as demonstrated by qPCR (Figure 2C).
In the experimental setup that is described above, the survival rate of the injected embryos was 95%. Similar results were obtained when the same method was used for other lentiviral vectors as summarized in Table 2. The percentage of embryos that survived the pronuclear injections was significantly lower (29–45%). Table 2 summarizes the representative results of the implantation efficiency of manipulated zygotes, considering the transfer of pseudopregnant vs. pregnant females. The use of non-manipulated embryos together with injected embryos was previously reported16. Our overall results suggest that pregnant female rats can be used as foster mothers with comparable efficiency. We obtained a similar percentage of implantation of foreign embryos in pregnant and pseudopregnant rats (overall average for several experimental setups: 15% vs. 16%). However, the implantation rate was higher when the embryos underwent more subtle manipulation, which means a subzonal injection (10% vs. 21%). Notably, the numerical data that were analyzed for individual rounds of microinjection indicated that the effectiveness of implantation depended on the number of injections of one embryo (Table 1, last column) and indirectly depended on viral load.
| vector | number of injections/embryo | number of embryos injected | number of pups | number of foster mothers | number of transgenic founders | Implantation efficiency for each variant |
| Syn-TDP-43WTLV | 1 | 48 | 20 | 3 | 0 | 42% |
| 10 | 45 | 0 | 3 | 0 | 0% | |
| 2 | 48 | 8 | 3 | 3 | 17% |
Table 1: Summary of number of subzonal injections of zygotes with Syn-TDP-43WT lentiviral vectors.
| Method | Vector | Titer/ Concentration | Number of injected embryos | Survived embryos | Survival rate | Number of foster mothers | Number of pups | Implantation efficiency | Pregnancy (P) /Pseudopregnancy (PP) |
| PNI | TTYH1-Thy1-EGFP | 1 ng/µL | 1083 | 424 | 39% | 16 | 54 | 13% | PP |
| PNI | H3mCherry | 0.5-2 ng/µL | 2229 | 647 | 29% | 29 | 67 | 10% | PP |
| PNI | Syn-TDP-43-A315T | 2 ng/µL | 1256 | 562 | 45% | 31 | 42 | 7% | PP |
| LV | Syn-TDP-43-A315T | 8.7 x 108 | 115 | 106 | 92% | 7 | 18 | 17% | P |
| LV | Syn-TDP-43 WT | 3.4 x 108 | 152 | 141 | 93% | 9 | 28 | 20% | P |
| LV | LVH3mcherry | 1.3 x 107 | 504 | 450 | 89% | 13 | 115 | 26% | PP |
Table 2: Embryo survival rate and implantation efficiency, depending on the injection method that was used and pregnancy versus pseudopregnancy induction. PNI, pronuclear injection; LV, lentiviral vector subzonal injection.

Figure 1: Microscopic photograph of one-cell-stage rat embryo that was prepared for subzonal lentiviral vector injection. The embryo was immobilized with a holding pipette. Two pronuclei that contained maternal and paternal genetic material and the polar body are visible. Scale bar = 20 μm. Please click here to view a larger version of this figure.
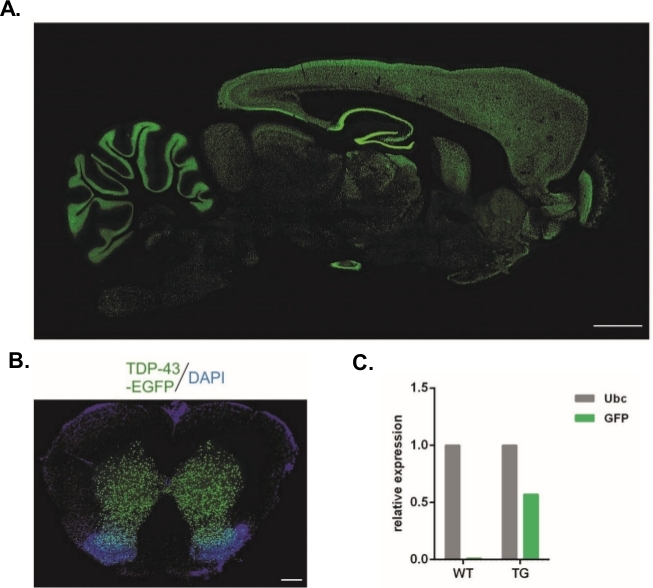
Figure 2: Generation of stable transgenic rat lines that expressed the TDP-43-eGFP fusion protein under control of the neuronal Synapsin-1 promoter throughout the entire central nervous system. (A) Synapsin-1 (Syn)-driven hTDP-43-eGFP expression pattern in a sagittal section of the transgenic rat brain. Scale bar = 3 mm. (B) Coronal section of the spinal cord of a transgenic rat where eGFP fluorescence, counterstained with DAPI, was restricted to gray matter of the spinal cord. Scale bar = 250 μm. (C) Relative expression of GFP transgene transcript compared with the ubiquitin C reference transcript. n = 2 wildtype. n = 2 transgenic. The figure was modified from14. Please click here to view a larger version of this figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Advances in transgenic technologies have made rodent models an invaluable tool in biomedical research. They provide the opportunity to study genotype-phenotype relationships in vivo. Here, we present a widely available alternative for conventional transgenesis by pronuclear injections. The use of lentiviral gene transduction bypasses the need for demanding microinjections because viral vectors can be injected under the zona pellucida. This approach does not affect embryo integrity, which essentially guarantees a 100% survival rate for injected zygotes. The transgene that is incorporated by means of lentiviral vectors is stably integrated into the host genome, allowing long-term expression and germline transmission. Additionally, we present two alternative techniques for modified embryo transfer to foster mothers. One technique utilizes embryo transfer to pseudopregnant females that are previously prepared by mating with vasectomized infertile males. The other technique is based on the use of naturally pregnant females that are mated with fertile males but with a different fur color (i.e., BN rats). This more physiological course of pregnancy allows the proper development of embryos that undergo challenging genetic modifications16.
