

Summary
In diesem Artikel beschreiben wir elektrochemische, Elektron-PARAMAGNETISCHE Resonanz und ultravioletten, sichtbaren und nahinfraroten Spectroelectrochemical Methoden für die Analyse organischer Verbindungen für Anwendung in der organischen Elektronik.
Abstract
Zyklischer Voltammetrie (CV) ist eine Technik, die in der Analyse von organischen Verbindungen verwendet. Wenn diese Technik mit Elektron-PARAMAGNETISCHE Resonanz (EPR) oder ultravioletten, sichtbaren und nahinfraroten (UV-Vis-NIR) Spectroscopies kombiniert wird, erhält man nützliche Informationen wie Elektron Affinität, Ionisation Potenzial, die Art der Bandlücke Energien Ladungsträger und Abbau-Informationen, die verwendet werden, um stabile organische elektronische Geräte zu synthetisieren. In dieser Studie stellen wir elektrochemische und Spectroelectrochemical Methoden für die Analyse der Prozesse, die in aktiven Schichten ein Bio-Gerät als auch die erzeugten Ladungsträger.
Introduction
Weltweit suchen Forscher ständig nach neuen organischen Materialien, die verwendet werden können, in der organischen Elektronik wünschenswert Leistung oder Stabilität, die aufgrund längerem Gebrauch fällt. Bei den Bio-Geräten ist es wichtig zu verstehen, das Verhalten der Ladungsträger vollständig die Regeln, die das Gerät Fahrverhalten kennen. Analyse der Wirkung von der molekularen Struktur auf die Generation der Ladungsträger und die Dynamik und Erhaltung des Gleichgewichts der injizierte Ladungsträger, sowohl positiv (Löcher) und negative (Elektronen), ist entscheidend für die Effizienz und Stabilität zu verbessern der Bio-Geräte. Dies sorgt für die effektive Rekombination dieser individuellen Gebühren und folglich verbessert deutlich die Photolumineszenz Effizienz der organische lichtemittierende Dioden (OLEDs)1,2. Für Organische Photovoltaik (OPV)3,4 , sowie Bio-Feld-Effekt Transistoren (OFETs)5,6ist es notwendig, um Materialien mit hohen Ladung Träger Mobilität haben. Neben der Analyse der Ladungsträger, mehrere wichtige Parameter der organischen Elektroaktive Materialien helfen, vorherzusagen, wo das Material verwendet werden könnten: Ionisation Potenzial (IP), Elektron-Affinität (EA)-Energieniveaus und Bandabstand zwischen ihnen7 ,8,9,10.
In dieser Arbeit präsentieren wir eine Methode für die effiziente Messung der zyklische Voltammetrie (CV), die in der Analyse aller Arten von Elektroaktive Materialien verwendet werden kann. Diese Technik bietet Informationen über Redox-Eigenschaften, doping/dedoping-Mechanismus, die Stabilität, die Umwandlung und Speicherung von Energie, etc.. Es ermöglicht auch für die Abschätzung des Elektrons Affinität und Ionisierung Energie der Testverbindungen gewissermaßen viel billiger und schneller im Vergleich zu anderen hohen Vakuum. Die oben genannten Parameter korrelieren mit den Energieniveaus des höchsten besetzten molekularen Orbital (HOMO) und niedrigsten unbesetzten molekularen Orbital (LUMO).
Die Methode in diesem Artikel vorgestellten kann verwendet werden, um alle Arten von konjugierte Verbindungen z. B. mit delokalisierte π-Elektronen in ihren Strukturen zu analysieren. Konjugierte Verbindungen möglicherweise kleine Moleküle mit großer polymerer Ketten. Kleine Moleküle können Monomere; während die erste Reaktion können (photochemische, elektrochemische oder chemische) Monomeren Polymere bilden. In OLED-Anwendung sind notwendig, das Energieniveau Werte ermöglichen die Verwendung von den korrekten Host für den Sender in eine thermisch aktivierte verzögerte Fluoreszenz (TADF)-Gast-Host-System oder zu entscheiden, mit denen die Exciplex-Donor-Akzeptor-Schicht Verbindungen könnte sein gebildet und welche zusätzliche Schichten (Electron Transport Layer (ETL) Transport Layer (HTL), Elektron-Sperrschicht (EBL) und Loch Sperrschicht (HBL) Loch) synthetisieren stabilen effizient berechnet werden müssen ausgewogen OLED-Geräte11 , 12 , 13 , 14 , 15 , 16 , 17. zusätzliche elektrochemische Messungen ermöglichen die Untersuchung der möglichen Nebenwirkungen während der Prozess des Abbaus der aktiven Ebene und die Bildung von niedrigen Mobile Ladungsträger (bipolaron)18,19 ,20,21,22.
Elektrochemischen Kopplung und Spectroelectrochemical Methoden erlaubt einfache, genaue und zuverlässige Bestimmung des Grades der Oxidation oder Reduktion konjugierte Verbindungen und ihren Abbau Potenzial, das entscheidend für Stabilität23 , 24 , 25 , 26 , 27 , 28. ultravioletten, sichtbaren und nahinfraroten (UV-Vis-NIR) Spektroskopie gepaart mit Elektrochemie kann chromatische Grundeigenschaften aller neuen konjugierte Verbindungen, z. B. das Ändern der Absorptionsbande bei doping charakterisieren 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30.
In einer Studie, die im Zusammenhang mit der doping-Mechanismus ist es wichtig, die Art der Ladungsträger zu definieren. In diesem Prozess zwei Klassen von geladenen Quasiteilchen teilnehmen, eine mit unkompensierten Spin (Polaronen) und der zweite diamagnetisch (bipolaron); Elektron-PARAMAGNETISCHE Resonanz (EPR)-Spektroskopie bietet unschätzbare Unterstützung, wodurch direkt zu beobachten und Nachverfolgen von Änderungen in den Bevölkerungen der paramagnetischen Polaronen29,30,31,32 . In kleine Moleküle es ist schwierig, Form bipolaron, aber diese Moleküle können ganz konjugiert und Bipolaron-induzierende Eigenschaften aufweisen; Es ist wichtig zu überprüfen, ob und an dem potenziellen Polaronen und bipolaron sind in der Struktur gebildet. Bipolaron sind mindestens einen Auftrag im Bereich der Mobilität unter dem Polaronen; Daher bipolaron in Arbeitsgeräte gebildet, könnte dann es zu einem unausgewogenen Verhältnis der Ladungsträger, führen die Hochstrom- und Überhitzung des OLED-Geräts führen würde oder möglicherweise auch die Zentren der Abbau33.
Die Methode der Messung in dieser Studie vorgeschlagenen ist billig und schneller und erlaubt die Bestimmung der wertvollsten operativen Parameter für eine große Anzahl von Elektroaktive Materialien ohne die Notwendigkeit für spezielle Geräte, die auf neu synthetisiert basieren Materialien, seine Leistung zu überprüfen. Durch die Anwendung der Elektrochemie und Spectroelectrochemistry, ist es möglich, ein Material auswählen, das wirklich vielversprechend aus Hunderten von neuen Materialien ist. Darüber hinaus ist es möglich, detaillierte Informationen über die Prozesse des Dopings zu erhalten und ihre Auswirkungen auf die chemische Struktur des Tests konjugiert mit elektrochemischen Systemen und Spectroelectrochemical Methoden, wodurch mehr bauen effiziente organische Elektronikgeräte.
Protocol
1. Vorbereitung des Experiments
- Bereiten Sie 25 mL 0,1 M-Elektrolyt-Lösung.
Hinweis: Abhängig von der untersuchten Verbindungen, verwenden verschiedene Elektrolyte als die Mischung aus organisches Salz und Lösungsmittel: die meisten gemeinsamen Salze verwendet in der Analyse von organischen Molekülen sind Tetrabutylammonium Hexafluorophosphate (Bu4NPF6 ) oder Tetrabutylammonium Tetrafluoroborate (Bu4NBF4); die am häufigsten verwendete Lösungsmittel in der Analyse verwendeten sind Tetrahydrofuran, Dichlormethan oder Acetonitril.- Wählen Sie anhand der Löslichkeit der Testverbindungen Elektrolyt-Lösungsmittel. Es sollte eine Lösung während der Untersuchung aber in einen festen Zustand zu sein, wenn das Material bei der Herstellung des Geräts, als dünner Film auf der Arbeitsfläche Elektrode abgeschieden verwendet wird.
