1. DsiRNA design and reconstitution
- Use Safari, Firefox, or Microsoft Edge to access the IDT website (https://eu.idtdna.com). From the home page, select the Products and Services tab followed by RNA interference. To generate custom dicer-substrates (DsiRNA) constructs targeting the Fut8 gene, select Design tool followed by the Generate Custom DsiRNA tab.
- Enter the Fut8 Accession Number or manually enter the gene sequence to begin. In this study, the proposed Fut8 sequences from both "CHO-K1" and "Chinese Hamster" entries were obtained from https://chogenome.org and used to generate DsiRNA. Most genes have several transcript variants, and it is important to verify that DsiRNA would be active on all reported variants in the current assembly.
- Select the option to perform a "Manual BLAST" search as the CHO cell genome is currently not available as a "reference genome" on the IDT website. This step searches for matches between custom DsiRNA sequences and the genome of interest.
- Click Search to generate DsiRNA sequences.
- In turn, assess the specificity of each DsiRNA using the "Manual BLAST" function with the tax id:10029 (CHO cell lines and Chinese hamster). Query coverage results indicate DsiRNA constructs match Fut8 (including Fut8 transcript variants) at 100%, whilst complementarity against unintended targets is ≤76%35.
- Check that the DsiRNA specifically targets Fut8 to ensure the GC content is between 30%-50%. A low GC content is associated with weak and unspecific binding, whilst a high GC content inhibits siRNA unwinding and loading into the RISC complex36.
- Select three DsiRNA for use in transfection studies (Table 1), each targeting a different location of the Fut8 gene. Purchase a predesigned non-targeting or scrambled DsiRNA from IDT for control experiments.
- Upon arrival, centrifuge the DsiRNA at 13,300 x g for 1 min to pellet before opening the tube. Reconstitute each lyophilized DsiRNA in nuclease-free duplex buffer (provided by IDT) to a 100 μM stock solution. For example, add 20 μL of buffer to 2 nmol of DsiRNA to obtain a 100 μM stock.
- To ensure sufficient mixing, incubate the stock solutions at room temperature for 30 min whilst shaking gently on an orbital shaker (50 rpm, 16 mm orbit).
- Create a master mix by combining 20 μL of each DsiRNA construct that targets Fut8 (100 μM of each DsiRNA). Prepare aliquots and store at −20 °C.
| DsiRNA target | Sequence | GC (%) | ||
| Structure A | 5' GAGAAGAUAGAAACAGUCAAAUACC 3' | 36% | ||
| 5’ GGUAUUUGACUGUUUCUAUCUUCUCUC 3' | ||||
| Structure B | 5' AGAAUGAGAAUGGAUGUUUUUCCTT 3' | 32% | ||
| 5' AAGGAAAAACAUCCAUUCUCAUUCUGA 3' | ||||
| Structure C | 5' AGAGAAGAUAGAAACAGUCAAAUAC 3’ | 32% | ||
| 5' GUAUUUGACUGUUUCUAUCUUCUCUCG 3' | ||||
Table 1. DsiRNA sequences used for Fut8 knockdown. Sequences generated by IDT that target Fut8 in Chinese hamster and CHO K1 cell genomes. The sense and antisense sequences for each construct are shown (respectively), and the GC content of each structure is displayed. Reprinted from Kotidis et al.52.
2. DsiRNA transfection
- Revive CHO cells expressing an IgG monoclonal antibody37 using culture conditions suitable for the cell line of interest.
NOTE: The cells used in this study were generated using the glutamine synthetase (GS) system, where endogenous GS is inhibited by L-methionine sulfoximine (MSX) to enhance the selection of rare high-producing clones. Therefore, only use MSX if required by the cell line of choice.- Defrost a vial of cells for 2-3 min in a water bath set to 37 °C. Clean the vial exterior with 70% (v/v) ethanol and continue all work in a class II biosafety cabinet.
- Transfer the cell suspension to a 15 mL centrifuge tube containing 9 mL of prewarmed medium appropriate for the cell line of choice. Pellet the cells by centrifugation at 100 x g for 5 min.
- Carefully remove and discard the medium without disturbing the cell pellet. Then, resuspend the cell pellet in 10 mL of prewarmed medium and take an aliquot for counting.
- Stain the cells with Trypan blue if counting with a hemocytometer, or an appropriate stain to distinguish live/dead cells when using an automated cell counter.
