Room temperature (RT) macromolecular crystallography is popular again within the structural biology community. The development of X-ray Free Electron Laser (XFEL) light sources has spurred the development of RT sample delivery approaches1,2,3,4, and these methods have now been applied to synchrotrons5,6,7,8. Not only do RT methods open up the possibility of pump-probe experimental strageties9,10,11,12, but there is also mounting evidence that they promote alternative conformational states within proteins13,14,15,16,17.
However, the principal reason why cryo-methods gained traction over RT approaches in the late 1990s was the slowing of radiation damage by sub-zero crystal temperatures18. Cryo-methods19 began to allow for the collection of a complete dataset from a single protein crystal. Modern RT methods at XFELs and synchrotrons solved the problem of single-crystal radiation damage by the development of rapid (> 100 Hz) crystal delivery strategies1,2,3,4. These methods allow for the collection of a complete dataset from thousands of individually exposed crystals. These RT delivery approaches therefore require the production of large quantities of solutions containing homogenous micro-crystals (> 100 µL of < 50 µm crystals). However, since cryo-methods tend to only require single crystals, methods to create such micro-crystalline slurries are currently not ubiquitous across protein crystallography laboratories.
There are examples in the literature of how to do parts of the micro-crystallization optimization procedure for serial crystallography samples. Here, a distinction should be made between membrane and soluble proteins. Protocols to optimize the growth of micro-membrane protein crystals grown in monoolein (or some other lipid), for lipidic cubic phase (LCP), have been well described20,21,22. However, methods for the micro-crystallization of soluble proteins, including membrane proteins grown in non-LCP conditions, are generally lacking. Previous studies have focused on specific parts of the process, such as micro-crystal screening23,24, enhancing nucleation24, and scaling using free-interface diffusion25, but not a complete method.
However, a method was recently described26 that attempts to offer a complete protocol. Like many aspects of protein crystallography, it is not new. Many of the ideas proposed were already described by Rayment (2002)27. The method aims to show crystallographers how to perform the conversion from a single, crystal grown using vapor diffusion, to a batch methodology to grow thousands of micro-crystals. The method focuses on vapor diffusion as a common starting point, as 95% of all Protein Data Bank (PDB) depositions come from crystals grown in vapor diffusion plates26. Vapor diffusion is, however, not the ideal method for micro-crystallization26, so a methodology is described to convert vapor diffusion to batch crystallization. Once crystals can be grown in batch, scaling routes to larger volumes become more practicable. Given the vagaries of protein crystallization, the authors would stress that this method is not failsafe. However, the protocol should, at least, provide an insight into the 'crystallization space' of a protein.
This method relies on the protein crystallization phase diagram and how an understanding of that diagram can act as a guide during micro-crystallization optimization. A protein phase diagram is commonly depicted as an x/y plot with precipitant and protein concentrations on the x and y axes, respectively (Figure 1A). From the pure water point (bottom left corner – Figure 1A), the concentration of both protein and precipitant increases until the solubility line is reached. The solubility line marks the point of supersaturation (purple line – Figure 1A). When a protein is supersaturated, the solution becomes thermodynamically unstable and will begin separating into two phases: 'protein-rich' and a stable saturated solution. This separation can occur anywhere beyond the solubility line and its kinetics are dependent upon the properties of the protein and the components of the solution.
When the protein and precipitant concentrations are too great, the protein will decompose unstably out of solution and result in amorphous precipitate (pink region – Figure 1A). However, ordered phase separation can occur in the nucleation region [see Garcia-Ruiz (2003)28 for detailed description] and crystal nucleants have the propensity to form (green region – Figure 1A). Nucleation and growth removes protein from the solution and moves the drop into the metastable region where growth can continue until the solubility line is reached [see McPherson and Kuznetsov (2014)29 for detailed discussion]. The diagram is, for the vast majority of crystallization conditions, a gross oversimplification30. Regardless of this however, the diagram is still of great utility for micro-crystallographers as the mapping of the diagram allows for the solubility line and the kinetics of nucleation to be determined.
In terms of creating micro-crystals, the two factors during crystallization that need to be optimized are the number of crystals (Xn) and their mean, longest dimension (Xs). Xn will be proportional to the number of nucleation events (n) (Eq. 1).
 Eq. 1
Eq. 1
Xs is proportional to the concentration of free protein above the solubility line (Ps) divided by Xn (Eq. 2).
