

Summary
Dans cet article, nous décrivons électrochimique, résonance paramagnétique électronique et les méthodes de spectroélectrochimiques ultraviolet-visible et proche infrarouge pour l’analyse des composés organiques pour application en électronique organique.
Abstract
Voltamétrie cyclique (CV) est une technique utilisée dans l’analyse de composés organiques. Lorsque cette technique est combinée avec la résonance paramagnétique électronique (RPE) ou ultraviolet-visible et proche infrarouge spectroscopies (UV-Vis-NIR), nous obtenons des informations utiles telles que le type de, énergies de bande interdite, potentiel d’ionisation, affinité électronique porteurs de charge et les informations de dégradation qui peuvent être utilisées pour synthétiser des dispositifs Electroniques organiques stables. Dans cette étude, nous présentons électrochimiques et spectroélectrochimiques méthodes pour analyser les processus qui se produisent dans les couches actives d’un dispositif organique ainsi que les porteurs de charge générée.
Introduction
Dans le monde entier, chercheurs cherchent continuellement de nouvelles matières organiques qui peuvent être utilisés dans l’électronique organique performant souhaitable ou stabilité, ce qui diminue en raison d’une utilisation prolongée. Dans le cas de dispositifs organiques, il est important de comprendre le comportement du porteur de la charge à bien connaître les règles du comportement du dispositif de conduite. Analyse de l’effet de la molecular structure sur la génération de porteur de la charge et la dynamique et le maintien de l’équilibre injecté des porteurs de charge, les deux résultats positifs (trous) et négatives (électrons), est cruciale pour améliorer l’efficacité et la stabilité des dispositifs organiques. Cela garantit la recombinaison efficace de ces charges individuelles et par conséquent améliore considérablement l’efficacité de la photoluminescence de l’électroluminescent organique à diodes (OLED)1,2. Pour le photovoltaïque organique (pollinisation)3,4 , ainsi que le champ organique effet transistors (OFETs)5,6, il est nécessaire de disposer de matériaux avec mobilité de porteur de charge élevée. Outre l’analyse des porteurs de charge, plusieurs paramètres importants de matériaux électroactifs organique aident à prédire où le matériau pourrait être utilisé : potentiel d’ionisation (pi), les niveaux d’énergie électronique affinité (EA) et bande interdite entre eux7 ,8,9,10.
Dans ce travail, nous présentons une méthode pour la mesure efficace de voltampérométrie cyclique (CV) qui peut être utilisé dans l’analyse de tous les types de matériaux électroactifs. Cette technique fournit des informations sur les propriétés redox, le mécanisme de dopage/dedoping, la stabilité, la conversion et le stockage d’énergie, etc.. Il permet également l’estimation de l’énergie des électrons affinité et ionisation des composés test une manière beaucoup moins cher et plus rapide par rapport aux autres méthodes sous vide élevés. Les paramètres susmentionnés sont corrélés avec les niveaux d’énergie de l’orbitale moléculaire occupée plus élevé (HOMO) et plus bas inoccupé orbitale moléculaire (LUMO).
La méthode présentée dans cet article peut être utilisée pour analyser tous les types de composés conjugués tels que ceux avec les électrons π délocalisés dans leurs structures. Composés conjugués peuvent être petites molécules ayant des grandes chaînes polymériques. Petites molécules peuvent également être des monomères ; au cours de la réaction initiale des monomères (photochimiques, électrochimiques ou chimiques) peuvent former des polymères. En application de l’OLED, les valeurs de niveau d’énergie sont nécessaires pour permettre l’utilisation de l’hôte correct pour l’émetteur dans un thermiquement activées retardé système chef invité par fluorescence (TADF) ou pour décider avec qui des composés la couche de donneur-accepteur exciplexe pourrait être formé et quelles épaisseurs supplémentaires (electron transport couche (ETL), trou transport couche (HTL), couche de blocage électron (EBL) et couche de blocage trou (HBL)) sera nécessaires pour synthétiser stable chargé efficacement équilibrés dispositifs OLED11 , 12 , 13 , 14 , 15 , 16 , 17. autres mesures électrochimiques permettent l’étude des réactions secondaires possibles au cours du processus de dégradation de la couche active et la formation de basse mobile porteurs de charge (bipolarons)18,19 ,20,21,22.
Couplage électrochimique et spectroélectrochimiques méthodes permettant la détermination facile, précise et fiable du degré d’oxydation ou réduction de composés conjugués et leur dégradation potentielle, qui est cruciale pour la stabilité23 , 24 , 25 , 26 , 27 , 28. ultraviolet-visible et proche infrarouge spectroscopie (UV-Vis-NIR) couplée à l’électrochimie peut caractériser les propriétés chromatiques fondamentales de tous les nouveaux composés conjugués, tels que le changement de la bande d’absorption au cours de dopage 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30.