The first successful attempts to generate transgenic rats were reported in 19907. However, because of difficulties in rat transgenesis17, a relatively small number of transgenic rat lines have been generated in recent decades9. Several main differences are observed between mouse and rat transgenesis using microinjections. For rats, mainly outbred lines (e.g., Wistar and SD) are used for transgenesis. For mice, researchers mainly use the F1 crossbreed of inbred strains because of their higher fertility, better response to hormonal superovulation, and relatively easy development of embryos in vitro from the one-cell stage to blastocysts18. The induction of superovulation in rats is much less efficient than in mice using standard PMSG/hCG hormone stimulation. For this reason, attempts have been made to develop alternative protocols to administer these hormones in rats that utilize continuous FSH infusion instead of a single PMSG administration19. However, superovulation that is caused by PMSG/hCG or FSH/hCG has been shown to have comparable efficiency20. In our opinion, the most critical factor that affects the effectiveness of superovulation is the age of selected females. Nevertheless, the exact parameters should be tested for each rat strain, laboratory, etc.
The procedure for injecting DNA solution into the pronucleus of a single-cell embryo is similar for both rodent species. However, the pronuclei of rat zygotes do not have such regular shapes as in mice and tend to be more difficult to define in the cytoplasm of the cell. Additionally, the rat zygote cell membrane and pronuclear membrane are more elastic and viscous, thus complicating the insertion of a glass micropipette that is loaded with DNA solution. These factors lead to lower rat egg survival rates after the microinjection (31–65% vs. 80% in mice) and explain the lower transgenesis efficiency in rats9. Moreover, intensive, mechanical manipulation of the embryo can also affect implantation efficiency, which in many laboratories, including ours, reaches a maximum of 10%. This relatively low yield is observed even after the implantation of an appropriate number of embryos21.
One method that overcomes the aforementioned difficulties is the infection of single-cell embryos with retroviruses. Retroviruses contain genetic material in the form of RNA, which upon entry into the infected cell is transcribed into DNA by reverse transcriptase of the virus. The DNA is then transported through the nuclear pores to the cell nucleus, where it integrates into the genome of the cell in the form of a provirus. Lentiviral vectors have been used to generate transgenic mice and rats12,14,22. Single-cell embryos that lack a zona pellucida can be incubated in a solution with a lentiviral vector, or the vector can be injected under the zona pellucida into the perivitelline space. The main advantage of this method is its extremely high efficiency, reaching more than 80% of transgenic offspring. After infection with the lentiviral vector, many copies at different sites may integrate into the zygote genome, in contrast to the transgenesis method by pronuclear microinjection, in which one integration site is usually observed12. In offspring of the transgenic founder that is made using lentiviral vectors, individual copies of the transgene are segregated, which may be manifested by different expression profiles of the transgene in each of the progeny. However, this can increase the chance of receiving a subject with the desired expression profile that is derived from the transgene. The restrictions mainly apply to the size of the transgene, which is limited to approximately 8 kb23.