 Eq. 2
Eq. 2
In a perfect situation, every nucleation event would yield a possible crystal and every one of these crystals, would have equal access to the available protein in solution. Figure 2 is a graphical representation from an ideal scenario of the relationship between Xn and Xs. Practically, the principal control a crystallographer has over Xn and Xs is by influencing the amount of nucleation or by the addition of seed crystals. The micro-crystallographer must judge how to increase Xn such that a suitable crystal concentration and crystal size can both be created.
The majority of crystallization techniques require a 'transitionary period' (Figure 1B). For example, in a vapor diffusion experiment, upon mixing the protein and precipitant solutions, the concentrations of each will change as the drop equilibrates with the well solution. One hopes that these changes will gradually transition the drop into the nucleation zone where the propensity for crystallization will increase. As crystals begin to nucleate and grow, the amount of protein in solution will begin to fall, decreasing the probability of further nucleation. The ultimate amount of nucleation will be protein and condition specific, and also dependent upon the depth of penetration of into the nucleation zone. Given the limited nucleation zone penetration of methods that require a transitionary step, the level of nucleation will ultimately be limited to the rate of nucleation at the metastable-nucleation region boundary.
Due to the importance of being able to enhance the level of nucleation for a micro-crystallographer, it is important to move to a batch crystallization methodology. Batch can take greater advantage of the whole nucleation region (Figure 1C). In batch methods, the idea is to mix the protein and precipitant together such that a supersaturated solution is created without the need of any changes in component concentrations. Nucleation should be possible immediately upon mixing. Batch methods therefore allow for the entire nucleation zone to be theoretically reached. Any increase in nucleation kinetics beyond the metastable-nucleation boundary can then be utilized.
If the basal-level of crystal nucleation is not enough to generate a large Xn, micro-seeding methods can be used. In micro-seeding, pre-grown crystals are broken up to create a slurry of crystalline fragments which can act as a scaffold for fresh crystal growth31,32. Micro-seeding has been widely used in serial crystallographic sample preparation as a way to increase Xn without the need of increasing crystal nucleation (Figure 1C).
The transition from vapor diffusion to batch can be visualized on a phase diagram as moving the experimental starting point from either the non-supersaturated or metastable regions to the nucleation zone. This can be done by increasing the protein and/or precipitant concentrations, and/or the ratio of the two within the drop (Figure 1D), and observing which conditions yield crystals appearing rapidly (< 24 h)26. Complete vapor diffusion drop equilibration can take days or weeks33. Therefore, by looking for conditions that show rapidly appearing crystals, batch conditions can be found without having to move to alternative crystallization screening formats such as micro-batch34,35,36,37.
Once the nucleation zone has been found, a batch condition has been found and a morphogram – here, a rough phase diagram – can be created. The morphogram is of great utility when contemplating whether to use a seeded-batch or straight batch protocol. By plotting the Xn as a function of the protein and precipitant concentration, an assessment of the nucleation kinetics can be made26. If Xn remains low across the whole nucleation region, seeded-batch may be required to make Xn large enough to limit crystal growth. This assessment is the first step in the process of scaling to larger volumes (> 100 µL).
This method was designed such that it could be conducted in the majority of crystallization laboratories by using standard vapor diffusion crystallization equipment. Many studies have also been conducted which describe techniques to facilitate many parts of this process, should the equipment be available. These include, but are not limited to, dynamic light scattering (DLS)25,27, non-linear imaging20,24,25, powder diffraction20,24,27, and electron microscopy26 [see Cheng et al. (2020)40 for a nice review].
The aim of this work is to provide a visual demonstration of the method to transition from small volume (< 500 nL) vapor diffusion crystallization to large volume (> 100 µL) batch crystallization. Endothiapepsin from Cryphonectria parasitica has been used as an example system for demonstrating this translation. The type of experiment and sample delivery method that the micro-crystals are required for will influence the ideal Xs output26. For mixing experiments requiring a millisecond time resolution41 or gas-dynamic virtual nozzles42, a final Xs of < 5 µm may be desirable. In this case, the goal was to produce protein crystals that diffract to approximately 1.5 Å, for a photon-activated pump-probe experiment, and using a fixed-target delivery approach.
To give an illustration of the sample requirements of such a serial crystallography experiment using endothiapepsin, Table 1 shows the experimental parameters of a hypothetical experiment. The sample information was based upon the protocol described below. Given some conservative estimates on hit rates and data collection requirements, 50 mg is the total sample consumption estimate for the whole experiment.