Dans une étude concernant le mécanisme de dopage, il est important de définir le type de porteurs de charge. Dans ce processus, deux classes de quasi-particules chargées participent, un avec spin sans compensation (polarons) et le second étant diamagnétique (bipolarons) ; par spectroscopie de résonance paramagnétique (EPR) fournit une aide indispensable, ce qui permet directement d’observer et de suivre les changements dans les populations de paramagnétique polarons29,30,31,32 . Dans les petites molécules, il est difficile de forme bipolarons, mais ces molécules peuvent être conjugués assez et ont des propriétés inductrices de bipolaron ; Il est important de vérifier si et au cours de laquelle les potentiels polarons et bipolarons se forment dans la structure. Bipolarons sont au moins un ordre inférieur en mobilité à celui de polarons ; par conséquent, si bipolarons se forment dans les dispositifs de travail, alors il pourrait conduire à un ratio déséquilibré les porteurs de charge, qui se traduirait par un courant élevé et une surchauffe de l’appareil OLED ou peut-être les centres de dégradation33.
La méthode de mesure proposé dans cette étude est bon marché et plus rapide et permet la détermination des paramètres du dispositif plus précieux pour un grand nombre de matériaux électroactifs sans besoin de dispositifs spéciaux basés sur nouvellement synthétisé matériaux pour vérifier ses performances. En appliquant l’électrochimie et spectroélectrochimie, il est possible de choisir un matériau qui est vraiment prometteur parmi la centaine de nouveaux matériaux. En outre, il est possible d’obtenir des informations détaillées sur les processus de dopage et leurs effets sur la structure chimique de l’essai conjugué systèmes électrochimiques et spectroélectrochimiques méthodes, ce qui permet de construire plus dispositifs de l’électronique organique efficace.
Protocol
1. préparation de l’expérience
- Préparer 25 mL de solution d’électrolyte 0,1 M.
Remarque : Selon les composés incriminés, utiliser différents électrolytes comme le mélange de sel organique et solvant : le plus commun de sels utilisés dans l’analyse de molécules organiques sont hexafluorophosphate de tétrabutylammonium (Bu4FNP6 ) ou le tétrafluoroborate de tétrabutylammonium (Bu4NBF4) ; le solvant couramment utilisé dans l’analyse sont dichlorométhane, l’acétonitrile ou le tétrahydrofuranne.- Choisissez l’électrolyte à base de solvant sur la solubilité des composés test. Il devrait être une solution au cours de l’enquête, mais être dans un état solide lorsque le matériau est utilisé dans la fabrication de l’appareil, comme une couche mince déposée sur la surface de l’électrode de travail.
- Nettoyer les électrodes pour être utilisé dans l' expérience29.
- Pour les mesures électrochimiques, utiliser une électrode de platine disque diamètre 1 mm sous l’électrode de travail (WE), une platine bobine ou les fils comme électrode auxiliaire (AE) et une électrode d’argent/chlorure d’argent (Ag/AgCl) comme l’électrode de référence (RE).
- Pour des mesures spectroélectrochimiques EPR, utiliser fil de platine comme le WE, une bobine de platine ou les fils comme l’AE et Ag/AgCl comme le réemploi.
- Pour les mesures spectroélectrochimiques UV-Vis-NIR, utiliser un quartz indium tin oxyde (ITO) ou l’oxyde d’étain dopé au fluor (FTO) comme le WE, un fil de platine comme l’AE et une électrode Ag/AgCl comme le réemploi.
- Polir l’électrode de platine disque en frottant du côté incliné de l’électrode (la zone de travail de l’électrode de platine) sur un plateau de polissage recouvert de 1 µm de boue alumine pendant 3 min.
- Rincer l’électrode à l’eau déionisée pour enlever tous les boues de l’électrode et nettoyer dans un bain à ultrasons (320 W, 37 kHz) avec de l’eau désionisée pendant 15 min.
- Rincer l’électrode à l’aide d’une seringue de 1 mL avec de l’isopropanol (3 x 1 mL), puis avec de l’acétone (3 x 1 mL).
- Utilisez une serviette en papier pour enlever tous les résidus solides et sécher à l’air pendant 3 min.
- Nettoyer les électrodes ITO et la FTO à l’eau désionisée. Placez-les dans un bain à ultrasons (320 W, 37 kHz) dans l’acétone pendant 15 min, puis dans l’alcool isopropylique pour 15 min suivantes.
- Brûler les électrodes de platine, les fils et bobines à l’aide d’un chalumeau à gaz haute température (> 1000 ° C) pour 1 min. attention ! Utiliser les pinces pour tenir les électrodes. Attendre 5 min après la combustion et avant d’utiliser les électrodes.
- Nettoyer les cellules électrochimiques et spectroélectrochimiques (Figure 1) avec de l’eau, puis avec de l’acétone. Utilisez une serviette en papier pour enlever tous les résidus solides. Nettoyer tous les autres éléments (pièces PTFE env.) avec de l’acétone et sécher à l’air avant de l’utiliser.
2. analyse de CV
- Allumez le potentiostat et l’ordinateur.
- Remplir une cellule électrochimique avec 1,5 mL de la solution d’électrolyte à analyser.