- Reinigen Sie die Elektroden in der Experiment-29verwendet werden.
- Verwenden Sie für elektrochemische Messungen ein 1 mm Durchmesser Platin Disk Elektrode als Arbeitselektrode (WE), Platin Spule oder Draht als Hilfselektrode (AE) und einer Silber/Silberchlorid (Ag/AgCl) Elektrode als Bezugselektrode (RE).
- Verwenden Sie für EPR Spectroelectrochemical Messungen Platindraht als am WE, Platin Spule oder Draht als AE und Ag/AgCl als der RE.
- Verwenden Sie für UV-Vis-NIR Spectroelectrochemical Messungen eine Quarz Indium-Zinn-Oxid (ITO) oder Fluor-dotierte-Zinn-Oxid (FTO) als am WE, einem Platindraht als AE und einer Ag/AgCl-Elektrode als RE.
- Polieren der Platin Disk-Elektrode durch Reiben den Nachteil der Elektrode (Platin-Elektrode Arbeitsbereich) auf ein Polierpad mit 1 µm Aluminiumoxid Gülle für 3 min abgedeckt.
- Spülen Sie die Elektrode mit entionisiertem Wasser, entfernen alle die Schlämme aus der Elektrode und säubern in ein Ultraschallbad (320 W, 37 kHz) mit entionisiertem Wasser für 15 Minuten.
- Spülen Sie die Elektrode mit einer 1 mL Spritze mit Isopropanol (3 x 1 mL) und dann mit Aceton (3 x 1 mL).
- Verwenden Sie ein Papiertuch, um alle festen Rückstände zu entfernen und an der Luft für 3 min trocknen.
- Reinigen Sie die ITO und FTO Elektroden mit entionisiertem Wasser. Legen Sie sie in ein Ultraschallbad (320 W, 37 kHz) in Aceton für 15 min und dann in Isopropanol für die nächsten 15 Minuten.
- Brennen die Platin-Elektroden, Drähte und Spulen mit einem Hochtemperatur-Gasbrenner (> 1000 ° C) für 1 min. vorsichtig sein! Benutzen Sie eine Pinzette, um die Elektroden zu halten. 5 min. warten Sie, nach dem Brennen und vor der Verwendung der Elektroden.
- Reinigen Sie elektrochemischen und Spectroelectrochemical Zellen (Abbildung 1) mit Wasser und dann mit Aceton. Verwenden Sie ein Papiertuch um die festen Rückstände zu entfernen. Alle anderen Elemente (ca. PTFE Teile) mit Aceton reinigen und Trocknen in der Luft vor dem Gebrauch.
2. Lebenslauf, Analyse
- Schalten Sie die potentiostaten und Computer.
- Füllen Sie eine elektrochemische Zelle mit 1,5 mL Elektrolytlösung analysiert werden.
- Alle drei Elektroden (WE, AE und RE) in einer Zelle (die Zelle Kappe ist mit der PTFE-Elektrodenhalter, also stecken Elektroden durch die Löcher in diesem Halter ausgestattet) und verbinden Sie es mit einem potentiostaten. Halten Sie am WE und RE als nahe beieinander wie möglich (Abbildung 1). Folgen Sie Informationen über die Verbindung von jedem Draht von dem potentiostaten an seinen jeweiligen Elektrode.
- Bei Bedarf (z. B. beim Abbau-Analyse), Argon (oder Stickstoff) Rohr (durch ein zusätzliches Loch in den Elektrodenhalter) und beginnen Sie sprudeln die Lösung mindestens 5 min lang. Danach bewegen Sie das Argon (oder Stickstoff) Rohr ü. die Lösung und halten Sie die Gaszufuhr für die Messung läuft.
Hinweis: Die Zelle sieht geschlossen aber nicht völlig auslaufsicher sein; Daher muss eine Methode zum Entfernen der redundanten Menge von Gas aus der Zelle verfügbar sein (zB., durch ein zusätzliches kleines Loch in den Elektrodenhalter). Der Druck hängt das Rohr verwendet; Stellen Sie der Druck zu hoch ein, wie es erforderlich ist, um den langsamen Gasstrom eingestellt. Wenn leicht flüchtige Lösungsmittel in das Experiment (z. B. Dichlormethan) verwendet wird, oder das Experiment sehr lange (mehr als 30 min dauert), dann verwenden Sie die Drechsel Flasche Gas Argon (oder Stickstoff) mit dem Lösungsmittel verwendet bei der Messung zu sättigen. - Die potentiostaten Software ausführen, wählen Sie das CV-Verfahren und verwenden Sie die folgenden Einstellungen: start Potenzial von 0.00 V, oberen Scheitelpunkt Potenzial von 2,0 V, untere Scheitelpunkt Potenzial von 0.00 V (wenn der Prozess der Reduktion, dann einen niedrigeren Scheitelpunkt Potenzial der −2.5 V untersuchen), Potenzial von 0.00 V, Anzahl der Stop-Kreuzung von 6 zu stoppen, und Abtastrate von 0,05 V/s.
- Im Abschnitt exportieren ASCII-Datendefinieren einen Dateinamen und wählen Sie einen Ordner zum Speichern der Daten in, und drücken Sie die Starttaste (erhöhen oder verringern der unteren und oberen Scheitelpunkt Potenzial, das elektrochemische Fenster oder die Probe passen Elektrochemische Aktivität)
- Wenn jeder Gipfel in den positiven Bereich Potenzial sichtbar sind, wiederholen Sie den Reinigungsvorgang. Potenzial im negativen Bereich gibt es ein Höhepunkt, dann setzen Sie die Argon (oder Stickstoff) Leitung in der Lösung und für eine zusätzliche 5 min sprudeln.
- Einen Tropfen von 1 mM Ferrocen (in dem Lösungsmittel zur Vorbereitung des Elektrolyts verwendet) auf die Elektrolyt-Lösung mithilfe einer Spritze.
- Das Start-Potenzial auf 0.00 V, der oberen Scheitelpunkt zu 0,85 V, der untere Scheitelpunkt potenzielle auf 0.00 V, das Stop-Potenzial auf 0.00 V, die Anzahl der Stop-Kreuzung bis 10 und die Abtastrate auf 0,05 V/s festgelegt. Im Abschnitt exportieren ASCII-Datenändern Sie den Dateinamen und drücken Sie die Starttaste .
- Nach der Messung abschließend die Reinigungsverfahren zu wiederholen, wie in den Schritten 1.2 und 1.3 erwähnt.
- Bereiten Sie 4 mL 1 mM Testverbindung in den vorbereiteten Elektrolyten (Schritt 1.1).
- Füllen Sie die elektrochemische Zelle mit zusammengesetzten Testlösung (1,5 mL); Legen Sie alle drei Elektroden in eine Zelle und verbinden mit dem potentiostaten (Schritt 2.4). Um den Prozess der Reduktion zu untersuchen, entfernen Sie den Sauerstoff (Schritt 2.5).
- Voraussichtlich Anfang potenzielle 0.00 V, den oberen Scheitelpunkt potenzielle 0,50 V (oder 0.00 V bei Untersuchung der Reduktionsprozess), der untere Scheitelpunkt potenzielle auf 0.00 V (−0.50 V bei Untersuchung der Reduktionsprozess), das Stop-Potenzial auf 0.00 V , die Anzahl der Stop-Kreuzung bis 10 und die Abtastrate auf 0,05 V. Im Abschnitt exportieren ASCII-Datenändern Sie den Dateinamen und drücken Sie die Starttaste .
- Wiederholen Sie Schritt 2.13 durch eine Erhöhung der oberen Scheitelpunkt Potentiale (oder durch eine Verringerung der untere Scheitelpunkt-Potenzial) um 0,1 V bis Gipfel Registrierung (Abbildung 2a). Wenn der aufeinander folgenden Scan Potenzial (Abb. 2 b) verschoben wird, dann reinigen Sie das Rück und lassen Sie ihn in der Elektrolytlösung. Wiederholen Sie die Messung.
