



Overview
Fonte: Laboratório do Dr. Paul Bower - Universidade purdue
O método de adições padrão é um método de análise quantitativa, que é frequentemente utilizado quando a amostra de interesse tem múltiplos componentes que resultam em efeitos matriciais, onde os componentes adicionais podem reduzir ou melhorar o sinal de absorção de analitos. Isso resulta em erros significativos nos resultados da análise.
Adições padrão são comumente usadas para eliminar efeitos matriciais de uma medição, uma vez que se presume que a matriz afeta todas as soluções igualmente. Além disso, é usado para corrigir as separações de fase química realizadas no processo de extração.
O método é realizado lendo a intensidade experimental (neste caso fluorescente) da solução desconhecida e, em seguida, medindo a intensidade do desconhecido com quantidades variadas de padrão conhecido adicionado. Os dados são plotados como intensidade de fluorescência versus. a quantidade do padrão adicionado (o desconhecido em si, sem padrão adicionado, é plotado no eixo y). A linha de quadrados menos cruza o eixo x no negativo da concentração do desconhecido, como mostra a Figura 1.
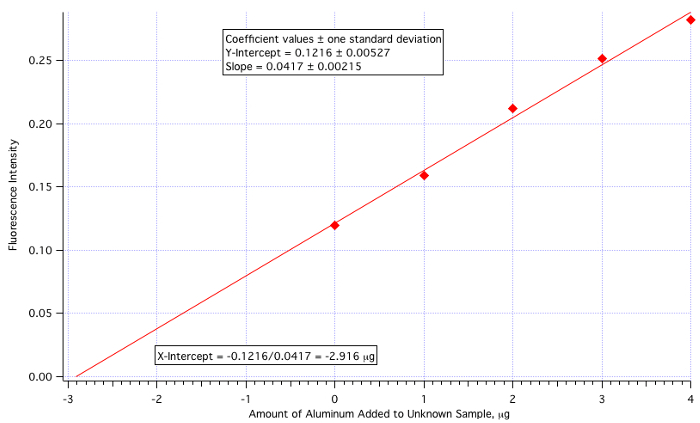
Figura 1. Representação gráfica do método de adição padrão.
Principles
Neste experimento, o método de adições padrão é demonstrado como uma ferramenta analítica. O método é um procedimento para a análise quantitativa de uma espécie sem a geração de uma curva de calibração típica. A análise de adição padrão é realizada medindo a intensidade espectroscópica antes e depois da adição de alíquotas precisas de uma solução padrão conhecida do analito.
Este experimento estuda espécies não fluorescentes reagindo-as de forma a formar um complexo fluorescente. Esta abordagem é comumente usada na investigação de íons metálicos. Íons de alumínio (Al3+) serão determinados formando um complexo com 8-hidroxiquinolina (8HQ). O Al3+ é precipitado por 8HQ a partir de solução aquosa e, em seguida, é extraído em clorofórmio; a fluorescência da solução de clorofórmio é medida e relacionada à concentração da solução Original Al3+. A sensibilidade na faixa de parte por milhão (ppm ou μg/mL) é esperada para este experimento.
A reação é

A quantidade de alumínio em cada amostra durante este experimento é calculada da seguinte forma:
| Em branco | 0 | ||
| Padrão desconhecido + 0 mL | VDesconhecido(CDesconhecido) = 25 mL(CDesconhecido) | ||
| Padrão desconhecido + 1 mL | VDesconhecido(CDesconhecido) +PadrãoV(PadrãoC ) = 25 mL (CDesconhecido) + 1 mL (1 μg/mL) | ||
| Padrão desconhecido + 2 mL | VDesconhecido(CDesconhecido) +PadrãoV(PadrãoC)= 25 mL(CDesconhecido)+ 2 mL (1 μg/mL) | ||
| Padrão desconhecido + 3 mL | VDesconhecido(CDesconhecido) +PadrãoV(PadrãoC)= 25 mL(CDesconhecido)+ 3 mL (1 μg/mL) | ||
| Padrão desconhecido + 4 mL | VDesconhecido(CDesconhecido) +PadrãoV(PadrãoC)= 25 mL(CDesconhecido)+ 4 mL (1 μg/mL) | ||
Subscription Required. Please recommend JoVE to your librarian.