- Following either method of enumeration, transfer an appropriate volume of cell suspension to a 125 mL Erlenmeyer shake flask at a viable cell density of 3 x 105 cells·mL-1 in 30 mL of medium (with optional supplementation of 50 μM MSX).
- Transfer cells to an incubator set to 36.5 °C, 5% CO2 and place on a shaking platform at 150 rpm (16 mm orbit).
- Passage cells every 3-4 days at a seeding density of 2 x 105 cells·mL-1 and a working volume of 50 mL in a 250 mL Erlenmeyer shake flask. If using MSX, discontinue supplementation after the first passage.
- Passage cells 2x in addition to thawing.
- Transfection
- Assess the cell density and ensure the cell viability is greater than 90%.
- Carefully clean the biosafety cabinet and all equipment with 70% (v/v) ethanol and an RNase inhibitor solution to avoid contamination.
- Pellet cells at 100 x g for 5 min and resuspend in prewarmed medium to a viable cell density of 5 x 106 cells·mL-1.
- Transfer 8 μL (equivalent to 1 μM) of the DsiRNA master mix or control to a sterile (0.4 cm) electroporation cuvette. Then, transfer 800 μL of the cell suspension (equivalent to 4 x 106 cells) to the same cuvette and ensure both components are mixed.
- Deliver the following pulse conditions: 1200 V, 0.1 ms, square waveform.
- Transfer the cell suspension from the cuvette to one well of a 6-well plate, taking care to avoid foam-like material. Recover the cells in the incubator (36.5 °C, 5% CO2) without shaking for 10 min.
- Add 800 μL of prewarmed media to make a final volume of 1.6 mL per well and return the transfected cells to the incubator for growth (36.5 °C, 5% CO2) while shaking at 150 rpm (16 mm orbit).
- Harvest the supernatants and cells at 48 h post-transfection.
Stopping point: The cell culture supernatant can be kept at -20 °C; however, it is advisable that cell lysis should be performed immediately after cell pellet collection to avoid protein degradation. Supernatants are used in Step 3, Step 4, and Step 5, while cell pellets are used in Step 6.
3. IgG quantification and purification
- Measure IgG concentration using biolayer interferometry or another method of choice.
- Hydrate protein A biosensor tips in sample diluent for 10-30 min. In the meantime, transfer cell suspensions into 15 mL centrifuge tubes and pellet cells at 100 x g for 5 min or remove cells by filtration through a 0.45 µm nitrocellulose filter.
- Without disturbing the pellet, carefully transfer the isolated supernatant containing IgG into a clean centrifuge tube.
- Use the following settings for biolayer interferometry: shaker speed, 2200 rpm; run time, 60 s. These parameters will be used to measure all samples and controls.
- Once the biosensor tips are hydrated, create a standard curve using an IgG standard (4 μL of each concentration).
- Quantitate sample concentrations by loading 4 μL of the cell supernatant. Clean the apparatus with lint-free wipes between each sample.
- Link the standard curve to the unknown samples to interpolate the binding rate of the unknown samples. Save the data and export as a csv or PDF file.
- IgG purification
- Prepare elution buffer containing 0.2 M glycine (pH 2.5) and sterilize by passing through a 0.22 μm filter.
- Filter 1 mL of the isolated supernatant at room temperature using 0.22 μm microcentrifuge filter tubes until the entire supernatant flows through.
- Pellet 150-200 μL of protein A agarose beads in 1.5 mL centrifuge tubes at 100 x g for 3 min and discard the supernatant. Then, wash the protein A beads with 150-200 μL of cell culture medium and repeat centrifugation. Discard the supernatant.
- Resuspend the equilibrated protein A beads with 1 mL of the prepared supernatants from Step 3.2.3. and load into a 1 mL polypropylene tube. Affix the polypropylene tube to a rotary mixer (15 mm orbit) and spin at 30 rpm for 60-90 min at room temperature or overnight at 4 °C.
- After incubation is complete, collect the flow-through and wash the beads with 1 mL of 1x PBS to remove any unbound proteins. Collect the wash fraction as well.
- Elute IgG from the protein A beads by adding 3 mL of elution buffer to the polypropylene column. Collect sequential fractions that drain from the column (500 μL each) into labeled tubes.
- Optional: If the purified antibody is to be stored, neutralize the elution buffer by adding 25 μL of 1 M Tris pH 9.5. Skip this step if the antibody will be processed immediately via buffer exchange, etc.