- Oxidation und Reduktion Prozesse auf den gleichen Lebenslauf für die Ermittlung der IP-Adresse und EA zu messen: das Start-Potenzial auf 0.00 V, das Potenzial der oberen Scheitelpunkt auf 1,00 V, das untere Scheitelpunkt Potenzial, −2.70 V und das Stop-Potenzial auf 0.00 V. Wählen Sie die obere und untere Vert gesetzt Ex-Potenzial in einer Weise registriert, die vollständige Reduktion und Oxidation Gipfeln und vermeidet weitere Redox-Schritte (falls einer vorhanden ist) (Abbildung 2a).
Hinweis: Schätzen IP und EA durch die Messung der potenziellen Ausbruch. Es gibt ein paar Möglichkeiten, um diese Parameter von CV berechnen aber am nützlichsten ist, mit den möglichen Ausbruch zu berechnen. Manchmal ist es nicht möglich, reversible Redox-Paar, vor allem für n und p doping zu beobachten. Mischen von zwei verschiedenen Techniken ist auch unerwünscht, da es falsche Ergebnisse und Schlussfolgerungen bieten kann.- Um den möglichen Ausbruch zu messen, markieren Sie die Tangente an die CV-Peak (Abbildung 3). Berechnen Sie die gewünschten Werte vom Beginn Potenzial für die Minderung oder Oxidationsprozess mit Gleichungen (Gleichungen 1 und 2)10,18,19,36,37 , 38.
 (1)
(1)
wo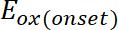 ist das Auftreten von Oxidationspotential kalibriert mit Ferrocen*
ist das Auftreten von Oxidationspotential kalibriert mit Ferrocen*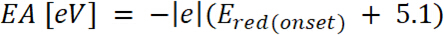 (2)
(2)
wo ist das Auftreten von Reduktionspotenziale kalibriert mit Ferrocen*
ist das Auftreten von Reduktionspotenziale kalibriert mit Ferrocen*
* Potenzial mit Ferrocen kalibriert bedeutet, dass der Wert des Potentials von der Messung durch das Oxidationspotential von Ferrocen reduziert wird.
- Um den möglichen Ausbruch zu messen, markieren Sie die Tangente an die CV-Peak (Abbildung 3). Berechnen Sie die gewünschten Werte vom Beginn Potenzial für die Minderung oder Oxidationsprozess mit Gleichungen (Gleichungen 1 und 2)10,18,19,36,37 , 38.
- Ziehen Sie die Elektroden und Gießen Sie der Probelösung in den richtigen Abfallbehälter aus.
- Wiederholen Sie den Reinigungsvorgang (Schritte 1.2 und 1.3).
- Wiederholen Sie die Schritte 2,9 – 2.12 mit 0,25 0,10 und 0,20 V/s Abtastraten.
- Geben Sie einen Tropfen von 1 mM Ferrocen (in dem Lösungsmittel zur Vorbereitung der Elektrolytlösung), die Testlösung mit einer Spritze. Set Start-Potenzial auf 0.00 V, der oberen Scheitelpunkt potenzielle auf 0,60 V, der untere Scheitelpunkt Potenzial, 0.00, das Stop-Potenzial auf 0.00 V, die Anzahl der Stop-Kreuzung 6 und die Abtastrate auf 0,05 V. die START -Taste drücken. Nachdem die Messung abgeschlossen ist, ändern Sie den oberen Scheitelpunkt potenzielle auf einen Wert, der das Ferrocen-Paar mit dem ersten Höhepunkt der Oxidation von der Testverbindung registriert.
- Ziehen Sie die Elektroden und Gießen Sie der Probelösung in den richtigen Abfallbehälter aus.
- Tritt der Electropolymerization bei elektrochemischen Oxidation, die wir sorgfältig waschen mit der Elektrolyt-Lösung mit der Spritze (3 ´ 0,5 mL). Reinigen Sie RE, AE und elektrochemische Zelle mit dem Standardverfahren (Stufen 1,2-1,3).
- Elektrochemische Produkte hinterlegt auf wir untersuchen, fahren Sie mit Schritt 2.23. Um eine Lösung zu untersuchen, gehen Sie zu Schritt 1.2.1 und starten Sie erneut ab Schritt 2.3.
- Füllen Sie die elektrochemische Zelle mit einem Elektrolyt-Lösung. Dann legen Sie alle drei Elektroden in eine Zelle und verbinden Sie diese mit einem potentiostaten (Schritt 2.4). Um den Abbauprozess, Deoxidate Lösung (Schritt 2.5) zu untersuchen.
- Wiederholen Sie die Schritte 2.10, 2.12 und dann Schritt 2.16.
 (1)
(1)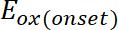 ist das Auftreten von Oxidationspotential kalibriert mit Ferrocen*
ist das Auftreten von Oxidationspotential kalibriert mit Ferrocen*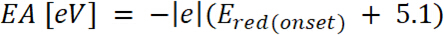 (2)
(2) ist das Auftreten von Reduktionspotenziale kalibriert mit Ferrocen*
ist das Auftreten von Reduktionspotenziale kalibriert mit Ferrocen*(3) UV-Vis-NIR Spectroelectrochemical Analyse
- Vorbereitung der Messung
- Schalten Sie das Spektrometer und potentiostaten.
- Füllen Sie die Spectroelectrochemical-Zelle mit 0,5 mL Elektrolytlösung (Schritt 1.1).
- Setzen Sie die PTFE-Dichtung auf der einen Seite der Zelle; dann legen Sie die ITO-Elektrode auf die Zelle auf diese Weise, eine leitende Seite der Elektrode berühren die PTFE-Dichtung. Setzen Sie den Rest der PTFE-Teile, wie in Abbildung 1dargestellt. RE und AE durch die PTFE-Elektrodenhalter und bedecken Sie die Oberseite der ITO-Elektrode mit Kupferfolie wie in Abbildung 1 dargestellt, für bessere Leitfähigkeit.
- Setzen Sie die montierte Zelle in der Halterung des Spektrometers und verbinden Sie die Elektroden mit einem potentiostaten. Folgen Sie Informationen über die Verbindung von jedem Draht von dem potentiostaten an seinen jeweiligen Elektrode.
- Starten Sie die Software potentiostaten und Spektrometer.
- Wählen Sie in der Spektrometer-Software Datei | Neu | Absorption Messung.
- Wählen Sie eines der aufgelisteten Detektoren.
- Stellen Sie sicher, dass die Schaltfläche auf die Detektoren in der "offenen" Position befindet. Klicken Sie auf die leuchtende Glühbirne; Schließen Sie den Detektor (bewegen Sie den Knopf in die geschlossene Position) und klicken Sie dann auf die dunkle Glühbirne.
- Wählen Sie den zweiten Detektor und wiederholen Sie Schritt 3.1.7.
- Schließen Sie Elektroden aus dem potentiostaten und Elektroden und anderen Elementen der Spectroelectrochemical Zelle herausziehen.
- Wiederholen Sie die Reinigungsverfahren (Schritte 1.2 und 1.3).
- Füllen Sie die Spectroelectrochemical-Zelle mit einer verdünnten Lösung (1´10-5 M) der Testverbindung in der Elektrolyt-Lösung, wenn das kleine Molekül zu testen. Füllen Sie die Spectroelectrochemical Zelle mit dem Elektrolyt, wenn die Polymerschicht (Oligomere) auf die ITO-Elektrode abgeschieden zu testen.
- Montieren Sie die Spectroelectrochemical-Zelle (Schritt 3.1.3).
- Klicken Sie auf Speichern.
- Wählen Sie eine der aufgeführten Detektoren.
- Definieren Sie die speichern Speicherort und den Namen der Datei (einschließlich den ausgewählten Detektor).
- Wählen Sie den zweiten Detektor und wiederholen Sie Schritt 3.1.15.
- Potentiostatischer Analyse18.
- Ein neutrales Potenzial (d. h. 0.00 V) gelten. Dieses Spektrum wird die starten-Spektren. Erhöhen Sie Potenzial von 0,1 V und warten Sie ca. 10 s, bis der Prozess stabilisiert und Absorptionsspektren (Schritte 3.1.13–3.1.16) erfasst. Fortsetzen Sie die Messung der ersten Änderung im UV-Vis-NIR-Spektren; warten Sie 10 s und dann speichern die Spektren (Schritte 3.1.13–3.1.16).
