Cryo-SXT can play a central role in biological imaging research as it provides 3D medium resolution (25-30 nm half pitch) volumes of hydrated whole cells1,2,3,4,5,6. In the water window energy range, between the carbon and the oxygen absorption K edges (4.4-2.3 nm), carbon-rich cellular structures absorb 10 times more than the oxygen-rich medium that permeates and surrounds them. In this energy range, vitrified cells up to 10 µm thickness can be imaged without the need for sectioning or staining, leading to quantitative high absorption contrast projections, which, combined with sample rotation capabilities, allow for the tomographic reconstruction of the cellular structure. Cryo-SXT fills a niche in terms of specimen dimensions and spatial resolution that is not easily accessible by any other imaging technique.
In brief, the absorption contrast of cryo-SXT is quantitative, as the attenuation of the photons through the specimen of thickness t obeys the Beer-Lambert law as follows:  , where I0 represents the incident intensity and µl the linear absorption coefficient, which depends on the wavelength λ and the imaginary part β of the refractive index of the specimen (
, where I0 represents the incident intensity and µl the linear absorption coefficient, which depends on the wavelength λ and the imaginary part β of the refractive index of the specimen ( ). The attenuation is a function of the biochemical composition and the thickness of the structures being imaged, with each biochemical component having a specific X-ray linear absorption coefficient µl (LAC). This means that each tomography voxel value depends on the chemical elements and their concentration in that voxel7. This allows for the natural discrimination of different organelles such as nuclei, nucleoli, lipid bodies or mitochondria, or different compaction states of chromatin solely based on their inherent LAC values reconstructed2,8,9.
). The attenuation is a function of the biochemical composition and the thickness of the structures being imaged, with each biochemical component having a specific X-ray linear absorption coefficient µl (LAC). This means that each tomography voxel value depends on the chemical elements and their concentration in that voxel7. This allows for the natural discrimination of different organelles such as nuclei, nucleoli, lipid bodies or mitochondria, or different compaction states of chromatin solely based on their inherent LAC values reconstructed2,8,9.
In addition, cryo-SXT is a high throughput technique with tomograms being collected in few minutes. This specifically enables mesoscale imaging of cell populations that can be captured at key time points such as division, differentiation, and apoptosis, but also at different response states such as those induced by chemical exposure to specific drug therapies or to pathogenic infections. Data collected at those key points will deliver 3D description of the system with a faithful record of the spatial organization of the different cellular organelles at those specific moments.
Usually, cryo-SXT is used in combination with other techniques following correlative approaches that allow locating specific features, events, or macromolecules within the 3D cellular environment4,10,11,12,13,14,15,16, or hard X-ray fluorescence data17,18. Correlative approaches at cryogenic conditions are of paramount importance in order to obtain the most complete and valuable picture of the system of interest. A succinct summary of the typical workflow at the Mistral (Alba) and B24 (Diamond) cryo-SXT beamlines is sketched in Figure 1.
Moreover, taking advantage of the wavelength tuning capability at synchrotron facilities, spectroscopic information can be obtained in addition to the structural one using the specific differential absorption of particular elements contained in the sample. An example of this would be the location of calcium in the study of biomineralization processes in cells19,20,21. By taking 2D images at different photon energies (spectra) or tomograms below and at the x-ray absorption edge of interest, the pixels or voxels containing the selected element can be identified. Spectra also permits differentiating chemical states (i.e., the evolution of amorphous calcium to hydroxyapatite as in the previous biomineralization example20). Quantification of different elements is possible in 2D and 3D. Spectroscopic imaging of vitrified cells is typically done in the water window, but is also possible at other energy ranges if the water content is low enough or if other sample preparation protocols, including dehydration, are used22. A detailed spectroscopy step-by-step protocol is beyond the focus of the protocol herein.
In what follows, the protocol focuses on briefly summarizing the major sample preparation steps, although each system might need individual refinement, followed by a detailed step-by-step data collection procedure for cryo soft X-ray tomography.
Preparing samples for cryo-SXT can be challenging. Even within the same sample grid, it is possible to have areas that are ideal, and areas that are non-ideal, as can be seen in Figure 5, which shows two squares from the same Au finder grid. The ideal sample should have single cells at the center of a square mesh, embedded in a thin layer of ice and surrounded by well-dispersed Au fiducial markers used for the alignment of tilt projections prior to tomography reconstruction. Figure 5A shows a fibroblast-like cell (NIH 3T3) that complies with many of these criteria. A single slice from a 3D reconstruction using ART27 of the area marked with the red box indicating the field of view (FoV) is shown in Figure 5B. Many different organelles such as mitochondria (M), endoplasmic reticulum (ER), vesicles (V) and the nucleus (N) can be distinguished thanks to the quantitative reconstruction of the LACs. In addition, the signal-to-noise ratio of the reconstruction is very high allowing to achieve high contrast of the cellular features. On the other hand, Figure 5C shows a square with higher cell density. Because of this, the blotting is usually less efficient, leading to a thicker ice layer, or even vitrification issues. In some cases, this can already be observed when screening the grid using epifluorescence mapping prior to the X-ray imaging, and those grids should be avoided at any cost. In Figure 5C, a crack within the grid and the vitrified ice can be observed going through the entire mesh square (marked by the red arrows). Any imaging near cracks should be avoided due to probable instability of the grid when exposed to the beam. In addition, cracks can be a sign of thick ice, as was the case in this area. A tilt series was recorded in the area marked with the red box. In Figure 5D, a single slice from the corresponding 3D reconstruction is shown. Even though some larger structures can be recognized, fine details are lost within the noise and grainy texture due to the poor vitrification quality of the thick ice, as can be seen specifically, for instance, on the upper mitochondria pointed by the arrow.