Figure 3 shows a flow-chart of the complete optimization process from initial small volume vapor diffusion crystallization to large scale batch. For the majority of serial crystallography projects, this protocol will begin at Step 2: 'transitioning to batch', since the target protein will already have been crystallized. However, Step 1 has been included for completeness and to remind readers of its importance. Finding a condition that gives rise to a well diffracting, single, large crystal is the best starting point for micro-crystal optimization. In Step 2, this condition can then be optimized from vapor diffusion to batch, and a morphogram of the nucleation and metastable regions can be plotted. Once this has been done, scaling the batch condition to larger volumes can be performed in Step 3. By the end of the flow-chart, a crystallographer will have created a repeatable, large-volume (> 100 µL), micro-crystallization, batch protocol for endothiapepsin. This method can then be applied to their particular protein of interest.
Optimizing crystal morphology
Step 1, optimizing crystal morphology, has been included to remind reader of its importance. It may be possible to create perfect micro-crystals from poorly diffraction needle-balls; however, the authors would suggest that it is better to optimize the two separately. First, find conditions that give rise to well-diffracting, single crystal via vapor diffusion, and then convert these conditions into batch rather than trying to do combine the two steps together. Discovering highly nucleating conditions, at this stage, is not necessary; morphology and diffraction quality are the principal goals.
Before beginning the micro-crystallization of endothiapepsin, an analysis of deposited structure crystallization conditions from the PDB was conducted. Crystallization conditions and approximate protocols could be obtained for 47 of the 48 depositions of enthothiapepsin. These were broadly all based upon the first crystallization of endothiapepsin conducted by Moews and Bunn (1970)46. Given the similarities of these conditions and their 'classical' origin, a 96-well, vapor diffusion, sparse-matrix screen was performed to explore a wider variety of crystallization conditions. Endothiapepsin was concentrated to 70 mg/mL and a PACT sparse-matrix screen47 was performed in a 96-well sitting-drop plate at 20 °C mixing 100 nL of protein with 100 nL of well solution. Every condition from this experiment after 36 h gave rise to crystals. However, an analysis of the crystal morphology indicated that some conditions might prove better for micro-crystallization optimization.
Figure 4A shows a drop from the PACT screen that was broadly representative of those observed in the majority of the plate. At first glance, it may be tempting to think that these crystals might be worth optimizing further for micro-crystallization. The crystals are large and there appears to be significant nucleation. However, the overall crystal morphology is not ideal. Firstly, the crystals are not observably singletons as it appears that multiple crystals are growing from single nucleation points. Secondly, the crystal size is highly asymmetric with growth principally occurring down a single axis. Such crystals are theoretically more likely to preferentially align when delivered to the X-ray beam. Both characteristics present problems during collection and processing of serial crystallographic data.
Figure 4B, however, shows endothiapepsin crystals grown in the presence of MgCl2. This morphology was consistent across all conditions that contained MgCl2 and therefore suggested that their morphology was due to MgCl2. The MgCl2 conditions produced single, more box-like crystals that represented a better target for the ultimate serial experiments.
There were four conditions within the PACT screen that contained MgCl2. To better understand the influence of all the different components of these conditions on endothiapepsin crystallization, a random optimization was performed. A screen was created containing a random combination of the buffers and precipitants at a range of concentrations and pHs. The MgCl2 concentration was also varied and then the resulting drops were arbitrarily graded from 0-5 (0 being no crystals or precipitation) in terms of their visual crystal quality and precipitation level.
Figure 5A shows a heatmap of the results from a Pearson's correlation analysis between the precipitation level and crystal quality, and the screen variables (examples of the drops from this experiment are shown in Figure 5B, C and D). The results indicated that the pH of the solution was highly correlated to the level of precipitation, with alkaline buffers resulting in more precipitation. MgCl2 concentration was slightly correlated to the level of precipitation, as was the pH and precipitant concentration to crystal quality.
Based on these results, the decision was taken to take the crystals grown in 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2, 20% (w/v) PEG 6,000 to the next step of the protocol - Transitioning to batch. The morphology of crystals was acceptable and an analysis of the X-ray diffraction and data quality metrics from these crystals suggested that there was no significant difference between the crystals grown in and out of the presence of Mg2+ (Figure 9).
Transitioning to batch
For many serial crystallography micro-crystallization optimizations, Step 2 will be the starting point. The protein of interest will have already been crystallized for cryo-crystallography and the crystallization protocol will now need transforming to create micro-crystal slurries. This protocol has only used 96-well vapor diffusion plates to perform the transformation to batch since vapor diffusion is the crystallization method used by 95% of PDB entries26. The protocol has avoided moving into microbatch34,35,37 since this transition might still incur a similar optimization. This is not to say that this protocol can only be done in vapor diffusion plates. All of the steps presented, would also work in microbatch if this was the original crystallization method.