Procedure
1. Preparando os Reagentes
- Solução Al3+ padrão de 100 ppm: Dissolver 0,9151 g de nitrato de alumínio (Al(NO3)3•9H2O) em um frasco volumoso de 1 L com água DI.
- Solução 8HQ em ácido acético de 1 M (2% wt/vol): Adicione 2,0 g de 8-hidroxiquinolina a um frasco volumoso de 100 mL.
- Adicione cuidadosamente 5,74 mL de ácido acético glacial ao frasco de 100 mL e, em seguida, dilua para a marca com água DI. Isso permite que a hidroxiquinolina 8 se dissolva em fase aquosa.
- 1 M NH4+/NH3 tampão (pH~8): Adicione 20 g de acetato de amônio (NH4OAc) a uma garrafa de 100 mL.
- Adicione 7 mL de hidróxido de amônio de 30% a esta garrafa de 100 mL e dilua à marca com água DI. Isso ajuda a neutralizar o ácido na solução 8HQ quando combinado.
- Outros reagentes incluem sulfato de sódio anidro (Na2SO4) e clorofórmio (grau spec).
2. Preparando as Amostras
- Prepare uma solução Al 3+ padrão de1,00 ppm adicionando 1,0 mL da solução Al3+ de estoque de 100 ppm com uma pipeta a um frasco volutrico de 100 mL.
- Coloque seis funis separados de 125 mL nos anéis que estão em um grande suporte de anel localizado no capô. Eles devem ser rotulados da seguinte forma: BL, 0, 1, 2, 3, 4. Certifique-se de que todos os vidros estão escrupulosamente limpos, pois é difícil obter resultados quantitativos se pequenas contas de clorofórmio grudarem nas paredes dos vidros.
- Adicione 25,00 mL da solução Al3+ desconhecida aos cinco funis separados rotulados 0, 1, 2, 3 e 4. Neste exemplo, a concentração desconhecida é de 0,110 ppm.
- Adicione 0, 1,00, 2,00, 3,00 e 4,00 mL da solução Al3+ padrão de 1,00 ppm, respectivamente, aos 5 funis com uma pipeta de 1 mL.
- Prepare um BLANK adicionando 25,00 mL de água destilada ao funil separador rotulado BL.
- Adicione 1,0 mL da solução de 8 hidroxiquinolina com uma pipeta para cada uma das seis soluções.
- Adicione 3,0 mL de solução tampão com uma pipeta a cada uma das seis soluções.
- Extrair cada solução duas vezes com 10 mL de clorofórmio, tremendo vigorosamente por 1 min cada vez. Lembre-se de ocasionalmente ventilar o funil separador para liberar o acúmulo de pressão. (NOTA: Uma boa extração só ocorre quando há muito contato líquido-líquido entre as fases).
- Colete o clorofórmio em um béquer limpo e seco de 100 mL rotulado. O clorofórmio tem uma densidade de cerca de 1,5 g/cm3,por isso é a camada inferior. Não deve haver vestígios de cor amarela deixados na fase aquosa após uma extração completa.
- Transfira o extrato combinado de clorofórmio de cada béquer em seu respectivo frasco volumoso de 25 mL e dilua para a marca com clorofórmio. Certifique-se de colocar rolhas em cada frasco volumoso para evitar que qualquer clorofórmio evapore.
- Adicione ~1 g de sulfato de sódio anidro (Na2SO4) a cada um dos seis béquers de 100 mL do passo 2.9. O sulfato de sódio ajuda a remover qualquer traço de água que possa estar presente no extrato de clorofórmio.
- Transfira as soluções de volta para seus respeitados béquers. Gire cuidadosamente para facilitar a desidratação de qualquer água da amostra.