- Mettre tous les trois électrodes (WE, AE et RE) dans une cellule (cap de la cellule est équipé du porte-électrode PTFE, alors mettez des électrodes dans les trous de cet titulaire) et connectez-le à un potentiostat. Garder le WE et RE comme rapprochées que possible (Figure 1). Suivre les informations concernant la connexion de chaque fil de la potentiostat à son électrode respectif.
- Si nécessaire (par exemple lors de l’analyse de réduction), mettre le tuyau argon (ou azote) (à travers un trou supplémentaire dans le porte-électrode) et bouillir la solution pendant au moins 5 min. Après cela, déplacez le tuyau argon (ou azote) au-dessus du niveau de la solution et maintenir le flux de gaz en cours d’exécution pour la mesure.
Remarque : La cellule semble fermée mais ne peut pas être complètement étanches ; par conséquent, une méthode pour enlever la superflu quantité de gaz de la cellule doit être disponible (e.g., par un petit trou supplémentaire dans le porte-électrode). La pression dépend du tuyau utilisé ; Réglez la pression élevée tel qu’il est nécessaire de régler le débit de gaz lente. Si un solvant très volatil est utilisé dans l’expérience (p. ex., dichlorométhane) ou l’expérience prend beaucoup de temps (plus de 30 min), puis utilisez la bouteille de Drechsel pour saturer avec le solvant utilisé dans la mesure du gaz argon (ou azote). - Lancez le logiciel potentiostat, choisissez la procédure de CV et utilisez les paramètres suivants : commencer à potentiel de 0,00 V, potentiel sommet supérieur de 2,0 V, faible potentiel de vertex de 0,00 V (si l’enquête sur le processus de réduction, puis un faible potentiel de vertex de −2, 5 V), arrêter le potentiel de 0,00 V, nombre d’arrêt croisement de 6 et scan de 0,05 V/s.
- Dans la section données ASCII à l’exportation, définir un nom de fichier et choisissez un dossier pour enregistrer les données dans et appuyez sur le bouton Démarrer (diminuer ou augmenter le potentiel sommet inférieur et supérieur pour pouvoir afficher la fenêtre électrochimique ou l’échantillon activité électrochimique)
- Si les sommets sont visibles dans la gamme positive du potentiel, puis répétez la procédure de nettoyage. Si il y a un pic dans la gamme négatif potentiels, puis mettre le tuyau argon (ou azote) dans la solution et bouillir pendant un 5 min supplémentaire.
- Ajouter une goutte du ferrocène 1 mM (dans le solvant utilisé pour préparer l’électrolyte) de solution électrolytique à l’aide d’une seringue.
- Définir le potentiel de départ à 0,00 V, le vertex haut potentiel à 0,85 V, le vertex plus faible potentiel à 0,00 V, le potentiel d’arrêt à 0,00 V, le nombre d’arrêt passage à 10 et la vitesse de balayage à 0,05 V/s. Dans la section données ASCII à l’exportation, changer le nom de fichier et appuyez sur le bouton Démarrer .
- Après la mesure, terminer en répétant les opérations de nettoyage, tel que mentionné à l’étape 1.2 et 1.3.
- Préparer 4 mL de substance dans l’électrolyte préparé (étape 1.1) 1 mM d’essai.
- Remplir la cellule électrochimique avec la solution d’essai composé (1,5 mL) ; mettre tous les trois électrodes dans une cellule et se connecter à la potentiostat (étape 2,4). Pour étudier le processus de réduction, retirer l’oxygène (étape 2.5).
- Régler la marche potentielle à 0,00 V, le vertex haut potentiel à 0,50 V (ou 0,00 V dans le cas d’investigation des processus de réduction), le vertex plus faible potentiel à 0,00 V (−0.50 V dans le cas d’investigation des processus de réduction), le potentiel d’arrêt à 0,00 V , le nombre d’arrêt passage à 10 et la vitesse de balayage à 0,05 V. Dans la section données ASCII à l’exportation, changer le nom de fichier et appuyez sur le bouton Démarrer .
- Répétez l’étape 2.13 en augmentant le potentiel sommet supérieur (ou en diminuant le potentiel sommet inférieur) par 0,1 V jusqu’au enregistrement PIC (Figure 2 a). Si l’analyse successive est décalée en potentiel (Figure 2 b), puis nettoyer le RE et le laisser dans la solution d’électrolyte. Puis, recommencez la mesure.
- Mesurer l’oxydation et réduction des processus sur le même CV pour la détermination de la propriété intellectuelle et EA : définir le potentiel de départ à 0,00 V, le potentiel sommet supérieur à 1,00 V, le potentiel sommet inférieur à −2.70 V et le potentiel d’arrêt à 0,00 V. choisir le vert supérieur et inférieur ex le potentiel d’une manière qui enregistre toute réduction et oxydation des pics et évite de nouvelles mesures de redox (le cas échéant) (Figure 2 a).