To assess the crystallization of endothiapepsin in the chosen condition, a morphogram – or a rough phase diagram – was created. The purpose of the morphogram experiment is threefold. Firstly, an analysis of the morphogram is of great utility when assessing scaling routes in Step 3 - Scaling. Secondly, the morphogram acts as an optimization tool, helping to discover vapor diffusion conditions that give rise to crystals via batch [i.e., rapidly appearing crystals (< 24 h)]. Thirdly, if crystals have not appeared rapidly, an analysis of the seeded drops can give the crystallographer an idea of the approximate location of the current condition on the phase diagram. For example, if the seeded conditions give crystals but the unseeded do not, those conditions are likely to be in the metastable region.
The morphogram experiment of endothiapepsin was performed based on the 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2, 20% (w/v) PEG 6,000 condition. The protein and PEG concentrations were varied from 100 to 12.5 mg/mL and 5 to 40% (w/v), respectively. The drops were analyzed and results plotted using the worksheet provided (Figure 6A).
It was also already clear from the Optimizing crystal morphology stage that endothiapepsin crystal growth in this condition, and at these protein concentrations, would result in crystals grown in under 24 h. This indicated that crystallization was occurring via a batch rather than a vapor diffusion driven process. The crystal grown in these conditions were, therefore, suitable for scaling to larger volumes.
If crystals had not been visible in the unseeded-drops after 24 h, then it would have been likely that crystallization was still dependent upon a transition (Figure 1B) and, therefore, not batch. In this case, the results from the morphogram experiment are still of interest. They give an indication of the probable starting point for crystallization on the phase diagram and hence, how the subsequent optimization should proceed. Look at the seeded drops. The seeds will allow for crystal growth in the metastable region regardless of nucleation. For example, if crystals appear within 24 h in the seeded-drops but not the unseeded drops, this indicates part of the metastable region can be observed. If no crystals are observed in either the seeded or unseeded-drops, all wells remain undersaturated.
Scaling
Looking at the morphogram (Figure 6A), a number of observations could be made. The amount of nucleation did appear to be affected by both the protein and precipitant concentrations. There was also a very clear demarcation of drops that lead to protein precipitation, with drops either containing: nothing, crystals or precipitate (Figure 6B). The addition of seeds (Figure 6D) also greatly increased Xn when compared to the drops without seeds (Figure 6C).Taking all these results together, it was decided to attempt to scale both a batch and seeded-batch protocol at 30% (w/v) PEG 6,000 and 100 mg/mL endothiapepsin.
The initial test scaling was done in 24-well hanging drop plates. The drop volumes were gradually increased so that any changes in crystallization behavior could be observed (Figure 7). As can be seen, in both the unseeded and seeded drops crystal growth has occurred. All the unseeded drops grew a range of crystal sizes, but predominantly large crystals (100-200 µm – longest dimension). The seeded drops, however, produced smaller crystals (5 – 50 µm – longest dimension). These initial tests suggested that seeds would be required to decrease Xs, but also, that this condition should be suitable for larger volumes.
When the volume was increased in 200 µL, the crystallization volume was continually agitated during crystal growth. The principal reason for this agitation was to ensure that the crystallization solution remains homogenous and that growing crystals do not settle on the bottom or sides of the tubes. Settling of crystals can lead to a heterogenous crystal population with both very large and small crystals. Agitating the crystallization solution can also promote nucleation44,45.
Unfortunately, the unseeded 30% (w/v) PEG 6,000 produced no crystals, so the PEG concentration was increased to 35% (w/v). This increase improved the crystallization markedly, with a final Xn and Xs range of 3.6 ± 1.2 x 106 crystals·mL-1 and 42 ± 4.1 µm, respectively (Figure 8A and B – black). Although a significant improvement and an acceptable crystal concentration, the final crystals were too large for the planned experiment, so further optimizations were undertaken. To reduce the size of the final crystals two avenues were explored (Figure 1E): decreasing the protein concentration to try and limit the final crystal growth (Figure 8A and B – hot pink), and increasing the PEG concentration to try and increase nucleation (Figure 8A and B – green).
The reduction of the protein concentration unfortunately also dramatically reduced the Xn, which ultimately produced even larger crystals. Increasing the PEG concentration to 40% yielded a final Xn and Xs range of 3.1 ± 0.7 x 106 crystals·mL-1 and 39 ± 2.3 µm, respectively. These were not significantly different to the 35%, but since the final crystal size was reduced, this condition was continued with the further optimizations.