- Decantar os extratos de clorofórmio em uma célula fluorímetro de quartzo (Nota: Clorofórmio dissolverá uma célula de poliestireno plástico).
3. Selecionando o comprimento de onda de excitação
Determine os comprimentos de onda de excitação e emissão executando varreduras, em seguida, basta ler e registrar a intensidade de fluorescência de todas as amostras nesses valores. As larguras de banda de excitação e emissão são predefinidas a 5 nm. O complexo absorve no UV próximo, então o comprimento de onda de excitação deve ser de cerca de 385 nm. Inicialmente, monitore a fluorescência a 500 nm no ramo de emissões.
- No Fluoriímetro, verifique se tanto os ventiladores de resfriamento interno quanto externo no fluorímetro estão ligados antes de ligar a lâmpada de xenônio. A lâmpada de xenônio fica muito quente, e requer resfriamento contínuo.
- Ligue a alta tensão (HV) ao detector PMT a 400 V.
- Abra as duas janelas.
- Abra o programa de aquisição de dados no computador, aqui é "100nmFluorScan".
- Coloque a solução "Amostra + 2 mL adicionada" (2) na célula de quartzo para usar para determinar os melhores comprimentos de onda de excitação e emissão.
- Com o comprimento de onda de emissão inicialmente definido em 500 nm, execute uma varredura de excitação de 335-435 nm com uma velocidade de varredura de 2 nm/s.
- A partir do enredo de fluorescência resultante, determine o comprimento de onda de fluorescência máxima(EXλmax),e defina o instrumento para esse valor.
4. Selecionando o comprimento de onda de emissão
- Defina o comprimento de onda de emissão de fluoriímetro para 450 nm.
- Defina a faixa de comprimento de onda de emissão para executar uma varredura de 100 nm de 450-550 nm.
- Para iniciar a varredura, clique no botão "iniciar a prova" no programa ao mesmo tempo em que pressionar o botão "START" no painel frontal do fluorímetro.
- A partir do enredo de fluorescência resultante, determine o comprimento máximo de onda de emissão de fluorescência(EMλmax),e defina o instrumento para esse valor(Figura 2).

Figura 2. Determinando os comprimentosde onda máximo EXλ e EM λ.
5. Medindo a fluorescência das amostras
- Todas as amostras são executadas em EMλmax e EXλmax. Exames não são necessários para cada amostra, mas apenas o valor da fluorescência nessas condições. Começando com a amostra mais diluída (em branco), coloque em uma célula de quartzo e, em seguida, em instrumento. Regisso recorde a intensidade fluorescente no caderno de laboratório.
- Repita para todas as outras amostras.
- Lembre-se de que a Intensidade Relativa Em Branco deve ser subtraída das Intensidades Relativas de cada solução antes de criar o gráfico de calibração.
6. Criando o gráfico de adição padrão
- Plote a intensidade da fluorescência vs. μg de Al3+ adicionado.
- Determine o valor de menor quadrado do enredo resultante e registo a inclinação e intercepte.
- Determine o μg de Al3+ na amostra desconhecida da equação μg de Al3+ = -b/m
- Sabendo que o alumínio desconhecido tinha um volume de 25,0 mL adicionado a cada amostra, determinar a concentração do alumínio no desconhecido.
O método de adição padrão é uma técnica de análise quantitativa utilizada para minimizar efeitos matriciais que interferem com sinais de medição de analitos.
Concentrações de componentes desconhecidas são frequentemente elucidadas através de uma série de técnicas analíticas, como espectroscopia de luz, espectrometria de massa e eletroquímica. No entanto, a medida pode ser afetada por outros componentes da amostra, chamado de matriz, e causar a redução inadvertida ou aprimoramento do sinal, chamados efeitos matriciais. Esses efeitos podem distorcer resultados e causar erros significativos na análise.
O método de adição padrão pode ser usado para minimizar os efeitos da matriz nos sinais de medição. Isso é realizado adicionando volumes precisos de uma solução analito conhecida à amostra.