NOTE : Estimer IP et EA en mesurant l’apparition potentielle. Il y a deux façons de calculer ces paramètres de CV, mais est le plus utile de calculer à l’aide de l’apparition potentielle. Parfois, il n’est pas possible d’observer le couple redox réversibles, en particulier pour n et p dopage. Mélange des deux techniques différentes est aussi indésirable qu’il peut fournir des conclusions et des résultats erronés.- Pour mesurer l’apparition potentielle, marquez la ligne tangente au pic de CV (Figure 3). Calculer les valeurs souhaitées de la possibilité d’apparition de la réduction ou le procédé d’oxydation à l’aide d’équations (équations 1 et 2)10,18,19,,du3637 , 38.
 (1)
(1)
où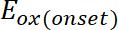 est l’apparition du potentiel d’oxydation calibré avec ferrocène*
est l’apparition du potentiel d’oxydation calibré avec ferrocène*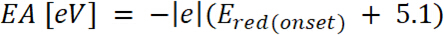 (2)
(2)
où est l’apparition de la réduction du potentielle calibrée avec ferrocène*
est l’apparition de la réduction du potentielle calibrée avec ferrocène*
* Potentiel calibré avec ferrocène signifie que la valeur du potentiel de la mesure est réduite par le potentiel d’oxydation du ferrocène.
- Pour mesurer l’apparition potentielle, marquez la ligne tangente au pic de CV (Figure 3). Calculer les valeurs souhaitées de la possibilité d’apparition de la réduction ou le procédé d’oxydation à l’aide d’équations (équations 1 et 2)10,18,19,,du3637 , 38.
- Retirer les électrodes et videz la solution d’essai dans le conteneur de déchets approprié.
- Répétez la procédure de nettoyage (étapes 1.2 et 1.3).
- Répétez les étapes 2,9 – 2.12 avec 0,25, 0,10 et 0,20 vitesses de balayage V/s.
- Ajouter une goutte du ferrocène 1 mM (dans le solvant utilisé pour préparer la solution d’électrolyte) dans la solution d’essai à l’aide d’une seringue. Définir le potentiel de départ à 0,00 V, le vertex haut potentiel à 0,60 V, le potentiel de vertex inférieur à 0,00, le potentiel d’arrêt à 0,00 V, le nombre d’arrêt passage à 6 et la vitesse de balayage à 0,05 V. Appuyer sur le bouton Démarrer . Une fois la mesure terminée, modifiez le vertex haut potentiel en une valeur qui enregistre le couple ferrocène avec le premier pic d’oxydation de la substance d’essai.
- Retirer les électrodes et videz la solution d’essai dans le conteneur de déchets approprié.
- Si l’électropolymérisation se produit pendant l’oxydation électrochimique, Lavez soigneusement le WE avec la solution d’électrolyte à l’aide de la seringue (3 ´ 0,5 mL). Nettoyez le RE, AE et cellule électrochimique à l’aide de la procédure standard (étapes 1,2-1,3).
- Afin d’étudier les produits électrochimiques déposés sur nous, passez à l’étape 2.23. Afin d’étudier une solution, passez à l’étape de 1.2.1 et recommencer à l’étape 2.3.
- Remplir la cellule électrochimique avec une solution d’électrolyte. Ensuite, mettre tous les trois électrodes dans une cellule et connectez-les à un potentiostat (étape 2,4). Pour étudier le processus de réduction, deoxidate la solution (point 2.5).
- Répétez les étapes 2.10, 2.12 et alors l’étape 2.16.
 (1)
(1)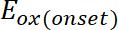 est l’apparition du potentiel d’oxydation calibré avec ferrocène*
est l’apparition du potentiel d’oxydation calibré avec ferrocène*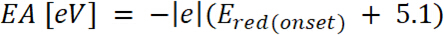 (2)
(2) est l’apparition de la réduction du potentielle calibrée avec ferrocène*
est l’apparition de la réduction du potentielle calibrée avec ferrocène*3. UV-Vis-NIR spectroélectrochimiques analyse
- Préparation de la mesure
- Allumez le spectromètre et le potentiostat.
- Remplir la cellule spectroélectrochimiques avec 0,5 mL de solution d’électrolyte (point 1.1).
- Appliquez le joint PTFE sur un côté de la cellule ; Ensuite, mettre l’électrode ITO à la cellule de cette manière d’avoir un côté conducteur de l’électrode toucher le joint PTFE. Mettez le reste des pièces PTFE comme illustré à la Figure 1. Mettre le RE et les AE dans le porte-électrode PTFE et couvrir la partie supérieure de l’électrode ITO avec le clinquant de cuivre tel qu’illustré à la Figure 1 pour une meilleure conductivité.
- Mettre la cellule Assemblée dans la porte du spectromètre et connecter toutes les électrodes à un potentiostat. Suivre les informations concernant la connexion de chaque fil de la potentiostat à son électrode respectif.
- Lancez le logiciel potentiostat et spectromètre.
- Dans le logiciel de spectromètre, choisissez fichier | Nouveau | Absorbance mesure.
- Choisissez l’un des détecteurs cotées.