- Potenziale von 0,05 V zu erhöhen; warten Sie 10 s und speichern (Schritte 3.1.13–3.1.16).
- 3.2.2 zu wiederholen, bis das Potenzial der ersten oder zweiten Oxidationspotential (diese Potentiale sind bekannt aus der CV-Messung); dann das Potenzial umkehren und zurück zum Ausgangspunkt Potenzial.
- Nach Abschluss der Messungen Messen Sie den Lebenslauf der Testlösung. Fügen Sie 1 mM Ferrocen (10 μl) und Messen Sie den Lebenslauf wieder. Der Wert des Ferrocen hilft schätzen das Potenzial des Prozesses und die Daten zu vereinheitlichen.
- Diese Analyse gibt Auskunft über die optische Bandlücke, maximale Wellenlänge undotierter und dotierter Staaten und Isosbestic Punkten des elektrochemischen Prozesses. Berechnen Sie die optische Bandlücke von Beginn Werte der π-π* Bands auf den UV-Vis-Spektren und mit Gleichung 318,19,23,24.
 (3)
(3)
wo h die PLANCKsche Konstante, ist λBeginn ist zusammengesetzte Aufnahme auftreten, und c ist die Lichtgeschwindigkeit im Vakuum. - Zeitaufgelösten Potentistatic Analyse19,32,33
- Bereiten Sie die Messung im 3.1.1–3.1.16 beschriebene Methode ähnlich und setzen Sie die Argon (oder Stickstoff) Leitung (durch ein zusätzliches Loch in den Elektrodenhalter) und sprudelt die Lösung mindestens 5 min vor der Messung. Danach bewegen Sie das Argon-Rohr höher als Lösung und halten Sie die Gaszufuhr für die Messung läuft.
- Die potentiostaten Software ausführen, wählen Sie das Chronoamperometry-Verfahren mit den folgenden Einstellungen: Potenzial start Potenzial auf 0.00 V, obere Potenzial auf 1,0 V, niedrigeren Potential, −1.00 V, Dauer 5 s, Intervall-Zeit als 0,010 s, Anzahl der Wiederholungen auf 10 und Verzögerung Zeit für 10 s. Definieren Sie im Abschnitt exportieren ASCII-Dateneinen Dateinamen und wählen Sie einen Ordner zum Speichern der Daten.
- Wählen Sie in der Spektrometer-Software Datei | Neu | High-Speed-Übernahme. Es erscheint ein neues Fenster; richten Sie die folgenden Parameter: Integrationszeit – 10 ms, Scans, Durchschnitt — 1, Boxcar Wirth — 0, Anzahl der Scans – 10000. Klicken Sie nicht auf die Schaltfläche " Go ".
- Drücken Sie in der Software potentiostaten die Starttaste . Wenn der Countdown fällt auf 1 s, drücken Sie die Schaltfläche " Go " in der Spektrometer-Software.
- Beachten Sie, dass es nicht möglich, Absorption Ergebnisse zu sehen, bis der Prozess beendet ist.
- Analysieren Sie die Daten, um Parameter liefern. Verwenden Sie die Informationen in der Datei gespeichert solche Durchlässigkeit, Strom, Spannung und Stromdichte Arbeitsparameter zu bieten:
- Berechnen Sie die optische Dichte (ΔOD), die Absorption der Testverbindung beim doping (dedoping) mit Gleichung 4:
 (4)
(4)
wo ist Tb Durchlässigkeit in dotierten Form, ein "Bleach" Zustand [%] und Tc Durchlässigkeit im undotierten Form, einem "farbigen" Zustand [%]. - Berechnen Sie Färbung Effizienz (CE, η), die Menge der elektrochromen Farbe gebildet durch die verwendete Menge kostenlos und als Verbindung zwischen der injiziert/ausgeworfen kostenlos als Funktion der Elektrodenfläche (Qd) und die Veränderung der optischen Dichte (ΔOD) Wert bei einer bestimmten Wellenlänge (λMax.) mit Gleichung 5. Sein Wert hängt die Wellenlänge für die Studie, so der Messwert bei λMax. der optischen Absorption Band des farbigen Staates gewählt.
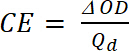 (5)
(5)
wo ΔOD ist die optische Dichte [-] und Qd ist die Ladungsdichte [C/cm2]. - Der Kontrast Verhältnis (CR), die gemessene Intensität der Farbänderung während elektrochemische doping zu berechnen und in der Regel gekennzeichnet durch λMax. der farbigen Form mit Gleichung 6:
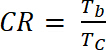 (6)
(6)
wo ist Tb Durchlässigkeit in dotierten Form, "bleach" Zustand [%] und Tc Durchlässigkeit im undotierten Form, "farbigen" Zustand [%].
- Berechnen Sie die optische Dichte (ΔOD), die Absorption der Testverbindung beim doping (dedoping) mit Gleichung 4:
 (3)
(3) (4)
(4)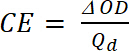 (5)
(5)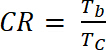 (6)
(6)(4) EPR-Spectroelectrochemical-Analyse
- Vorbereitung der Messung
- Aktivieren Sie das Spektrometer und dem potentiostaten.
- Füllen Sie die Spectroelectrochemical-Zelle mit Elektrolyt (Schritt 1.1).
- Satz die Macht zu 1 mW und Melodie der Resonator, ein scharfes, zentriert Signal im Q-Dip (verwenden Sie die Option Autotuned) zu erreichen.
- Die Mn-Marker (Mangan standard) auf 600 festgelegt und schließen Sie das Q-Dip. Besteht kein Mn-Marker, dann überspringen Sie diesen Schritt und gehen Sie zu Schritt 4.8.
- 2.5´ "fegen Breite ±" voraussichtlich 10 mT, "mod Breite ±" 1.0´0.1 mT, "Amplitude", 5´100, "Zeitkonstante" 0,03 s und "Mittelfeld" Wert auf ca. 338 mT und starten Sie die Messung.
- Wenn sechs Spektrallinien von Mn Marker nicht zu registrieren, ändern Sie den Wert "Mittelfeld".
- Bei der Registrierung sechs Spektrallinien von Mn Marker (Abbildung 4) legen Sie den Wert "Mittelfeld" zwischen den dritten und vierten Zeile und verringern Sie den Wert von "Sweep Breite ±" auf einen Wert, der nur diese zwei Zeilen umfasst. Starten Sie die Messung erneut.
- Gehen Sie in die Q-Dip-Option, die Mn-Markierung auf 0 gesetzt, und schließen Sie das Q-Dip.
- Messen Sie das reine Elektrolyt als Hintergrund zu und prüfen Sie, ob gibt es keine Signale. Wenn die Signale sichtbar sind, kann das Signal von der Kontamination, geringe Oxidation mögliche Verbindungen, oder aus dem Glas sein.
- Reinigen Sie die Spectroelectrochemical-Zelle (Schritt 1.3).
- Untersuchung von kleinen Molekülen (Lösungen).
- Füllen Sie die Spectroelectrochemical-Zelle mit einer verdünnten Lösung (1´10-5 M) von der Testverbindung im Elektrolyten; der Elektrodenhalter in die Zelle auf diese Weise alle drei Elektroden durchgestellt damit am WE und RE befinden sich innerhalb der Spirale AE (Abbildung 1).
- Legen wir nahe am unteren Rand der Zelle und der RE an der Oberseite des aktiven (Metall nicht abgedeckt von PTFE) Teil am WE (Abbildung 1). Legen Sie die Zelle mit einer Elektrode im Inneren der Probe Hohlraum des Spektrometers ausgestattet und verbinden Sie die Elektroden mit einem potentiostaten (folgen Angaben über die Verbindung der einzelnen Draht der die potentiostaten an seinen jeweiligen Elektrode).
- Wenden Sie das Potenzial entspricht dem Beginn des ersten Redox-Peak (Wert aus CV Messung bekannt). Wenn ein Signal angezeigt wird, ist das Gerät ordnungsgemäß eingerichtet.
- Gehen Sie in die Q-Dip-Option festlegen des Mn-Markers auf 600, das Q-Dip-Fenster zu schließen und starten Sie die Messung. Die Registrierung des Signals mit dem Signal des Mn Marker erlaubt die Berechnung der g-Faktor des empfangenen Signals.