Figure 1: Workflow. Schematic workflow followed prior to cryo-SXT data collection. Please click here to view a larger version of this figure.
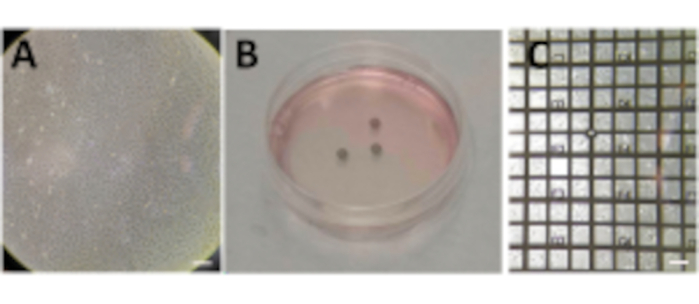
Figure 2: Growing cells on grids. (A) Cells growing in a P100 Petri dish with a confluence around 80%-90%. (B) P60 Petri dish with several grids after seeding the cells. (C) Cells growing on top of a grid after 24 h. Scale bars: 100 µm. Please click here to view a larger version of this figure.

Figure 3: Loading grids on the sample holders and into the transfer chamber. (A) Workstation filled with liquid nitrogen with the shuttle and the cryoboxes ready for loading the grids. (B) Sample holder inserted into the loader with the grid loaded. (C) Shuttle with the sample holder in position 3 without the cover. (D) Workstation with the transfer chamber attached. Please click here to view a larger version of this figure.

Figure 4: Loading samples into the TXM (A) Attaching the transfer chamber to the TXM. (B) Shuttle inside the TXM. (C) TXM robot arm inserting the sample holder into the sample stage. Please click here to view a larger version of this figure.

Figure 5: Example of cryo soft X-ray tomograms. Upper row: ideal sample, (A) 2D mosaic view of a grid square showing an isolated cell at the center. (B) One slice from the reconstructed 3D volume showing the marked area with the red box (A). Compared to (D) the image is much smoother and more details are visible. Lower row: non-ideal samples, (C) 2D mosaic view of a grid square showing too high cell confluency and cracks in the ice and grid foil (red arrows). (D) One slice from the reconstructed 3D volume showing the area marked with the red box in (C). The poor or suboptimal vitrification can be identified by the grainy texture of the image. N: Nucleus; M: Mitochondria; ER: Endoplasmic Reticulum; MV: Multivesicular bodies; V: Vacuole; Scale bars: A & C 20 µm; B & D 2 µm.VPlease click here to view a larger version of this figure.