Another difficulty in rat transgenesis is the generation of females that serve as surrogate mothers for genetically modified embryos. In the standard procedure, females are crossed with sterile vasectomized males to induce pseudopregnancy. In rats, the pseudopregnancy assessment technique is much more difficult than in mice, so stimulation with the gonadotropin releasing hormone agonist is sometimes used a few days before mating with males. For these reasons, in the described protocol we provide two alternative approaches to obtain foster mothers. The overall implantation efficiency of manipulated zygotes when pregnant or pseudopregnant females are used is similar. However, the presence of natural, non-manipulated embryos together with manipulated ones can improve the pregnancy rate16. Although the main difference in implantation rate is the manipulation technique (i.e., PNI vs. LV, 10% vs. 20%; see Table 2), the use of pseudopregnant females as foster mothers may be beneficial for some experiments.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
The author (W.K.) has rights to the patent, “Method of producing of a transgenic animal,” from the patent office of the Republic of Poland (no. P 355353; 21.03.2008).
Acknowledgments
This study was supported by the ANIMOD project within the Team Tech Core Facility Plus program of the Foundation for Polish Science, co-financed by the European Union under the European Regional Development Fund to WK.
Materials
| Name | Company | Catalog Number | Comments |
| 7500 Real Time PCR System | Applied Biosystems | ||
| Aerrane (isoflurane) | Baxter | FDG9623 | |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma | A5177-5EA | |
| Atipam 5 mg/ml | Eurovet Animal Health BV | N/A | 0.5 mg/kg |
| Baytril 25 mg/ml (enrofloksacin) | Bayer | N/A | 5-10 mg/kg |
| Borosilicate glass capillaries with filament GC100TF-15 | Harvard Apparatus Limited | 30-0039 | injection capillary |
| Bupivacaine 25 mg/ml | Advanz Pharma | N/A | 0.25% in 0.9% NaCl |
| Butomidor 10 mg/ml (butorphanol tartrate) | Orion Pharma | N/A | 1 mg/kg |
| CELLSTAR Tissue Cell Culture Dish 35-mm | Greiner Bio-One | 627160 | |
| CELLSTAR Tissue Cell Culture Dish 60-mm | Greiner Bio-One | 628160 | |
| CellTram Oil | Eppendorf | 5176 000.025 | |
| Cepetor (Medetomidine) 1 mg/ml | cp-pharma | N/A | 0.5 mg/kg |
| Chorulon, Human Chorionic Gonadotrophin | Intervet | N/A | 150 IU/ ml ml 0.9% NaCl |
| DMEM low glucose | Sigma Aldrich | D6048 | |
| DNase, RNase-free | A&A Biotechnology | 1009-100 | |
| EmbryoMax Filtered Light Mineral Oil | Sigma | ES-005-C | |
| Envelope protein coding plasmid for lentiviral vectors (VSVg plasmid) | ADDGENE | 14888 | |
| FemtoJet | Eppendorf | 4i /5252 000.013 | |
| Fetal Bovine Serum | Sigma Aldrich | F9665-500ML | |
| Folligon, Pregnant Mare’s Serum Gonadotropin | Intervet | N/A | 125 IU/ml in .9% NaCl |
| HEK 293T cells | ATCC | ATCC CRL-3216 | |
| Hyaluronidase from Bovine Testis | Sigma | H4272-30MG | 0.5 mg/ml in M2 medium |
| Inverted Microscope | Zeiss | Axiovert 200 | |
| Ketamine 100mg/ml | Biowet Pulawy | N/A | 50 mg/kg |
| Liquid Paraffin | Merck Millipore | 8042-47-5 | |
| M16 medium EmbryoMax | Sigma | MR-016-D | |
| M2 medium | Sigma | M7167 | |
| Magnesium Chloride 1M | Sigma Aldrich | 63069-100ML | |
| Microforge | Narishige | MF-900 | |
| Mineral Oil | Sigma | M8410-500ML | |
| NaCl 0.9% | POLPHARMA OTC | N/A | sterile, 5ml ampules |
| Operation microscope | Inami Ophthalmic Instruments | Deca-21 | |
| Packaging system coding plasmid for lentiviral vectors (delta R8.2 plasmid) | ADDGENE | 12263 | |
| PEI reagent (Polyethylenimine, Mw ~ 25,000,), | Polysciences, Inc | 23966-1 | |
| Penicilin-streptomycin | Sigma Aldrich | P0781-100ML | |
| Phosphate Buffered Saline, pH 7.4, liquid, sterile-filtered, suitable for cell culture | Sigma Aldrich | 806552-500ML | |
| Puller | Sutter Instrument Co. | P-97 | |
| Reflex Clip Applier/Reflex Clips | World Precision Instruments | 500345/500346 | |
| Safil, polyglycolic acid, braided, coated, absorbable threads | B.Braun Surgical | 1048029 | |
| Stereomicroscope | Olympus | SZX16 | |
| Surgical Sewing Thread | B.Braun | C1048040 | |
| SYBR Green PCR Master Mix | Applied Biosystem | 4334973 | |
| Tolfedine 4% (tolfenamic acid) | Vetoquinol | N/A | 2 mg/kg |
| TransferMan NK2 | Eppendorf | N/A | |
| Trypsin EDTA solution | Sigma Aldrich | T3924-500ML | |
| Ultracentrifuge | Beckman Coulter | Optima L-100 XP | |
| VacuTip | Eppendorf | 5175108.000 | holders capillary |
| Vita-POS | Ursapharm | N/A | eye ointment |
| Warming Plate | Semic | N/A | |
| Watchmaker Forceps | VWR | 470018-868 |
References
- Lazar, J., Moreno, C., Jacob, H. J., Kwitek, A. E. Impact of genomics on research in the rat. Genome Research. 15 (12), 1717-1728 (2005).