- Gehen Sie in die Q-Dip-Option, die Mn-Markierung auf 0 setzen, schließen Sie das Q-Dip und warten ca. 5 min.
- Legen Sie den Wert "Mittelfeld" in der Mitte des Signals, verringern Sie den Wert von "Sweep Breite ±" auf einen Wert, der das Signal deckt, aber schneiden Sie den Anfang und das Ende des Signals (wenn "fegen Breite ±" geändert wird, gesamte Signal Check) und Abnahme der Wert von "m nicht Breite ± od "gut aufgelöste Spektren zu bekommen.
- Wenn das Signal klein ist, erhöhen Sie den Wert "Amplitude" und die "Zeit" (Acquisition Time) der Messung.
- Um das Signal-Rausch-Verhältnis zu verringern, legen Sie den Erwerb auf 4, 9 oder 16 je nachdem, wie laut das Signal ist. Wenn das Signal sehr laut ist, wählen Sie 16.
- Füllen Sie die Spectroelectrochemical-Zelle mit einer verdünnten Lösung (1´10-5 M) von der Testverbindung im Elektrolyten; der Elektrodenhalter in die Zelle auf diese Weise alle drei Elektroden durchgestellt damit am WE und RE befinden sich innerhalb der Spirale AE (Abbildung 1).
- Untersuchungen der Polymeren Schicht auf der Oberfläche der am WE hinterlegt.
- Füllen Sie die Spectroelectrochemical Zelle mit einer Elektrolytlösung (Schritt 1.1).
- Genommen Sie Elektroden in der Zelle. Achten Sie darauf, dass Sie nicht die Polymerschicht auf die WE zu zerstören. Verbinden Sie die Elektrode mit dem potentiostaten (Schritt 4.2.1).
- Gelten Sie neutrale Potenzial (d.h. 0,00 V) um sicherzustellen, dass die Verbindung in einem neutralen Zustand; Dieses Spektrum wird den starten EPR-Spektren.
- Erhöhen Sie Potenzial von 0,10 V. warten Sie ca. 10 s zu, bis der Prozess stabilisiert und nehmen Sie die EPR-Spektren auf.
- Wiederholen Sie Schritt 4.3.4, wenn das EPR-Signal angezeigt wird.
- Erhöhen Sie das Potenzial von 0,05 s ca. 10 V. warten zu, bis der Prozess stabilisiert und nehmen Sie die EPR-Spektren auf.
- Wiederholen Sie Schritt 4.3.6 bis zum ersten oder zweiten Oxidationspotential ist und so weiter erreicht (Werte dieser Potenziale sind bekannt aus CV-Messung); dann das Potenzial umzukehren und zurück zur möglichen ab.
- Wenden Sie das Potenzial, an dem der EPR Signal erschien im vorherigen Zyklus (4.3.5), gehen in die Q-Dip-Option festlegen des Mn-Markers auf 600, das Q-Dip-Fenster zu schließen und starten Sie die Messung. Registrieren Sie das Signal mit der dritten und vierten Spektrallinie von Mn Marker (4.1.7).
- Wenn die Messung abgeschlossen ist, analysieren Sie die Daten.
- Einige der Analyse wie z. B. eine Anzahl von Drehungen nur für die Verbindungen durchgeführt werden kann hinterlegt (unlösliche Polymere) auf die WE. Zur Berechnung der Anzahl der Drehungen, integrieren Sie Doppel-das EPR-Signal, gefolgt von einem Vergleich der erhaltene Wert mit kalibrierten Mn internem Standard. Berechnen Sie die Anzahl sich wiederholender Einheiten bilden das Polymer auf die Elektrode mit der Gleichung 724,32,33:
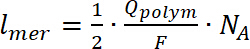 (7)
(7)
wo lMer – Anzahl sich wiederholender Einheiten im Polymer, Qerzielenden berechnet die Polymerisation, F die Faraday-Konstante ist und NA ist die Avogadro-Zahl. - Verwenden Sie den Lebenslauf der hinterlegten Polymer erfasst in einer Spectroelectrochemical Zelle vor der EPR-Messung mit Gleichung 8, um doping kostenlos berechnen:
 (8)
(8)
wo Qd ist das doping kostenlos i ist der Strom, E ist der Satz möglicher, E0 ist das Start Potenzial, V ist die Abtastrate und e ist die elementare kostenlos. - Verwenden Sie berechnete doping kostenlos doping, mit 9 Gleichungberechnen:
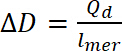 (9)
(9)
wo ∆D- doping Ebene, Qd die doping kostenlos und lMer – Anzahl der Einheiten in das Polymer zu wiederholen - Zum Berechnen der Anzahl von bipolaron übernehmen Sie eine anfängliche doping Ebene gleich NULL. Verwenden Sie die Anzahl der Polaronen pro Einheit aus der Konzentration von Spins, so dass die Anzahl der bipolaron berechnet werden soll, mit Gleichung 10. In den Berechnungen gehen davon aus, dass der ursprüngliche Betrag der bipolaron NULL ist und dass faradaic Strom viel höher als die der nicht-faradaic Strom ist.
 (10)
(10)
wo Zich der Ladungsträger elementare kostenlos, ist niist die Anzahl der Ladungsträger pro Polymer-Einheit (p) ist die Polaronen und (b) ist die bipolaron. - Extrahieren der g-Faktor Wert aus den EPR-Spektren. Bestimmen mit Mn als interner Standard (Schritte 4.2.3 und 4.3.8), wohl wissend, dass die vierte Zeile der Mangan drin einen g hat-Faktor des 2.03324. Berechnen Sie die Linienbreite des EPR-Signal als die Entfernung in mT zwischen Minimum und Maximum des EPR-Signals. Dieser Wert ist wichtig, da es sagen kann, wo (an welchem Atom) die radikale gebildet werden.
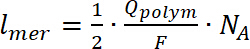 (7)
(7) (8)
(8)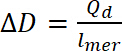 (9)
(9) (10)
(10)Representative Results
Die häufigste Anwendung der CV-Analyse ist die Schätzung von IP und EA. Obwohl es ein Paare Möglichkeiten gibt, CV-Daten zu erhalten, wird es dringend empfohlen, basierend auf den Beginn der Redox-Gipfel (Abbildung 3) zu berechnen. Dieser Ansatz ermöglicht es, das Berechnungsverfahren zu vereinheitlichen. Nicht alle der getesteten Materialien durchlaufen reversible Oxidation/Reduktion Prozesse (Abbildung 3); in solchen Situationen, es ist nicht möglich, auf der Grundlage im Durchschnitt berechnen Potentiale (Durchschnitt aus Potentialen entsprechen maximal kathodische Reduktion und anodische Oxidation Gipfel). Allerdings ist es fast immer möglich, die Tangente zu einer Spitze zu betreiben, wie in Abbildung 3dargestellt. An der Kreuzung mit der Hintergrund-Linie und die Verwendung der Gleichungen 1 und 2sind die IP und EA Werte geschätzt als 5,35 eV und −2.90 eV, beziehungsweise. Es gibt auch mehrere verschiedene Skalen verwendet, um IP- und EA basierend auf CV Messungen auszuwerten. Die am häufigsten verwendeten Skalen für organische Materialien −4.8 und −5.1 sind eV 0.00 V im Vergleich zu normalen Wasserstoff Elektrode (NHE) gleichgestellt. Allerdings sind alle von der Waage nur eine Näherung; Denken Sie daran, wenn Sie unterschiedliche Ergebnisse zu vergleichen. Entscheidend ist, anzugeben, welche Parameter für die Berechnung berücksichtigt wurden. In diesem Fall hat der Wert −5.1 eV wurde ausgewählt, da es die formale Potenzial des Ferrocen Redox-Paares entsprechen sollte; in der Fermi-Skala ist es 0.40 V gegenüber gesättigt Kalomel Elektrode (SCE) in Acetonitril, die im Einvernehmen mit der vorherigen Messung ist.
Es gibt viele Artikel über die Analyse der CV hierin veröffentlicht, wir zeigen, wenn der Prozess nicht geht, wie es erwartet wird. Die Analyse basiert auf Thiophen Ableitung: NtVTh (Struktur in Abbildung 4dargestellt), der erfährt Abbau bei der Oxidation (Abbildung 5).