- Assurez-vous que le bouton sur les détecteurs est en position « ouverte ». Ensuite, cliquez sur l’ampoule d’éclairage incandescent ; Fermez le détecteur (déplacer le bouton position fermée) et cliquez ensuite sur l’ampoule d’éclairage sombre.
- Sélectionnez le deuxième détecteur et répétez l’étape 3.1.7.
- Reconnecter les électrodes entre le potentiostat et retirer les électrodes et autres éléments de la cellule spectroélectrochimiques.
- Répéter les procédures de nettoyage (étapes 1.2 et 1.3).
- Remplir la cellule spectroélectrochimiques avec un dilué (1´10−5 M) solution de la substance dans la solution d’électrolyte si Testez la petite molécule d’essai. Remplir la cellule spectroélectrochimiques avec l’électrolyte si les tests de la couche (oligomère) polymère déposée sur l’électrode ITO.
- Assembler la cellule spectroélectrochimiques (étape 3.1.3).
- Cliquez sur Enregistrer.
- Sélectionnez un des détecteurs cotées.
- Définir l’enregistrement emplacement et le nom du fichier (inclut le détecteur sélectionné).
- Sélectionnez le deuxième détecteur et répétez l’étape 3.1.15.
- Potentiostatique analyse18.
- Appliquer un potentiel neutre (c.-à-d., 0,00 V). Cette partie du spectre sera les départ de spectres. Augmenter le potentiel de 0,1 V et attendez env. 10 s jusqu'à ce que le processus se stabilise et enregistre les spectres d’absorption (pas 3.1.13–3.1.16). Poursuivre la mesure à la première modification dans les spectres UV-Vis-NIR ; attendre 10 s et puis enregistrez les spectres (mesures 3.1.13–3.1.16).
- Potentiels d’augmentation de 0,05 V ; attendre 10 s et enregistrer (pas 3.1.13–3.1.16).
- 3.2.2 le répète jusqu'à réaliser le potentiel du potentiel d’oxydation de première ou deuxième (ces potentiels sont connus de l’évaluation de CV) ; puis, inverser le potentiel et revenir au potentiel de départ.
- Lorsque les mesures sont terminées, mesurer le CV de la solution d’essai. Ajouter 1 mM ferrocène (10 μL) et mesurer de nouveau le CV. La valeur du ferrocène aidera à estimer le potentiel du processus et d’unifier les données.
- Cette analyse fournit des informations concernant l’optique bande interdite, la longueur d’onde maximale des États non dopés et dopés et points isobestiques du processus électrochimique. Calculer les bande optiques-lacunes de valeurs de l’apparition des bandes de π-π* sur les spectres UV-Vis et en utilisant l’équation 318,19,23,24.
 (3)
(3)
où h est la constante de Planck, λapparition est composé d’absorption apparition et c est la vitesse de la lumière dans le vide. - Analyse de Potentistatic Time-Resolved19,32,33
- Préparer la mesure semblable à la méthode décrite dans 3.1.1–3.1.16 et mettez le tuyau argon (ou azote) (à travers un trou supplémentaire dans le porte-électrode) et bouillonne solution au moins 5 min avant la prise de mesure. Après cela, déplacez le tuyau argon au-dessus du niveau de la solution et maintenir le flux de gaz en cours d’exécution pour la mesure.
- Exécuter le logiciel potentiostat, choisissez la procédure chronoampérométrie avec les paramètres suivants : potentiel commencer potentiel à 0,00 V, un potentiel supérieur à 1,0 V, un potentiel inférieur à −1, 00 V, durée 5 s, intervalle de temps comme 0.010 s, nombre de répétitions à 10 et retard temps de 10 s. Dans la section données ASCII à l’exportation, définir un nom de fichier et choisissez un dossier pour enregistrer les données.
- Dans le logiciel de spectromètre, choisissez fichier | Nouveau | Acquisition rapides. Une nouvelle fenêtre apparaîtra ; Configurez les paramètres suivants : temps d’intégration — 10 ms, Scans à moyenne — 1, wagon couvert Wirth — 0, nombre de Scans — 10000. Ne cliquez pas sur le bouton OK .
- Dans le logiciel potentiostat, appuyez sur le bouton Démarrer . Lorsque le compte à rebours tombe à 1 s, appuyez sur le bouton aller dans le logiciel du spectromètre.
- Notez qu’il n’est pas possible de voir les résultats d’absorption jusqu'à ce que le processus se termine.
- Analyser les données pour fournir des paramètres. Utilisez les informations enregistrées dans le fichier de cette transmission, courant, tension et densité de courant pour fournir les paramètres de fonctionnement :
- Calculer la densité optique (ΔOD), l’absorbance de l’essai composé au cours de dopage (dedoping) avec l’équation 4:
 (4)
(4)
où Tb transmission forme dopé, un état de « bleach » [%] et Tc est la transmittance sous forme non dopé, un état de « couleur » [%]. - Calculer l’efficacité de la Coloration (CE, η), la quantité de couleur électrochromique formé par le montant utilisé gratuitement et que la connexion entre la redevance injecté/éjecté en fonction de la zone de l’électrode (Qd) et la variation de la densité optique (ΔOD) valeur à une longueur d’onde spécifique (λmax) avec l’équation 5. Sa valeur dépend de la longueur d’onde choisie pour l’étude, si la valeur est mesurée à λmax de la bande d’absorption optique de l’état coloré.