To increase the Xn, seeds were added. This dramatically increased the Xn (1.1 ± 1.8 x 108 crystals·mL-1) and lead to a smaller Xs (4.2 ± 4.0 µm) (Figure 8A and B – dashed purple). These crystals, although very suitable for some serial crystallography experiments, were deemed too small so the concentration of the added seeds was changed.
This tuning of the added seed stock, however, proved difficult to reliably repeat; therefore, quenching was attempted. After the addition of a seed stock, the crystal size was monitored and once a suitable crystal size was achieved (approximately 10 – 20 µm), the batch crystallization was quenched (Figure 8C and D). Quenching was proposed, with regard to micro-crystallization, in Kupitz et al. (2014)25. Although perhaps not an ideal method, as protein solution will ultimately be wasted26, the technique was very useful in this situation as crystal growth was difficult to control. The idea behind quenching is to rapidly return the crystallization mixture to a point just above the solubility line (Figure 1F). Once the solution has returned to the solubility line, the solution has returned to a stable saturated solution and no further crystal growth will occur.
Attempting to quench a crystallization reaction is not without risk. If too great a quenching solution is added, the protein in solution might be diluted so much that the solubility line is passed. In this case, the solution will become undersaturated and the crystals will start dissolving. To prevent this, it is possible to estimate the amount of required quenching solution based on the morphogram results. At the point of quenching, take the concentration of the protein solution. By comparing the protein concentration at the solubility line and the protein concentration in solution, an estimate of the required dilution can be made.
The quenched version of the 40% (w/v) PEG 6,000, 10 x diluted seed experiment gave a final crystal concentration and size range of 2.6 ± 3.1 x 106 crystals·mL-1 and 15 ± 3.9 µm, respectively.
Throughout the entire process, test X-ray data collections of the endothiapepsin crystals were collected at the Swiss Light Source PXII beamline using a 10 x 30 µm focus, an energy of 12.4 keV attenuated by 80%, and under cryo-conditions. The data were processed using dials and Figure 9 shows a comparison of CC½. No dramatic change in in CC½ was observed over the course of the optimization.
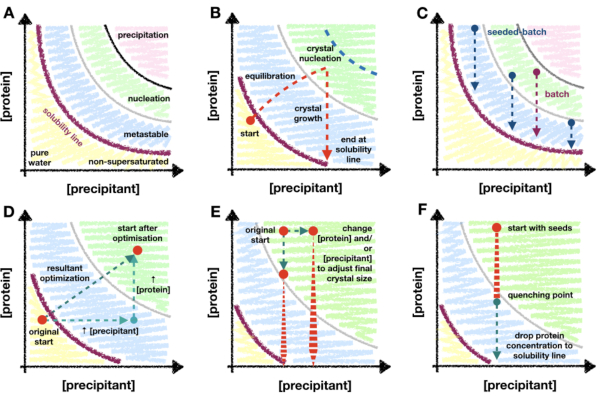
Figure 1: An overview of transitionary and batch crystallization, and scaling methods mapped onto a phase diagram. A. The zones and limits of the archetypical protein crystallization phase diagram. The precipitant and protein concentrations are plotted on the x and y axes, respectively, with the pure water point at the origin. The purple line indicates the protein supersaturation boundary, and the metastable, nucleation, and precipitation zones are shown in blue, green and pink, respectively. B. An example of the nucleation zone penetration limits of a 'transitionary phase' crystallization method, such as vapor diffusion. In this theoretical experiment, the drop precipitant and protein concentrations begin just below the solubility line – not yet supersaturated. While the drop equilibrates, the drop component concentrations increase such that the drop becomes supersaturated, and continues to move – or transition – into the nucleation zone. Upon crystal nucleation, the protein concentration in solution begins to drop. The concentration continues to fall as crystals grow until finally stopping at the solubility line. The blue dotted line marks a theoretical limit of the transition into the nucleation zone. As soon as nucleation begins, the protein concentration will drop, preventing further penetration. C. Example batch and seeded-batch crystallization trajectories. In batch, the mixing of the protein and precipitant must create a supersaturated solution within the nucleation zone so that crystal growth can occur. In seeded-batch, it is not strictly necessary to be in the nucleation zone due to the addition of micro-seeds, so locations in the metastable region can also be explored. D. A hypothetical optimization of the crystallization experiment shown in B from vapor diffusion to batch. The original vapor diffusion starting point has transitioned, via the resultant optimization vector, to the new start position; inside the nucleation zone. The resultant vector is the product of two optimizations: an increase in both protein and precipitant concentrations. E. Example optimizations when scaling batch conditions to tailor the final Xn and Xs. F. Quenching the crystallization experiment by the addition of crystallization buffer. It is essential that the quenching does not take the protein concentration out of the metastable region and, therefore, below the point of protein supersaturation. Otherwise, crystals will start to dissolve back into solution. B. and C. have been adapted from Beale et al. (2019)26 with the permission of the authors. Please click here to view a larger version of this figure.