NtVTh hat zwei Oxidation Potentiale: die erste bei 0,70 V und die zweite bei 0,84 V (Abbildung 5). Während des ersten Zyklus die Verringerung der Spitze wird nicht beobachtet, zeigt einen unumkehrbaren Prozess. Elektrochemische Eigenschaften der NtVTh zeigen, dass die Polymerisation tritt nicht auf, und nach der ersten Oxidationspotential einige Elektro-inaktive Schicht der Reaktionsprodukte Einlagen an der Elektrodenoberfläche, damit die Polymerisation zu behindern. Was sichtbar ist, ist die Reaktion mit dem radikalen kation auf der Vinyl-Bindung, wo das Molekül seine Konjugation und bilden Dimere auf der Elektrode verliert.
Es ist, zwar schwierig, Informationen über Ladungsträger aus dem Lebenslauf zu extrahieren ist es möglich, die Unterscheidung zwischen Polaronen und bipolaron Wenn Sie von einer UV-Vis-NIR-Spektrometer unterstützt. Die neutrale Poly (O-iPrThEE) zeichnete sich durch zwei breite Absorption π | π * Übergänge Bands mit Spitzen Maxima bei λmax1 = 363 nm und λmax2 = 488 nm, im Zusammenhang mit der aromatischen Form der Undotierte Polythiophen Ableitung. Während die oxidative Dotierung entstehen neue Polaronic und Bipolaronic Bands. Die UV-Vis-Spektrum erhalten bei der Oxidation von Poly (O-iPrThEE) offenbart die abnehmenden neutrale Polymer Absorptionsbande (300-550 nm) (Abbildung 6) zusammen mit der Bildung einer neuen Absorptionsbande (550-950 nm) von der radikal-Kationen der Bithiophene und p- Phenylenevinylene mit Maxima bei 692 nm. Der Isosbestic Punkt der Oxidationsprozess befand sich an 604 nm. Die Bipolaronic Band erschien zwischen den 950 nm und 1700 nm mit einem Maximum am 1438 nm befindet.
EPR-Spektroskopie ist die Technik, die Materialien mit einem ungepaarten Elektron erkennt, dazu gehören organische radikale39. Gibt es verschiedene Parameter, die aus EPR Spektren extrahiert werden konnten, aber eines der interessantesten schätzen, wo die radikalen lokalisiert werden soll. Elektronen, Protonen, ähnlich besitzen Spin. Mit der Abgabe eines Elektrons in einem äußeren Magnetfeld, kann diesem Spin zwei Arten aufgeteilt werden: parallel und antiparallel zum Magnetfeld, so dass zwei Energieniveaus. Dieses Phänomen nennt man die Zeeman Effekt40,41. Im Falle von organischen radikalen interagiert das ungepaarte Elektron nicht nur mit dem externen Magnetfeld, sondern auch mit magnetischen Kernen (Kerne, die eine ungleich NULL drehen; I≠0). Eine Reihe von entarteten Energieniveaus entsprechen 2I + 1, wo ich die Spin-Quantenzahl des Kerns das ungepaarte Elektron42 interagiertist. Das Zusammenspiel von das ungepaarte Elektron mit einer größeren Anzahl von magnetische Kerne führt die weitere Aufteilung der Energie-Ebenen und Hyperfein Struktur des EPR Spektren Registrierung43 (Abbildung 7).
Für Moleküle wo das ungepaarte Elektron Wechselwirkung mit einer noch größeren Zahl von Kernen, könnten die einzelne Spektrallinie überschneiden, welche Ergebnisse bei der Registrierung von einem einzigen, breites Signal44,45,46 (Abbildung 8). Dies ist typisch für konjugierte Polymere, wo das erzeugte radikale Ion in einem Redox-Prozess delokalisierte47,48 ist.
Die Kombination der EPR-Spektroskopie mit elektrochemischen Methoden ermöglicht die Charakterisierung der Ladungsträger (radikale Ion) erzeugt während der Redox-Prozess sowie die Bestimmung des Mechanismus der diese Prozesse49,50 . Wenn gut gelöst (Gipfel sind getrennt; nicht in Form von einem breiten Gipfel) Spektren sind registriert, wie im Falle der elektrochemische Reduktion von s -kurz-Derivat (Abbildung 7), die Analyse der Hyperfein Struktur der Spektren führt dann zu Rückschlüsse auf die Lokalisation der ungepaarte Elektron. Eine Möglichkeit, diese Art von Spektren zu analysieren ist, Simulation mit einer speziellen Software durchzuführen und simulierte Spektren mit den experimentellen ein51passen. Dies ist besonders hilfreich, wenn die Hyperfein-Struktur durch das Zusammenspiel von das ungepaarte Elektron mit einer großen Anzahl von Protonen ist. Bei der s- kurz-Derivat in Abbildung 7gezeigt zeigt Simulation von EPR Spektren (rote Linie) das Zusammenspiel von das ungepaarte Elektron mit vier Stickstoffatome von s- kurz.

Abbildung 1: elektrochemische und Spectroelectrochemical Zellen für Messungen verwendet. Die Abbildung zeigt das Schema-Setup der elektrochemischen/Spectroelectrochemical Zellen mit zyklischer Voltammetrie, Ultraviolett, sichtbar und nah-Infrarot (UV-Vis-NIR) und Elektron-PARAMAGNETISCHE Resonanz (EPR) Spectroelectrochemical Messungen. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 2: zyklischer Voltammetrie (CV) richtig gemessenen zusammengesetzten COPO1 (a) und die CV mit einer instabilen verweisen potenzielle Dibenzothiophene-S, S-Kohlendioxid mit Ferrocen (b)52. Die Abbildung zeigt zwei zyklische Voltammograms. (a) Geschenke richtig registriert CV und (b) zeigt eine Voltammogram mit einer Referenzelektrode mit keine stabile potenzielle registriert. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 3: zyklischer Voltammetrie (CV) von zusammengesetzten COPO1 in einer Vielzahl von Potenzialen. Schätzung der Beginn Potenziale bei EA und IP-Berechnungen für die COPO1 Verbindungen52. EA = −2.90 eV und IP = 5,35 eV. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 4. EPR sechs Spektrallinie von Mangan standard. Die paramagnetischen Signal von Mangan zur Kalibrierung der Signal-Schicht. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 5: zyklischer Voltammetrie (CV) von zusammengesetzten NtVTh. Zyklische Voltammograms 15 mm NtVTh in 0,1 M Bu4NBF4/CH3CN und relativ Ferrocen Standard präsentiert den Abbauprozess beteiligt an der Arbeitselektrode. Scan-Rate 0,05 V/s: (ein) im Bereich −1.4 V bis 0,8 V und (b) im Bereich −1.4 V bis 0,9 V. erfolgte Klicken Sie bitte hier, um eine größere Version dieser Figur.

Abbildung 6: ultravioletten, sichtbaren und nahinfraroten (UV-Vis-NIR) Spectroelectrochemistry der Poly (O-iPrThEE) Ableitung. UV-Vis-NIR-Spektren präsentiert die Evolution der Absorptionsbande durch die Erzeugung von Ladungsträgern auf Polymerstruktur. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 7: EPR-Spectroelectrochemical-Analyse der kurz Ableitung. (a) Struktur der s- kurz Ableitung; (b) EPR-Spektren registriert bei elektrochemische Reduktion von s- kurz-Derivat (schwarze Linie-experimentelle und rote Linie simulierte Spektrum). Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 8: EPR Spectroelectrochemical Polaron Signal des konjugierten Polymeren. Elektron-PARAMAGNETISCHE Resonanz (EPR) Spektren registriert während der erste Schritt der Oxidation von konjugierten Polymeren (EPR Spektren Polaron-Arten). Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 9: zyklischer Voltammetrie (CV) von Ferrocen und Decamethylferrocene. Vergleich der zwei elektrochemische Normen als reine sowie Mischung zeigt die Verschiebung des Potenzials. Bitte klicken Sie hier für eine größere Version dieser Figur.
Discussion
Elektrochemische und Spectroelectrochemical Techniken sind keine Grenzen gesetzt; Man kann solid-State und flüssigen Lösungen in einer Vielzahl von Temperatur und anderen Bedingungen mit diesen Techniken analysieren. Das wichtigste in all diesen Fällen ist, dass Verbindungen/Materialien unter der angewandten Potenzial, Replizieren von realen Bedingungen für organische Elektronikgeräte arbeiten analysiert werden. Der einzige Unterschied ist, dass in der Elektrochemie, die Bildung von Ladungsträgern, beobachtet.