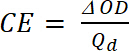 (5)
(5)
où ΔOD est la densité optique [-] et Qd est la densité de charge [C/cm2]. - Calculer le Ratio de contraste (CR), l’intensité mesurée de changement de couleur pendant le dopage électrochimique et généralement caractérisée par λmax de la forme colorée avec équation 6 :
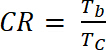 (6)
(6)
où Tb transmission forme dopé, « bleach » État [%] et Tc est la transmittance sous forme non dopé, état « couleur » [%].
- Calculer la densité optique (ΔOD), l’absorbance de l’essai composé au cours de dopage (dedoping) avec l’équation 4:
 (3)
(3) (4)
(4)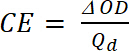 (5)
(5)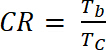 (6)
(6)4. EPR spectroélectrochimiques analyse
- Préparation de la mesure
- Allumez le spectromètre et le potentiostat.
- Remplir la cellule spectroélectrochimiques avec électrolyte (étape 1.1).
- Jeu de la puissance de 1 mW et tune la caisse de résonance pour obtenir un signal de forte, centré dans Q-dip (utilisez l’option autotuned).
- Définissez le Mn (manganèse standard) à 600 et fermez la fenêtre de Q-dip. S’il n’y a aucun marqueur de Mn, puis sautez cette étape et passez à l’étape 4,8.
- La valeur « balayer largeur ± » 2.5´ 10 TM, « mod largeur ± » 1.0´0.1 TM, « amplitude » à 5´100, la « constante de temps » à 0,03 s et « champ Centre » valeur à environ 338 mT et démarrer la mesure.
- Si non Inscrivez six raies spectrales du marqueur de Mn, puis modifiez la valeur de « champ Centre ».
- Lors de l’inscription de six lignes spectrales du marqueur Mn (Figure 4), définissez la valeur de « champ centre » entre la troisième et la quatrième ligne et diminuer la valeur de « balayage largeur ± » à une valeur qui couvre seulement ces deux lignes. Démarrer la mesure à nouveau.
- Allez dans l’option Q-Dip, définissez le Mn à 0, puis fermez la fenêtre de Q-dip.
- Mesurer l’électrolyte pur que le fond et vérifier si il n’y a aucun signal. Si les signaux sont visibles, le signal peut être de la contamination, composés de potentiel faible oxydation, ou de la vitre.
- Nettoyer la cellule spectroélectrochimiques (étape 1.3).
- Enquête de petites molécules (solutions).
- Remplir la cellule spectroélectrochimiques avec un dilué (1´10−5 M) solution du test composé de l’électrolyte ; mettre tous les trois électrodes à travers le porte-électrode à la cellule de cette manière afin que le WE et RE sont à l’intérieur de la spirale AE (Figure 1).
- Mettre le WE près du fond de la cellule et le RE à la face supérieure de l’actif (métal non couvert par PTFE) fait partie du WE (Figure 1). Placer la cellule équipée d’une électrode à l’intérieur de la cavité de l’échantillon du spectromètre et connecter les électrodes avec un potentiostat (suivre les informations concernant la connexion de chaque fil de la potentiostat à son électrode respectif).
- Appliquer le potentiel correspondant au début de la première apogée de l’oxydo-réduction (valeur connue de la mesure de la CV). Si un signal s’affiche, puis l’appareil est mis en place correctement.
- Allez dans l’option Q-Dip, définissez le Mn à 600, fermez la fenêtre de Q-trempette et démarrer la mesure. L’enregistrement du signal avec le signal du marqueur Mn permet le calcul de la g-facteur du signal reçu.
- Allez dans l’option Q-Dip, définissez le Mn à 0, fermez la fenêtre de Q-trempette et attendre ca. 5 min.
- Définissez la valeur de « champ centre » dans le centre du signal, de diminuer la valeur de « balayage largeur ± » à une valeur qui couvre le signal, mais ne coupe pas le début et la fin du signal (quand « sweep largeur ± » est changé, cocher tout signal) et diminution de la valeur de m « OD largeur ± » pour obtenir les spectres bien résolues.
- Si le signal est faible, puis augmentez la valeur de « amplitude » et « time » (temps d’acquisition) de la mesure.
- Pour diminuer le rapport signal sur bruit, définissez l’acquisition de 4, 9 ou 16, selon comment bruyant le signal est. Si le signal est très bruyant, sélectionnez 16.
- Remplir la cellule spectroélectrochimiques avec un dilué (1´10−5 M) solution du test composé de l’électrolyte ; mettre tous les trois électrodes à travers le porte-électrode à la cellule de cette manière afin que le WE et RE sont à l’intérieur de la spirale AE (Figure 1).
- Enquêtes de la couche de polymère déposés sur la surface du WE.
- Remplir la cellule spectroélectrochimiques avec une solution d’électrolyte (point 1.1).