NOTE: All parts of the purification (flow-through, washes, and all elution fractions) should be kept ensuring that the target protein is not lost. These samples can also help during troubleshooting if the purification is not successful. - Use the first elution (elution A) for downstream processing but keep all other fractions should they be required in the future.
4. Buffer exchange and sample concentration
- Exchange IgG elution buffer with 1x PBS.
- Load elution A onto a 3 kDa molecular weight cutoff centrifugal concentrator. Refer to the manufacturer's guide when selecting an appropriate MWCO column. In this experiment, a smaller pore size ensures maximal retention of secreted protein but also increases the centrifugation time.
- Centrifuge at 13,300 x g for 40-50 min at 4 °C. Centrifugation is complete once the residual volume is equal to or less than 50 μL.
- Discard the flow-through and add 500 μL of prechilled (4 °C) 1x PBS to dilute the residual supernatant. Centrifuge the sample again using the same conditions (13,300 x g for 40-50 min at 4 °C) until 50 μL of residual supernatant remains.
- Add 500 μL of prechilled (4 °C) 1x PBS to dilute the residual supernatant and repeat the centrifugation process again to obtain a 100x dilution of the supernatant.
- Concentrate the supernatant to a final concentration of ~2.5 g·L-1 in 40 μL or less to ensure compatibility with the glycan analysis method.
NOTE: 100 µg needs to be loaded for glycan analysis.
Stopping point: The concentrated IgG (in 1x PBS) can be stored at −20 °C and thawed prior to glycan analysis.
5. Glycan analysis
- Perform glycan analysis using capillary gel electrophoresis according to the manufacturer's instructions.
- Transfer 200 μL of the magnetic bead solution to a 0.2 mL PCR tube and place onto the magnetic stand to separate the beads from the supernatant.
- Carefully remove the supernatant and remove the tubes from the magnetic stand. Add the purified protein sample and vortex to ensure complete mixing.
- Add denaturation buffer (supplied) to the sample tube and incubate for 8 min at 60 °C. Keep the sample tubes open for optimal reaction performance.
- Add PNGase F (500 units per sample) and incubate for 20 min at 60 °C to cleave glycans from the purified antibodies.
- Following the release of N-glycans, close the sample tube and vortex. Add acetonitrile, vortex, and incubate at room temperature for 1 min.
- Place the sample tubes in the magnetic stand to separate the beads from solution. Use a pipette to carefully remove the supernatant without touching the beads.
- In a fume hood, add the glycan labeling solution containing a fluorophore to the sample. Vortex to ensure sufficient mixing and incubate at 60 °C for 20 min (open lids).
- Wash the sample 3x in acetonitrile to remove excess dye. Then, elute the labeled glycans in DDI water (supplied).
- Place the sample tube in the magnetic stand to separate the beads from the supernatant containing purified and labeled glycans.
- Prepare and load the glucose ladder standard, bracketing standards, and samples into the designated tray positions. Run the glycan analysis protocol.
- Use appropriate software to analyze and identify the glycans present in the sample.
NOTE: Ensure the temperature in the heat block is accurate for efficient incubation.
6. Western blot
- Quantify knockdown efficiency using western blot analysis with an anti-α-1,6-fucosyltransferase antibody. Polyacrylamide gels and buffer recipes are available from commercial suppliers38,39.
- At 48 h post transfection, count the cells using a hemocytometer or an automated cell counter and determine the volume of cell suspension equivalent to 5 x 106 cells.
- Transfer the appropriate volume of cell suspension from each experimental condition to a sterile 1.5 mL centrifuge tube and pellet at 13,200 x g for 10 min at 4 °C. Discard the supernatant.
- Lyse the cells with 200 μL of lysis buffer (suitable for the extraction of proteins from mammalian cells) containing 1% v/v protease inhibitor cocktail at room temperature for 10 min. Shake the mixture gently during incubation (50 rpm, 16 mm orbit).
- To remove the cellular debris, centrifuge the lysate at 13,200 x g for 10 min and then transfer the cleared lysate to a sterile 1.5 mL tube.
- Measure the protein concentration of each lysate using a spectrophotometer at 280 nm. Subsequently, prepare aliquots of each sample adjusted to have the same protein concentration.
- Denature the protein samples by incubation at 100 °C for 10-15 min in DTT-SDS sample loading dye; the final dye concentration is 1x.
- Load 15 μL of the denatured samples and 5 μL of prestained protein ladder into an SDS-PAGE gel with 12.5% resolving gel for efficient separation. Run the samples at 25 mA per gel for 90 min or until the dye front reaches the end of the gel.