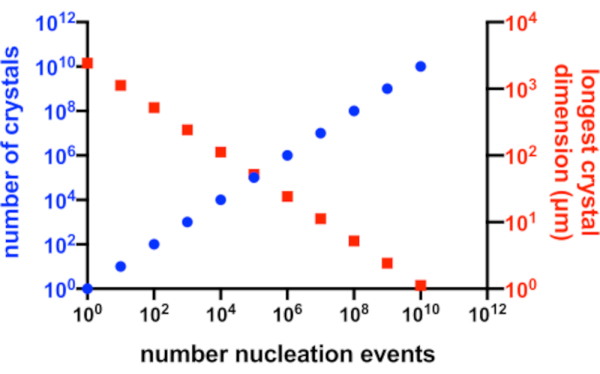
Figure 2: Increasing Xn and decreasing Xs. The idealized relationship between the number of crystals produced from a crystallization experiment and their mean longest dimension. To create this graph, the crystallization of a hypothetical 10 kDa model protein was used. The protein crystallized at a concentration of 10 mg/mL and yielded P212121 crystals with dimensions of 49x50x51 Å. Every nucleation event was assumed to yield a crystal. Crystal growth was assumed to be homogeneous from every face. Please click here to view a larger version of this figure.
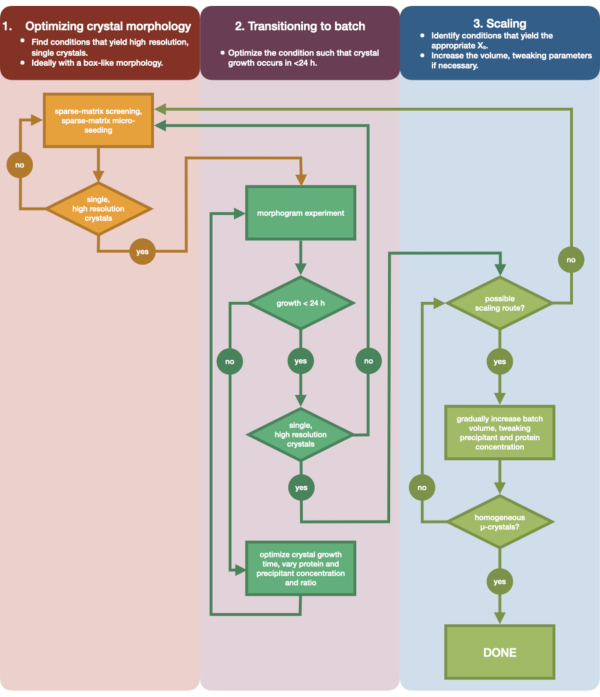
Figure 3: A flow-chart showing the steps to optimize a crystal grown in a small-volume (<500 nL), vapor diffusion experiment into a large-volume (> 100 µL) batch experiment. Crystal optimization is divided into three stages: (1) Optimizing crystal morphology. (2) Transitioning to batch. (3) Scaling. In Stage 1 it is important to identify suitable crystals for micro-crystallization. Some proteins only present in a single crystal morphology regardless of the crystallization condition. However, it is worth looking for conditions that give rise to single, cube-like crystals, or as close to these as humanly possible. Single, cube-like crystals, hypothetically and anecdotally, will generally give rise to better outcomes from serial crystallography experiments. Once a crystal morphology has been selected and the diffraction confirmed, it is then necessary to move the crystallization experiment from vapor diffusion to batch (Stage 2). Here, crystals should be optimized by their nucleation time. The goal is to find conditions that yield rapidly appearing crystals (> 24 h) as these conditions are likely to hit the nucleation zone immediately, and are therefore batch. Once a condition in the nucleation zone has been found, a morphogram can be created. The morphogram allows for the majority of the nucleation zone to mapped and potential scaling routes identified for Stage 3. The volume of an identified batch condition can then be either gradually or rapidly scaled in size to yield a final volume of >100 µL. Please click here to view a larger version of this figure.