- Placer les électrodes dans la cellule. Veillez à ne pas détruire la couche de polymère sur le WE. Connecter l’électrode avec le potentiostat (étape 4.2.1).
- Appliquer le potentiel neutre (c'est-à-dire 0,00 V) pour s’assurer que le composé est dans un État neutre ; cette partie du spectre sera les départ spectres rpe.
- Accroître le potentiel de 0,10 V. attendre env. 10 s jusqu'à ce que le processus se stabilise et enregistrer les spectres rpe.
- Répétez l’étape 4.3.4 lorsque le signal de l’EPR s’affiche.
- Augmenter le potentiel de 0,05 V. attendre env. 10 s jusqu'à ce que le processus se stabilise et enregistrer les spectres rpe.
- Répétez l’étape 4.3.6 jusqu’au premier ou second potentiel d’oxydation est réalisée et ainsi de suite (les valeurs de ces potentiels sont connus de mesure de la CV) ; puis, inverser le potentiel et revenir au départ potentiel.
- Appliquer le potentiel au cours de laquelle l’EPR signal est apparu lors du cycle précédent (4.3.5), allez dans l’option Q-Dip, définissez le Mn à 600, fermez la fenêtre de Q-trempette et démarrer la mesure. Enregistrer le signal avec la raie spectrale de troisième et quatrième du marqueur Mn (4.1.7).
- Lorsque la mesure est terminée, analyser les données.
- Certaines analyses telles qu’un nombre de tours est uniquement possible pour les composés déposés (polymères insolubles) sur le WE. Pour calculer le nombre de tours, double-intégrer le signal rpe, suivi d’une comparaison entre une valeur obtenue avec calibré étalon interne de Mn. Calculer le nombre d’unités formant le polymère sur l’électrode en utilisant l’équation 724,32,33répétitives :
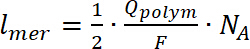 (7)
(7)
où lmer – nombre d’unités dans le polymère de répétitives, ferment de la Q est la charge de polymérisation, F est la constante de Faraday et NA est le nombre d’Avogadro. - Pour calculer les frais de dopage, utilisez le CV du polymère déposé enregistré dans une cellule de spectroélectrochimiques avant la mesure de l’EPR, en utilisant l’équation 8:
 (8)
(8)
où Qd correspond à l’accusation de dopage, i est le courant, E est l’ensemble du potentiel, E0 est le potentiel de départ, v est la vitesse de balayage et e est la charge élémentaire. - Charge calculée de dopage permet de calculer le niveau de dopage, à l’aide de l’équation 9:
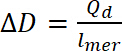 (9)
(9)
où ∆D- niveau, Qd le dopage est l’accusation de dopage et lmer – nombre d’unités dans le polymère répétitives - Pour calculer le nombre de bipolarons, supposent un initial dopage niveau égal à zéro. Utilisez le numéro de polarons unitaire de la concentration de tours, donc le nombre de bipolarons doit être calculé en utilisant équation 10. Dans les calculs, supposons que le montant initial de bipolarons est nulle et que le courant faradique est beaucoup plus élevée que celle du courant faradique-non.
 (10)
(10)
où zj’ai est la charge élémentaire de porteur de la charge, niest le nombre de porteurs de charge par unité de polymère, (p) est les polarons et (b) est le bipolarons. - Extrait de la g-facteur de valeur de spectres rpe. Déterminer à l’aide de Mn comme étalon interne (points 4.2.3 et 4.3.8), sachant que la quatrième ligne de manganèse dedans a un g-facteur de 2.03324. Calculer la largeur de la ligne de signal rpe comme étant la distance en mT entre minimum et maximum du signal rpe. Cette valeur est importante car elle peut dire où (sur quel atome), les radicaux sont forment.
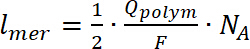 (7)
(7) (8)
(8)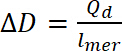 (9)
(9) (10)
(10)Representative Results
L’application la plus courante de l’analyse de CV est l’estimation de la propriété intellectuelle et EA. Bien qu’il y a deux manières d’obtenir des données de CV, il est fortement recommandé de calculer leur évolution l’apparition des pics d’oxydo-réduction (Figure 3). Cette approche permet d’unifier la méthode de calcul. Pas les matériaux testés sont soumis à des processus réversible oxydo-réduction (Figure 3) ; dans de telles situations, il n’est pas possible de calculer la base en moyenne potentiels (moyenne de potentiels correspondent à un maximum de pics cathodique-réduction et oxydation anodique). Toutefois, il est presque toujours possible de faire fonctionner la tangente à un sommet, tel qu’illustré à la Figure 3. À l’intersection avec la ligne de fond et l’utilisation des équations 1 et 2, les valeurs de la propriété intellectuelle et EA sont estimées à 5,35 eV et −2.90 eV, respectivement. Il y a aussi plusieurs différentes échelles utilisées pour évaluer les IP et EA basés sur des mesures de CV. Le plus couramment utilisé des échelles pour les matières organiques sont −4.8 et −5.1 eV comme équivalant à 0,00 V par rapport à l’hydrogène Normal électrode (NHE). Cependant, toutes les échelles ne sont que des approximations ; Rappelez-vous ceci lors de la comparaison des résultats différents. L’essentiel est de dire quels sont les paramètres ont été considérées pour les calculs. Dans ce cas, la valeur −5.1 eV a été choisi car il doit correspondre au potentiel formel du couple redox ferrocène ; dans l’échelle de Fermi, c’est de 0,40 V contre électrode de Calomel saturée (SCE) dans l’acétonitrile, qui est en accord avec la mesure précédente.