- Carefully remove the gel from the cassette and incubate in 1x transfer buffer containing methanol.
- Prepare the wet transfer system and activate a PVDF membrane with methanol. Assemble the gel and PVDF membrane for wet transfer, place an ice block in the transfer tank, and submerge the entire tank in ice to ensure the transfer conditions remain cold. Run at 350 mA/100 V for 60 min.
- Block the membrane by incubating in a blocking solution for 30 min at room temperature while shaking gently on an orbital shaker at 50 rpm (16 mm orbit).
- Rinse the membrane with sterile water for 5 min and repeat. Then, use a clean scalpel to carefully cut the membrane horizontally at ~50 kDa, using the visible protein ladder as a guide.
- Incubate the membrane harboring proteins greater than 50 kDa with anti-α-1,6-fucosyltransferase antibody at 1:1000 in an antibody diluent buffer. Incubate the membrane with immobilized proteins less than 50 kDa with anti-GAPDH at 1:10,000 in diluent buffer. Membranes may be incubated for 1 h at room temperature or overnight at 4 °C.
- Wash the membranes for 5 min (x3) and then incubate with an appropriate secondary antibody for at least 30 min at room temperature.
- Perform 3x washes with a wash buffer for 5 min, followed by 3x washes with water. Then, develop the membrane with a chromogenic substrate (or appropriate detection reagent) until bands appear (1-60 min).
- Rinse the membrane 2x with water, and then allow it to dry. Capture an image of the membrane and perform densitometric analysis40.
- To calculate the relative protein expression in each sample, first calculate the ratio of the GAPDH signal in samples. This is the normalization factor for each sample that corrects for discrepancies in sample loading.

- Divide the FUT8 signal intensity (for each lane) by the normalization factor of the corresponding lane to obtain the relative FUT8 protein expression.

Western blot analysis showed reduced FUT8 protein expression in cells transfected with a mixture of three Fut8 DsiRNA constructs. In control samples transfected with non-targeting DsiRNA, FUT8 appeared as a double band at ~65 and 70 kDa. Since the predicted molecular weight of FUT8 is 66 kDa, a reduction in the signal intensity of the lower molecular weight band is indicative of gene silencing. To confirm and quantify gene silencing, the level of FUT8 protein was normalized to the relative GAPDH protein level. Western blot analysis detected two bands for GAPDH at ~37 and 35 kDa. The higher molecular weight band corresponds to the predicted protein size and is, therefore, used in normalization calculations. When normalized against GAPDH protein levels, FUT8 protein expression was reduced by up to 60% (Figure 1).
In line with the observation of gene knockdown at 48 h post transfection, corresponding mAb samples were processed for analysis by CGE- LIF. Glycan structures from knockdown cells showed a decrease in fucosylation. This trend was most pronounced in agalactosylated structures (G0F) and observed to a lesser extent in galactosylated structures (G1F, G1F' and G2F). From this dataset, the total IgG core fucosylation decreased to ~75%, down from ~95% core fucosylation observed for the negative control condition (Figure 2). A greater reduction in core fucosylation was anticipated given the ~60% decrease in FUT8 protein levels. Upon reflection, it is noteworthy that the glycoprofile represents glycosylated mAbs that accumulated over a period of 48 h since transfection, while gene silencing represents protein levels present at the time of harvesting only.
Further scrutiny of this knockdown method involved varying the DsiRNA concentration, harvest time, and electroporation conditions. Each factor was individually probed to determine its relevance. The impact of electroporation pulse conditions on core fucosylation and cell viability is captured in Experiments B, C, D, and E. These results demonstrate a two-fold reduction in core fucosylation from electroporation using two square wave pulses (Experiment C) compared to a single square wave pulse (Experiment B), without significant differences in cell viability (Table 2). Electroporation condition e3 (Experiment D) led to the lowest cell viability (~90%) and IgG yield at this timepoint. However, cells that survived the electroporation event were moderately transfected, as evidenced by the ~10% decrease in core fucosylation (Table 2). Interestingly, Experiment D used electroporation conditions that provided the greatest reduction in core fucosylation (14.7%) but were evidently detrimental to cell viability (91%-93% viability). This limited set of experiments illustrates the need to determine electroporation settings that enable sufficient permeabilization of the cell membrane without causing irrevocable damage. It is also interesting to note the role of siRNA concentration and harvest time on core fucosylation. Overall, increasing siRNA concentration has a greater influence on core fucosylation than increasing the harvest time (Experiments B, F, G versus Experiments A, B, H). In future experiments, it would be interesting to titrate the siRNA concentrations delivered by the electroporation method e2.