- Tarkowski, A. K. Studies on mouse chimeras developed from eggs fused in vitro. National Cancer Institute Monographs. 11, 51-71 (1963).
- Gordon, J. W., Ruddle, F. H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science. 214 (4526), 1244-1246 (1981).
- Gill, T. J., Smith, G. J., Wissler, R. W., Kunz, H. W. The Rat as an Experimental Animal. Science. 245 (4915), 269-276 (1989).
- Aitman, T. J., et al. Progress and prospects in rat genetics: a community view. Nature Genetics. 40 (5), 516-522 (2008).
- Hammer, R. E., Maika, S. D., Richardson, J. A., Tang, J. P., Taurog, J. D. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 63 (5), 1099-1112 (1990).
- Mullins, J. J., Peters, J., Ganten, D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature. 344 (6266), 541-544 (1990).
- Menoret, S., Remy, S., Usal, C., Tesson, L., Anegon, I. Generation of Transgenic Rats by Microinjection of Short DNA Fragments. Rat Genomics: Methods and Protocols. 597, 81-92 (2010).
- Tesson, L., et al. Transgenic modifications of the rat genome. Transgenic Research. 14 (5), 531-546 (2005).
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: Technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Ritchie, W. A., Neil, C., King, T., Whitelaw, C. B. Transgenic embryos and mice produced from low titre lentiviral vectors. Transgenic Research. 16 (5), 661-664 (2007).
- Lois, C., Hong, E. J., Pease, S., Brown, E. J., Baltimore, D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 295 (5556), 868-872 (2002).
- Pfeifer, A., Ikawa, M., Dayn, Y., Verma, I. M. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proceedings of the National Academy of Sciences of the United States of America. 99 (4), 2140-2145 (2002).
- Koza, P., et al. Neuronal TDP-43 depletion affects activity-dependent plasticity. Neurobiology of Disease. 130, 104499 (2019).
- Scherr, M., Battmer, K., Blomer, U., Ganser, A., Grez, M. Quantitative determination of lentiviral vector particle numbers by real-time PCR. Biotechniques. 31 (3), 520 (2001).
- Canseco, R. S., et al. Gene transfer efficiency during gestation and the influence of co-transfer of non-manipulated embryos on production of transgenic mice. Transgenic Research. 3 (1), 20-25 (1994).
- Charreau, B., Tesson, L., Soulillou, J. P., Pourcel, C., Anegon, I. Transgenesis in rats: technical aspects and models. Transgenic Research. 5 (4), 223-234 (1996).
- Brinster, R. L., Chen, H. Y., Trumbauer, M. E., Yagle, M. K., Palmiter, R. D. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proceedings of the National Academy of Sciences of the United States of America. 82 (13), 4438-4442 (1985).
- Armstrong, D. T., Opavsky, M. A. Superovulation of immature rats by continuous infusion of follicle-stimulating hormone. Biology of Reproduction. 39 (3), 511-518 (1988).
- Popova, E., Krivokharchenko, A., Ganten, D., Bader, M. Comparison between PMSG- and FSH-induced superovulation for the generation of transgenic rats. Molecular Reproduction and Development. 63 (2), 177-182 (2002).
- Johnson, L. W., Moffatt, R. J., Bartol, F. F., Pinkert, C. A. Optimization of embryo transfer protocols for mice. Theriogenology. 46 (7), 1267-1276 (1996).
- van den Brandt, J., Wang, D., Kwon, S. H., Heinkelein, M., Reichardt, H. M. Lentivirally generated eGFP-transgenic rats allow efficient cell tracking in vivo. Genesis. 39 (2), 94-99 (2004).
- Remy, S., et al. The Use of Lentiviral Vectors to Obtain Transgenic Rats. Rat Genomics: Methods and Protocols. 597, 109-125 (2010).