Figure 4: An analysis of endothiapepsin crystallization conditions from a PACT sparse-matrix screen. A. and B. are photos after 24 h of wells A4 and C10, respectively from the PACT screen. The crystallization buffer components are highlighted on the figure. The SPG buffer is succinic acid, sodium dihydrogen phosphate, and glycine mixed in a 2:7:7 molar ratio. Please click here to view a larger version of this figure.
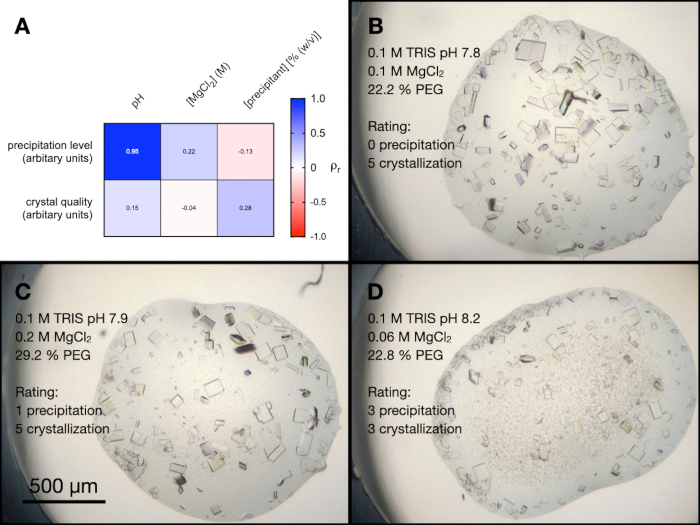
Figure 5: An analysis of the endothiapepsin crystallization optimization from the PACT MgCl2 conditions. A. A heat map of the results from a Pearson's correlation analysis between buffer pH, MgCl2 concentration, and precipitant concentration and the precipitation level and crystal quality. The precipitation level and crystal quality were both assessed arbitrarily on a scale of 0-5 (with 0 being no crystals or precipitation) after 24 h. B. C. and D. show examples of the crystallization and precipitation in three different drops. The crystallization condition and assessments of the precipitation level and crystal quality are also shown. Please click here to view a larger version of this figure.
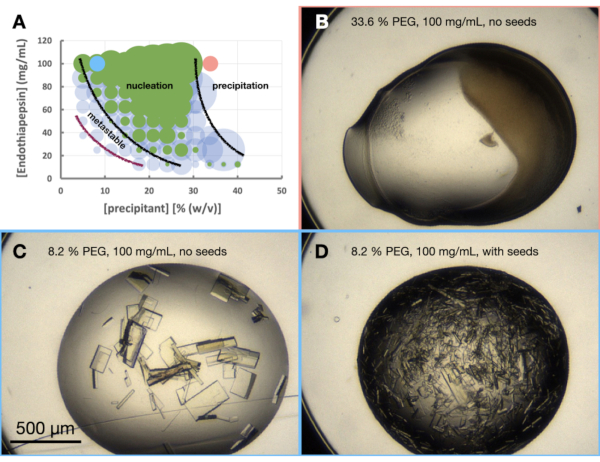
Figure 6: An endothiapepsin morphogram when crystallized in 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2 and PEG 6,000. A. A morphogram created from the "phase-diagram-generator" spreadsheet provided. The relative number of crystals in each drop is denoted by the size of the circles, and the results from drop 1 (protein and precipitant) and drop 2 (protein, precipitant and seeds) are highlighted in green and blue, respectively. The values of the protein and precipitant concentrations, on the x and y axis, respectively, denote pre-mixed values of each rather than final volumes. Based on the results, black lines and a purple line have been drawn to show the boundaries of the nucleation zone and metastable zone, respectively. B. C. and D. show some example results from the experiment. The red and blue dots marked on A. indicate the locations of B., and C. and D., respectively. Please click here to view a larger version of this figure.

Figure 7: Initial scaling trials of endothiapepsin in 24-well hanging drop plates. The same protein and precipitant concentrations were used for all trails: 100 mg/mL endothiapepsin in 0.1 M Na Acetate pH 4.6 and 0.1 M Tris-HCl pH 7.0, 0.15 M MgCl2, and 30% (w/v) PEG 6,000, respectively. All of the displayed images were taken after 24 h and the final drop volumes are labelled on each image. The left panel (A, D, and G) are a 1:1 mix of protein and precipitant, the middle panel (B, E, and H) are a 1:2:3 mix of seeds, precipitant, and protein and the right panel (C, F, and I) are magnified images of the middle panel. Please click here to view a larger version of this figure.