Este vídeo introduzirá o básico do método de adição de padrões e demonstrará como executar a técnica em laboratório usando uma medição de fluorescência.
Efeitos matriciais podem surgir em amostras complexas onde uma série de outras moléculas interagem com o analito. Por exemplo, isso pode ocorrer quando as moléculas se ligam ou aglomeram com o analito, alterando assim sua capacidade de fluoresce. Ou a matriz pode mudar a força iônica da solução global, mudando propriedades específicas do analito.
Para mitigar esses efeitos usando o método de adição padrão, uma gama de volumes de uma solução padrão analito é adicionada a volumes iguais da amostra. Os volumes da solução são então iguais usando solvente.
Em seguida, o sinal é medido para as amostras com e sem a adição padrão. Os dados são plotados como intensidade versus a quantidade do padrão adicionado à amostra, em vez de uma curva de calibração clássica. A concentração real do analito em qualquer frasco é definida pela seguinte equação. A resposta instrumental igualará alguns tempos constantes a concentração de analito. A equação resultante toma a forma linear y=mx+b. Assim, quando o enredo é extrapolado a absorção zero, a interceptação é igual à concentração desconhecida da amostra.
O gráfico do sinal deve ser linear sobre o intervalo de concentração de preocupação. Além disso, a interferência não deve variar como a razão de analito para alteração matricial amostral. Finalmente, a matriz em si não deve gerar sinais de medição por conta própria.
O experimento a seguir estuda o alumínio, uma espécie não fluorescente, reagindo-o com 8-hidroxiquinolina, ou 8HQ, para formar o complexo fluorescente ALQ3.
A fluorescência do complexo de alumínio em um solvente orgânico é medida e, em seguida, relacionada à concentração da solução original de alumínio. Essa abordagem é comum na análise de íons metálicos.
Agora que o básico do método de adição padrão foi delineado, e o básico do experimento explicou, vamos executar a técnica em laboratório.
Primeiro, prepare a solução de estoque de alumínio de 100 ppm na água e, em seguida, use-a para preparar uma solução padrão de 1 ppm.
Em seguida, adicione 2 g de 8-hidroxiquinolina, ou 8HQ, a um frasco volumoso de 100 mL.
Adicione cuidadosamente 5,74 mL de ácido acético glacial e, em seguida, dilua para a marca de 100 mL com água deionizada. Esta etapa permite que o 8HQ se dissolva na fase aquosa.
Em seguida, prepare o tampão adicionando 20 g de acetato de amônio e 7 mL de hidróxido de amônio de 30% a uma garrafa marcada de 100 mL e diluir. Verifique o pH com uma vara indicadora de pH. Este buffer ajuda a neutralizar o ácido na solução 8HQ quando combinado.
Outros reagentes necessários incluem sulfato de sódio anidro e clorofórmio de grau espectrofotmétrico.
Agora prepare as amostras, neste caso, extraindo a amostra aquosa na fase orgânica usando extração líquido-líquido. Coloque seis funis separados de 125 mL em anéis de suporte dentro do capô. Certifique-se de que todos os vidros estão escrupulosamente limpos, pois os vidros sujos distorcerão os resultados. Sequencialmente rotulam os funis como "em branco", "0", "1", "2", "3" e "4".
Utilizando uma pipeta, adicione 25 mL da solução de alumínio desconhecida a cada um dos cinco funis separados rotulados de "0" a "4". Prepare o espaço em branco adicionando 25 mL de água deionizada ao funil rotulado como "em branco".
Em seguida, adicione 1, 2, 3 e 4 mL da solução padrão de 1 ppm aos funis numerados correspondentes. Não adicione nenhuma solução padrão aos funis em branco ou 0.
Adicione 1 mL da solução 8HQ e 3 mL de solução tampão para cada um dos 6 funis.
Realize uma extração líquido-líquido adicionando 10 mL de clorofórmio a cada frasco. Agite o funil vigorosamente, e ocasionalmente desabastem o funil para liberar o acúmulo de pressão. Coloque o funil de volta no anel e deixe que as camadas líquidas se separem.