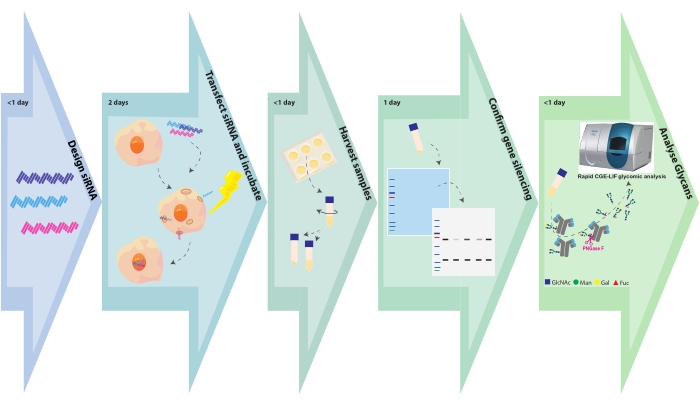
Figure 1. Experimental flow chart. Glycoengineering and sample analysis steps are depicted with the associated time required for each step. SiRNA design takes a few hours, depending on the number of gene targets or constructs per gene target. CHO cell transfection with siRNA is complete in a few hours, and transformed cells are left to grow for 48 h. Cell pellets and supernatants are harvested within a few hours. Cell pellets are lysed, and the intracellular proteins are separated on an SDS PAGE and subsequently blotted and probed with antibodies against the target gene. Glycans are cleaved from purified antibodies and analyzed by CGE-LIF. These assays may require 1 day each. Please click here to view a larger version of this figure.
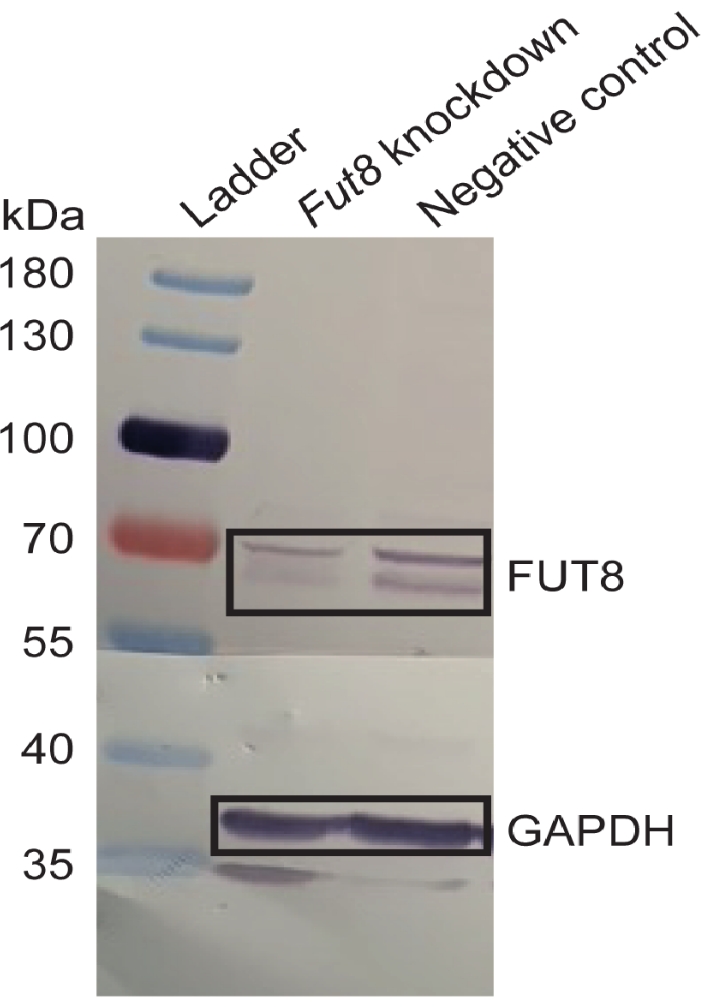
Figure 2. Confirmation of RNA interference. Western blot detection of α-1,6-fucosyltransferase (FUT8) protein levels in samples treated with Fut8 or non-targeting control DsiRNA. Bands corresponding to FUT8 are more intense in control than Fut8 knockdown samples. The GAPDH protein level was also assessed in order to normalize target gene expression. All samples were taken from Experiment G (see Table 2). Reprinted from Kotidis et al.52. Please click here to view a larger version of this figure.