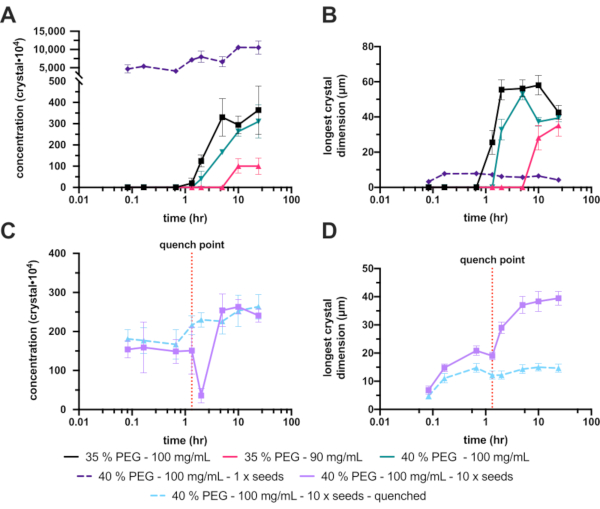
Figure 8: Analysis of the endothiapepsin micro-crystallization in 200-300 µL volumes. A. and C. show how Xn changed over the experiment time. B. and D. show how Xs (longest dimension) changed over time. The results of the experiments have been separated for clarity. The red dotted line on C. and D. show the point at which quenching was performed. Please click here to view a larger version of this figure.
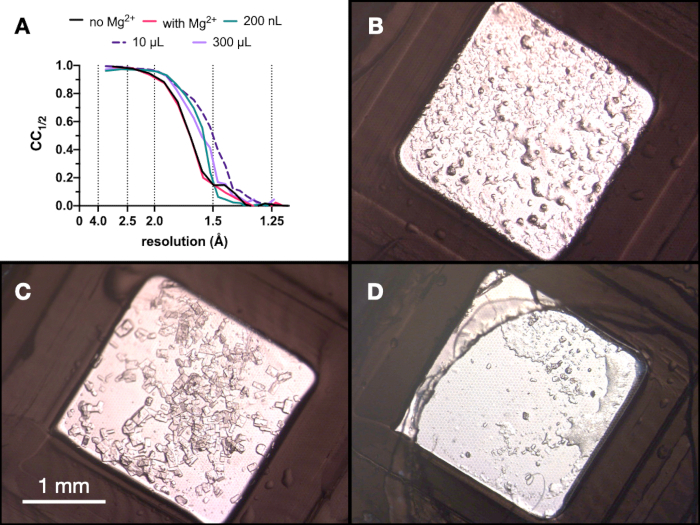
Figure 9: CC½ results and images of crystals obtained at each stage of the micro-crystallization process to assess diffraction quality. A. CC½ plotted against resolution from data collected from crystals grown: with and without Mg – part of the Stage 1 optimization, in a 200 nL volume, a 10 µL volume and the final 300 µL volume. B.C. and D. show the crystals from the 200 nL, 10 µL and 300 µL volume, respectively. Please click here to view a larger version of this figure.
| Protein Information | |
| Protein | Endothiapepsin |
| Molecular Weight (kDa) | 33.8 |
| Spacegroup | P1211 |
| a, b, c (Å) | 45.2, 73.3, 52.7 |
| α, β, ɣ (°) | 90.0, 109.2, 90.0 |
| Fixed-target parameters | |
| Volume loaded per chip (µL) | 150 |
| Aperatures per chip | 25,600 |
| Required crystal concentration (crystals/mL) | 500,000 |
| Sample Information | |
| Protein mass used to make 200 µL of sample (mg) | 10 |
| Crystal longest dimension (µm) | 15 |
| Crystal concentration (crystals/mL) | 2,500,000 |
| Experimental Variables | |
| Number of time points required | 5 |
| Number of images required per time point | 50,000 |
| Hit rate (integrated patterns/images collected) | 0.3 |
| Fixed-targets required per time point (rounded up) | 7 |
| Sample requirements | |
| Sample volume required per time point (µL) | 1,050 |
| Total sample volume required for experiment (mL) | 5.25 |
| Total mass of protein required (mg) | 52.5 |
Table 1: An example of the sample requirements for a hypothetical optical pump-probe experiment performed using fixed-targets. The protein used in this theoretical experiment was endothiapepsin. The fixed-target parameters were based on experiments reported in Ebrahim et al. (2019)48 and Davy et al. (2019)49. The sample information came from the protocol reported in this video article and the experimental variables were conservative estimates based on lived experience. The following sample requirements were subsequently calculated given the previous assumptions.