Die hier vorgestellten Methoden zeigen den Nutzen der Analyse der geladenen Träger erzeugt in organischen Verbindungen, die mit ihrer Anwendbarkeit in der organischen Elektronik korrelieren. Darüber hinaus die elektrochemischen und Spectroelectrochemical Techniken sind billiger und weniger anspruchsvoll als die typischen Methoden kostenlos Träger Analyse verwendet, aber es gibt einige wichtige Schritte und Änderungen des Protokolls, die je nach Bedarf sind die erzielten Ergebnisse.
Während elektrochemische Charakterisierung beginnen Sie immer mit einer bestimmten Konzentration. Vergleicht man eine Reihe von Verbindungen, dann müssen alle Materialien die gleiche molare Konzentration. Das beste ist, mit zu beginnen 1 mM Konzentration und 50 mV/s Abtastrate, wie im Protokoll in dieser Studie angegeben, aber es ist gut zu wissen, die Konzentration der Probe auf das beobachtete elektrochemischen Verhalten. Immer versuchen Sie, mindestens drei Scans zu messen. Die ersten beiden Scans sind in der Regel anders, weil die Startbedingungen (Gleichgewicht) unterschiedlich sind. Die zweite und die dritte Scans sollten übereinstimmen. Wenn die zweite und dritte Scans identisch sind, dann gibt es wahrscheinlich keine Nebenreaktionen beobachtet in diesem System (Abbildung 2a). In einer Oxidation erscheint ein neuer Peak auf einem niedrigeren Potential zeigt, dass das leitfähige Material auf den WE18,19,24,25,29,30 hinterlegt wurde , 31 , 32. wenn die Höhe der unteren Spitze in aufeinander folgenden Scans erhöht, dann wahrscheinlich konjugierten Polymeren hinterlegten18,19,war24,25,29 , 30 , 31 , 32. wenn die Ströme in aufeinander folgenden Scans zu verringern, dann nichtleitendem Produkt der Abbau auf die Elektrode abgelagert wurde. Wenn eine sehr kleine Spitze vor den wichtigsten Oxidation oder Reduktion Peak (vor allem für Polymere) beobachtet wird, dann ist dies wahrscheinlich kostenlos-Trapping Prozess19,23,31,34. Wenn ein sehr scharfer dedoping Höhepunkt der Oxidation oder Reduktion beobachtet wird, ist dies wahrscheinlich durch die Zersetzung von kristallinen Strukturen auf eine Elektrode, gebildet durch den Electrocrystallization-Prozess während der Oxidation35verursacht.
Überprüfen Sie immer das Verhalten der Testverbindung vor, während und nach der Redox-Gipfel. Es bedeutet, dass mindestens drei CV Scans registriert werden soll: mit oberen (in dem Fall Oxidation) oder untere Scheitelpunkt Potenzial höher oder niedriger, bzw. das Potenzial der Peak Maximum, mit oberen oder unteren Scheitelpunkt Potenzial legen Sie dann auf genau am Peakmaximum und mit Scheitelpunkt Potenziale höher (Oxidation) und unteren (Reduktion) als Potenzial der maximalen Höhepunkt. Der beobachtete Prozess variieren und manchmal zwei Prozesse können theoretisch unter einem Höhepunkt beobachtet werden. Vergleichen Sie immer die gesammelten zyklische Voltammograms des Elektrolyten (Schritt 2,6), Ferrocen (Schritt 2,9), die Verbindung (Schritt 2.13) und Ferrocen mit Compound (Schritt 2.19); Es gibt mehrere Probleme berücksichtigt werden.
Vergleichen Sie immer die CV-Signale von der Elektrolyt und die Testverbindung, wenn keine Signale aus dem Elektrolyten auf die zyklischen Voltammogram der gemessenen Verbindung sichtbar ist, dann das Elektrolyt muss geändert werden, da seine elektrochemische Fenster zu niedrig ist oder die Elektrolyt ist verschmutzt. Wenn das Signal (Redox-Paar) von Ferrocen (Schritt 2,9) und Ferrocen mit Compound (Schritt 2.19) an der gleichen Position sind, ist dann alles richtig durchgeführt. Wenn die Gipfel zwischen einander verschoben werden, dann überprüfen Sie das Rück und wiederholen Sie die Messung. Wenn das Signal (Oxidation, Reduktion oder Redox-paar potentielle) der Testverbindung mit zusätzlichen Ferrocen (Schritt 2.19) auf ein höheres Potential als das reine compound (Schritt 2.13), dann überlegen die Werte (Oxidation, Reduktion oder Redox-paar potentielle) aus die zyklischen Voltammogram der reinen Substanz. Die Verschiebung wird durch den höheren Betrag von Ferrocen in der Lösung verursacht. Wenn zwei Oxidationsprozesse eingehalten werden, kann der erste Prozess (Oxidation oder Reduktion) ist immer auf die WE die aktive Oberfläche auswirken; Dadurch kann eine Zunahme in der Oxidationspotential des zweiten Prozesses (Abbildung 9).
Disclosures
Die Autoren haben nichts preisgeben.
Acknowledgments
Die Autoren dankbar anerkennen die finanzielle Unterstützung der "Excilight" Projekt "Donor-Akzeptor Light Emitting Exciplexes als Material für die Easy-Schneider Hocheffiziente OLED-Lightning" (H2020-MSCA-ITN-2015/674990) finanziert durch Marie Skłodowska-Curie Aktionen innerhalb des Rahmenprogramms für Forschung und Innovation "Horizon 2020".
Materials
| Name | Company | Catalog Number | Comments |
| Potentiostat | Metrohm | Autolab PGSTAT100 | |
| EPR | JEOL | JES-FA200 | |
| UV-Vis detector | Oceanoptics | QE6500 | |
| NIR detector | Oceanoptics | NIRQuest | |
| Dichloromethane (DCM) | Sigma-Aldrich | 106048 | |
| Tetrabutylammonium tetrafluoroborate (Bu4NBF4) | Sigma-Aldrich | 86896 | |
| 2-propanol, 99.9% | Sigma-Aldrich | 675431 | |
| Acetone, 99.9% | Sigma-Aldrich | 439126 | |
| Ultrasonic Bath | Elma | S30H | |
| Tetrahydrofuran >99.9% | Sigma-Aldrich | 401757 | |
| ferrocene >98% | Sigma-Aldrich | F408 | |
| decamethylferrocene >97% | Sigma-Aldrich | 378542 |
References
- O'Brien, D. F., Baldo, M. A., Thompson, M. E., Forrest, S. R. Improved energy transfer in electrophosphorescent devices. Applied Physics Letters. 74, 442-444 (1999).
- Chin, B. D., et al. Effects of interlayers on phosphorescent blue organic light-emitting diodes. Applied Physics Letters. 86, 133505-133507 (2005).
- Cardon, C. M., Li, W., Kaifer, A. E., Stockdale, D., Bazan, G. C. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bang, A. -M., et al. A novel random terpolymer for high-efficiency bulk-heterojunction polymer solar cells. RSC Advances. 7, 1975-1980 (2017).
- Tybrandt, K., Kollipara, S. B., Berggren, M. Organic electrochemical transistors for signal amplification in fast scan cyclic voltammetry. Sensors and Actuators B: Chemical. 195, 651-656 (2014).
- Schaur, S., Stadler, P., Meana-Esteban, B., Neugebauer, H., Sariciftci, N. S. Electrochemical doping for lowering contact barriers in organic field effect transistors. Organic Electronics. 13, 1296-1301 (2012).
- Mullen, K., Scherf, U. Organic Light Emitting Devices: Synthesis, Properties and Applications. , Wiley-VCH. Weinheim. (2006).
- Monk, P. M. S., Mortimer, R. J., Rosseinsky, D. R. Electrochromism and Electrochromic Devices. , Cambridge University Press. Cambridge. (2007).
- Skotheim, T. A., Elsenbaumer, R. L., Reynolds, J. R. Handbook of Conducting Polymers. , Marcel Dekker. New York. (1998).