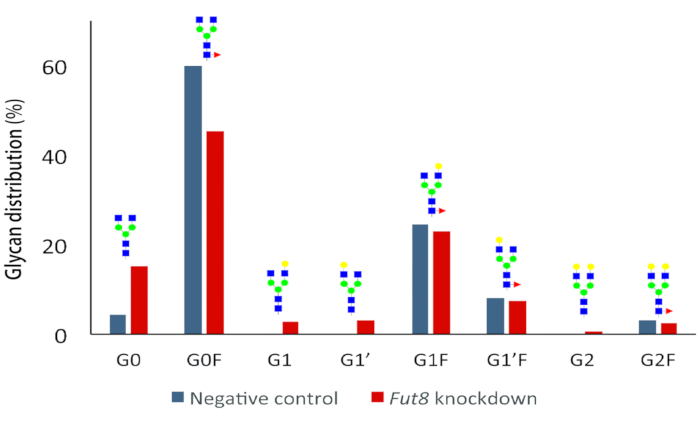
Figure 3. Effect of Fut8 knockdown on cumulative IgG glycosylation at 48 h. A shift in glycan distribution is detected in knockdown samples. In particular, the relative abundance of the main core-fucosylated structures (G0F) is reduced while the afucosylated species are increased in the knockdown experiment. Measurements were performed from Experiment G samples (see Table 2). Biological triplicates performed for each experiment were mixed after harvesting to reduce the burden of downstream analysis. Reprinted from Kotidis et al.52. Please click here to view a larger version of this figure.
| Experiment name | Electroporation method | DsiRNA concentration (nΜ) | Harvest time (h) | Viability (%) | Xv (106 cells·mL-1) | IgG titer (mg·L-1) | Difference in core-fucosylation (%) | ||||||
| ExpA_Negative | e1 | 500 | 24 | 98.3 | 4.71 | 122.5 | – | ||||||
| ExpA_Knockdown | e1 | 500 | 24 | 98.3 | 4.9 | 110.3 | 4.08 | ||||||
| ExpB_Negative | e1 | 500 | 48 | 95.6 | 9.55 | 453.3 | – | ||||||
| ExpB_Knockdown | e1 | 500 | 48 | 96.7 | 9.61 | 469 | 5.38 | ||||||
| ExpC_Negative | e2 | 500 | 48 | 96.3 | 9.91 | 449.3 | – | ||||||
| ExpC_Knockdown | e2 | 500 | 48 | 96.7 | 11 | 454.6 | 11.42 | ||||||
| ExpD_Negative | e3 | 500 | 48 | 90.6 | 6.25 | 318.5 | – | ||||||
| ExpD_Knockdown | e3 | 500 | 48 | 89.1 | 6.09 | 311.85 | 9.71 | ||||||
| ExpE_Negative | e4 | 500 | 48 | 91.1 | 7.2 | 380.3 | – | ||||||
| ExpE_Knockdown | e4 | 500 | 48 | 93.3 | 7.79 | 422.8 | 14.7 | ||||||
| ExpF_Negative | e1 | 750 | 48 | 96.2 | 9.7 | 501 | – | ||||||
| ExpF_Knockdown | e1 | 750 | 48 | 95.7 | 9.76 | 504.6 | 9.9 | ||||||
| ExpG_Negative | e1 | 1000 | 48 | 96.1 | 11.1 | 422.6 | – | ||||||
| ExpG_Knockdown | e1 | 1000 | 48 | 95.9 | 9.73 | 499.3 | 17.26 | ||||||
| ExpH_Negative | e1 | 500 | 72 | 94.4 | 14.3 | 925.8 | – | ||||||
| ExpH_Knockdown | e1 | 500 | 72 | 95 | 13.5 | 1018.4 | 7.37 | ||||||
Table 2. Transfection optimization. Iterative modifications of the electroporation method, DsiRNA concentration, and harvest time led to changes in cell viability, viable cell density, IgG titer at the harvest time, and differences in core fucosylation. Each experiment compared the knockdown and the respective negative control to determine if the modification produces the desired effect (i.e., a decrease in fucosylation). Electroporation settings were as follows: e1: 1200 V, 0.1 ms, square waveform; e2: 1200 V, 2x 0.1 ms, 5 s between pulses, square waveform; e3: 150 V, 20 ms, square waveform; e4: 250 V, 500 μF, exponential decay. Reprinted from Kotidis et al.52.