Em seguida, colete a fase do clorofórmio em um béquer limpo, seco e rotulado de 100 mL. Como o clorofórmio tem uma densidade maior que a água, é a camada inferior no funil.
Transfira o extrato de clorofórmio em um frasco volumoso de 25 mL e tampe cada frasco para evitar a evaporação.
Realize uma segunda extração líquido-líquido na solução aquosa restante, adicionando 10 mL de clorofórmio a cada funil. Agite o funil, como antes, para transferir qualquer analito restante para a fase do clorofórmio. Não deve sobrar cor amarela na fase superior aquosa.
Repita a segunda extração para cada funil e colete as fases de clorofórmio em béquers rotulados correspondentes. Despeje o clorofórmio coletado em seus respectivos frascos volumoscos e dilua até a marca com clorofórmio fresco.
Para remover a água do traço, adicione cerca de 1 g de sulfato de sódio anidro a cada um dos seis béquers de 100 mL. Transfira as soluções de volta para seus respectivos béquers, e gire para facilitar a desidratação da amostra.
Decante o extrato de clorofórmio em uma célula fluorímetro de quartzo.
Configure o fluorímetro de acordo com as instruções do fabricante e coloque a tensão em 400 V. Em seguida, abra o programa de aquisição de dados no computador.
Use a amostra 2 para determinar os melhores comprimentos de onda de excitação e emissão. Defina o comprimento de onda de emissão para 500 nm e execute uma varredura de excitação de 335-435 nm, com uma velocidade de varredura de 2 nm/s.
A partir do enredo de fluorescência, determine o comprimento de onda máximo para excitação. Defina o instrumento para esse valor de comprimento de onda de excitação, neste caso 399 nm.
Em seguida, determine o comprimento de onda de emissão realizando uma varredura de 450-550 nm. A partir do gráfico de fluorescência resultante, determine o comprimento máximo de onda e defina o comprimento de onda de emissão, neste caso 520 nm.
Meça cada amostra, incluindo o em branco no comprimento de onda de excitação e emissão selecionado. Registo cada leitura de intensidade de fluorescência.
Subtrair a fluorescência medida da amostra em branco de cada uma das outras 5 amostras.
Plote a intensidade da fluorescência de cada uma das cinco amostras versus a quantidade de alumínio adicionada à amostra. Determine o valor mínimo quadrado do enredo resultante e registo da inclinação e intercepte.
O enredo da intensidade da fluorescência versus quantidade de alumínio adicionado rendeu uma linha menos quadrada, como mostrado. A quantidade de alumínio na amostra pode então ser calculada usando esta linha. Como a quantidade de adicionados desconhecidos foi de 25 mL, o valor determinado, 2.916 μg é dividido por 25 mL. Isso dá um resultado final de 0,117 μg/mL, ou 0,117 ppm. Isso é bem próximo do valor conhecido de 0,110 ppm.
Agora, vamos olhar para algumas outras técnicas analíticas que podem ter resultados distorcidos devido aos efeitos matriciais.
Espectroscopia de absorção atômica é um método analítico que mede a absorção da luz por um analito alvo na fase gasosa. Para a maioria das amostras, uma simples curva de calibração relacionando a absorção à concentração amostral, pode servir como um método confiável para quantificar uma concentração desconhecida.
No entanto, essa técnica pode perder a precisão se outros componentes da mistura interagirem com o analito de destino e suprimir ou melhorar a absorção. O método de adição padrão pode ser usado neste caso para explicar os efeitos dessas interações, especialmente em amostras onde a matriz não pode ser removida antes da análise.
A calibração do instrumento desempenha um papel crucial na precisão de uma medição. O método de adição padrão é frequentemente utilizado para auxiliar na calibração de instrumentos como o ICP-MS. ICP-MS é um método comparativo, o que significa que a medição de uma amostra desconhecida baseia-se na medição de um padrão químico.
Assim, a incerteza de uma medição de um desconhecido não pode ser melhor do que a incerteza da calibração. O método de adição padrão pode, portanto, ser usado para criar uma curva de calibração mais precisa do que o método padrão, e explica as interações matricial na amostra.