- Data, P., et al. Kesterite Inorganic-Organic Heterojunction for Solution Processable Solar Cells. Electrochimica Acta. 201, 78-85 (2016).
- Data, P., Pander, P., Okazaki, M., Takeda, Y., Minakata, S., Monkman, A. P. Dibenzo[a,j]phenazine-Cored Donor-Acceptor-Donor Compounds as Green-to-Red/NIR Thermally Activated Delayed Fluorescence Organic Light Emitters. Angewandte Chemie International Edition. 55 (19), 5739-5744 (2016).
- Jankus, V., et al. Highly Efficient TADF OLEDs: How the Emitter-Host Interaction Controls Both the Excited State Species and Electrical Properties of the Devices to Achieve Near 100% Triplet Harvesting and High Efficiency. Advanced Functional Materials. 24 (39), 6178-6186 (2014).
- Data, P., et al. Exciplex Enhancement as a Tool to Increase OLED Device Efficiency. Journal of Physical Chemistry C. 120 (4), 2070-2078 (2016).
- Dias, F. B., et al. The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Advanced Science. , (2016).
- Okazaki, M., et al. Thermally Activated Delayed Fluorescent Phenothiazine-Dibenzo[a,j]phenazine-Phenothiazine Triads Exhibiting Tricolor-Changing Mechanochromic Luminescence. Chemical Science. 8, 2677-2686 (2017).
- Data, P., et al. Efficient p-phenylene based OLEDs with mixed interfacial exciplex emission. Electrochimica Acta. 182, 524-528 (2015).
- Goushi, K., Yoshida, K., Sato, K., Adachi, C. Organic light-emitting diodes employing efficient reverse intersystem crossing for triplet-to-singlet state conversion. Nature Photonics. 6, 253-258 (2012).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of heteroaryl group on electrochemical and spectroscopic properties of conjugated polymers. Electrochimica Acta. 83, 271-282 (2012).
- Data, P., Motyka, M., Lapkowski, M., Suwinski, J., Monkman, A. Spectroelectrochemical Analysis of Charge Carries as a Way of Improving Poly(p-phenylene) Based Electrochromic Windows. Journal of Physical Chemistry C. 119, 20188-20200 (2015).
- Enengl, C., et al. Explaining the Cyclic Voltammetry of a Poly(1,4-phenylene-ethynylene)-alt-poly(1,4-phenylene-vinylene) Copolymer upon Oxidation by using Spectroscopic Techniques. ChemPhysChem. 18, 93-100 (2017).
- Gudeika, D., et al. Hydrazone containing electron-accepting and electron-donaiting moieties. Dyes Pigments. 91, 13-19 (2011).
- Swist, A., Cabaj, J., Soloducho, J., Data, P., Lapkowski, M. Novel acridone-based branched blocks as highly fluorescent materials. Synthetic Metals. 180, 1-8 (2013).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of alkyl chain on electrochemical and spectroscopic properties of polyselenophenes. Electrochimica Acta. 87, 438-449 (2013).
- Laba, K., et al. Electrochemically induced synthesis of poly(2,6-carbazole). Macromolecular Rapid Communications. 36, 1749-1755 (2015).
- Pluczyk, S., et al. Unusual Electrochemical Properties of the Electropolymerized Thin Layer Based on a s-Tetrazine-Triphenylamine Monomer. Journal of Physical Chemistry C. 120, 4382-4391 (2016).
- Blacha-Grzechnik, A., Turczyn, R., Burek, M., Zak, J. In situ Raman spectroscopic studies on potential-induced structural changes in polyaniline thin films synthesized via surface-initiated electropolymerization on covalently modified gold surface. Vibrational Spectroscopy. 71, 30-36 (2014).
- Laba, K., et al. Diquinoline derivatives as materials for potential optoelectronic applications. Journal of Physical Chemistry C. 119, 13129-13137 (2015).
- Enengl, S., et al. Spectroscopic characterization of charge carriers of the organic semiconductor quinacridone compared with pentacene during redox reactions. Journal of Materials Chemistry C. 4, 10265-10278 (2016).
- Data, P., et al. Electrochemically Induced Synthesis of Triphenylamine-based Polyhydrazones. Electrochimica Acta. 230, 10-21 (2017).
- Brzeczek, A., et al. Synthesis and properties of 1,3,5-tricarbazolylbenzenes with star-shaped architecture. Dyes Pigments. 113, 640-648 (2015).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Electrochemistry and spectroelectrochemistry of a novel selenophene-based monomer. Electrochimica Acta. 59, 567-572 (2012).
- Data, P., et al. Electrochemical and spectroelectrochemical comparison of alternated monomers and their copolymers based on carbazole and thiophene derivatives. Electrochimica Acta. 122, 118-129 (2014).
- Data, P., et al. Evidence for Solid State Electrochemical Degradation Within a Small Molecule OLED. Electrochimica Acta. 184, 86-93 (2015).
- Data, P., et al. Unusual properties of electropolymerized 2,7- and 3,6- carbazole derivatives. Electrochimica Acta. 1282, 430-438 (2014).
- Etherington, M. K., et al. Regio- and conformational isomerization critical to design of efficient thermally-activated delayed fluorescence emitters. Nature Communications. 8, 14987 (2017).
- Trasatti, S. The absolute electrode potential: an explanatory note. Pure and Applied Chemistry. 58, 955-966 (1986).
- Cardona, C. M., et al. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bredas, J. -L. Mind the gap! Mater Horiz. 1, 17-19 (2014).
- Gerson, F., Huber, W. Electron Spin Resonance Spectroscopy of Organic Radicals. , (2003).
- Brustolon, M., Giamello, E. Electron Paramagnetic Resonance. A Practitioner's Toolkit. , John Wiley & Sons Inc. New Jersey. (2009).
- Weil, J., Bolton, J. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. , John Wiley & Sons, Inc. (2007).
- Rieger, P. H. Electron Spin Resonance. Analysis and Interpretation. , The Royal Society of Chemistry. (2007).
- Roznyatovskiy, V. V., Gardner, D. M., Eaton, S. W., Wasielewski, M. R. Radical Anions of Trifluoromethylated Perylene and Naphthalene Imide and Diimide Electron Acceptors. Organic Letters. 16, 16-19 (2013).
- Pluczyk, S., Kuznik, W., Lapkowski, M., Reghu, R. R., Grazulevicius, J. V. The Effect of the Linking Topology on the Electrochemical and Spectroelectrochemical Properties of Carbazolyl Substituted Perylene Bisimides. Electrochimica Acta. 135, 487-494 (2014).
- Ledwon, P., et al. A Novel Donor-acceptor Carbazole and Benzothiadiazole Material for Deep Red and Infrared Emitting Applications. Journal of Materials Chemistry C. 4, 2219-2227 (2016).
- Heinze, J., Frontana-Uribe, B. A., Ludwigs, S. Electrochemistry of Conducting Polymers-Persistent Models and New Concepts. Chemical Reviews. 110, 4724-4771 (2010).
- Kurowska, A., Kostyuchenko, A. S., Zassowski, P., Skorka, L., Yurpalov, V. L., Fisyuk, A. S., Pron, A., Domagala, W. Symmetrically Disubstituted Bithiophene Derivatives of Properties. Journal of Physical Chemistry C. 118, 25176-25189 (2014).
- Zotti, G., Schiavon, G., Zecchin, S., Morin, J. F., Leclerc, M. Electrochemical, Conductive, and Magnetic Properties of 2,7-Carbazole-Based Conjugated Polymers. Macromolecules. 35, 2122-2128 (2002).
- Kaim, W., Fiedler, J. Spectroelectrochemistry: The Best of Two Worlds. Chemical Society Reviews. 38, 3373-3382 (2009).
- Duling, D. R. Simulation of Multiple Isotropic Spin-Trap EPR Spectra. Journal of Magnetic Resonance, Series B. 104, 105-110 (1994).
- Benson, C. R., et al. Multiplying the Electron Storage Capacity of a Bis-Tetrazine Pincer Ligand. Dalton Transactions. 43, 6513-6524 (2014).
- Nobuyasu, R. S., et al. Rational Design of TADF Polymers Using a Donor-Acceptor Monomer with Enhanced TADF Effi ciency Induced by the Energy Alignment of Charge Transfer and Local Triplet Excited States. Advanced Optical Materials. 4, 597-607 (2016).