Muitas moléculas biológicas são analisadas usando cromatografia líquida de alto desempenho, ou HPLC. O HPLC é uma técnica que separa e analisa misturas complexas baseadas em propriedades de moléculas como polaridade, carga e tamanho. O momento em que o analito sai da coluna permite que o usuário identifique cada componente da mistura.
Moléculas biológicas podem frequentemente interagir em uma mistura, e são muito afetadas pela matriz em que estão suspensas. Muitas vezes, o método de adição padrão é usado para criar uma curva de calibração que explica esses efeitos.
Você acabou de assistir a introdução do JoVE ao método de adição padrão. Agora você deve entender como executar a técnica para explicar os efeitos matriciais na análise da amostra.
Obrigado por assistir!
Subscription Required. Please recommend JoVE to your librarian.
Results
Uma varredura do comprimento de onda de excitação de 335-435 mostrou a maior absorção em 399 nm, de modo que o monocromador de excitação foi definido para esse valor. Em seguida, a varredura de emissões foi realizada de 450 a 550 nm, e o sinal mais forte foi encontrado em 520 nm. Estes são os comprimentos de onda que são usados para todas as amostras.
| Amostra | Intensidade da fluorescência | Intensidade de fluorescência corrigida |
| Em branco | 0.008 | 0.000 |
| Amostra | 0.128 | 0.120 |
| Amostra + 1 mL | 0.167 | 0.159 |
| Amostra + 2 mL | 0.220 | 0.212 |
| Amostra + 3 mL | 0.260 | 0.252 |
| Amostra + 4 mL | 0.290 | 0.282 |
Um enredo de fluorescência (Figura 3) vs. μg de Al3+ adicionado(Figura 4) rendeu uma linha de menos quadrados de:
Intensidade de fluorescência = 0,0417 x (μg de Al3+ adicionado) + 0,1216
Quantidade de Al3+ = -(Y-Int)/Inclinação = -0,1216/0,0417 = -2.916 μg/mL
Como a quantidade de adicionados desconhecidos foi de 25 mL, então o valor de 2,916 μg/mL precisa ser dividido por 25.
Concentração de Alumínio Desconhecida = 2,916 μg/mL / 25,0 mL = 0,117 μg/mL = 0,117 ppm
que está bem próximo do valor real de 0,110 ppm (erro de 6,4%).

Figura 3. Fluorescência das amostras.

Figura 4. O gráfico de calibração de adição padrão.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
O método de adições padrão é frequentemente a técnica utilizada quando são desejados resultados quantitativos precisos, utilizados em análises analíticas como absorção atômica, espectroscopia de fluorescência, ICP-OES e cromatografia gasosa. Isso é frequentemente usado quando há outros componentes na amostra de interesse que causam uma redução ou aprimoramento da absorvância desejada para resultados quantitativos. Quando este é o caso, não se pode simplesmente comparar o sinal analitos com os padrões usando a abordagem tradicional da curva de calibração. Na verdade, a avaliação do efeito matricial deve ser uma parte obrigatória do procedimento de validação.
Outro exemplo onde as adições padrão podem ser usadas é ao extrair prata de resíduos fotográficos antigos. Os resíduos contêm halidos de prata, e podem ser extraídos para que a prata possa ser recuperada. Ao espiar o desconhecido "desperdício" com quantidades conhecidas de prata, este método pode prever a quantidade de prata obtida do filme fotográfico.
Os trabalhadores que estão expostos às fábricas de benzeno são frequentemente testados para verificar se estão seguramente abaixo dos níveis aceitos de benzeno. A urina deles é testada para o produto químico, e essa é a matriz biológica. Além disso, a quantidade de supressão de analitos varia para pessoas diferentes, de modo que um único kit de calibração não funcionará. Com o método de adição padrão, cada funcionário pode ser testado e avaliado com precisão.
Subscription Required. Please recommend JoVE to your librarian.