Il y a de nombreux articles publiés concernant l’analyse du CV dans les présentes, nous montrons que lorsque le processus ne va pas être comme il est prévu. L’analyse repose sur les dérivés de thiophène : NtVTh (structure, illustré à la Figure 4), qui subit une dégradation à oxydation (Figure 5).
NtVTh a deux potentiels d’oxydation : la première à 0,70 V et le second à 0,84 V (Figure 5). Durant le premier cycle, le pic de réduction n’est pas été respecté, ce qui indique un processus irréversible. Caractéristiques électrochimiques des NtVTh montrent que la polymérisation ne se produit pas après le premier potentiel d’oxydation, certains electro-inactif couche de produits de réaction dépose à la surface de l’électrode, entravant ainsi le processus de polymérisation. Ce qui est visible est la réaction avec le cation radical sur la liaison de vinyle, lorsque la molécule perd sa conjugaison et forme des dimères sur l’électrode.
S’il est difficile d’extraire des informations sur les porteurs de charge de la CV, il est possible de distinguer les polarons et bipolarons lorsque pris en charge par un spectrophotomètre UV-Vis-NIR. Le poly neutre (OiPrThEE) a été caractérisé par deux grande absorption π | bandes avec des maxima de pics à λmax1 les transitions π * = 363 nm et λmax2 = 488 nm, associé à la forme aromatique du dérivé polythiophène non dopé. Pendant le dopage oxydatif, nouvelles bandes polaronic et bipolaronic sont générés. Le spectre UV-Vis, obtenu au cours de l’oxydation de poly (OiPrThEE) a révélé la diminution de la bande d’absorption de polymère neutre (300 à 550 nm) (Figure 6) ainsi que la formation d’une nouvelle bande d’absorption (550-950 nm) des cations radicalaires de bithiophene et p- phenylenevinylene avec des maxima à 692 nm. Le point isobestique du processus d’oxydation se trouvait à 604 nm. Le groupe bipolaronic est apparu entre le 950 nm et 1700 nm avec un maximum situé à 1438 nm.
Spectroscopie RPE est la technique qui détecte les matériaux possédant un électron non apparié, ce qui inclut des radicaux organiques39. Il y a plusieurs paramètres qui peuvent être extraites de spectres rpe, mais une des plus intéressantes consiste à estimer où les radicaux sont localisées. Électrons, semblables à des protons, possèdent de spin. En plaçant un électron dans un champ magnétique externe, ce spin peut être fendu de deux façons : parallèle et antiparallèle au champ magnétique, ce qui donne deux niveaux d’énergie. Ce phénomène est connu comme l’effet Zeeman40,41. Dans le cas des radicaux organiques, l’électron interagit non seulement avec le champ magnétique externe, mais aussi avec des noyaux magnétiques (les noyaux qui ont un spin non nul ; I≠0). Un certain nombre de niveaux d’énergie dégénérés est égal à 2I + 1, où I est le nombre quantique de spin du noyau que l’électron interagit42. L’interaction de l’électron non apparié avec un plus grand nombre de noyaux magnétiques conduit à diviser davantage de niveaux d’énergie et à la structure hyperfine de l’enregistrement des spectres rpe43 (Figure 7).
Pour les molécules où l’électron interagit avec un nombre encore plus grand de noyaux, la raie spectrale individuel pourraient se chevauchent, qui se traduit par l’enregistrement d’un signal unique, large44,45,46 (Figure 8). Ceci est typique pour des polymères conjugués, où l’ion radicale générée lors d’un processus d’oxydo-réduction est délocalisé47,48.
La combinaison de spectroscopie RPE avec méthodes électrochimiques permet la caractérisation des porteurs de charge (ion radicalaire) générés pendant le processus d’oxydo-réduction, mais aussi la détermination du mécanisme de ces processus49,50 . Si bien résolues (pics sont séparés ; pas sous la forme d’un pic large) spectres sont enregistrés, comme dans le cas de la réduction électrochimique du s -tetrazine dérivé (Figure 7), puis l’analyse de la structure hyperfine des spectres conduit à conclusions au sujet de la localisation de l’électron non apparié. Une façon d’analyser ce genre de spectres est d’effectuer la simulation avec un logiciel spécial et pour s’adapter à des spectres simulés avec l’expérimental un51. Ceci est particulièrement utile lorsque la structure hyperfine est complexe en raison de l’interaction de l’électron non apparié avec un grand nombre de protons. Dans le cas du dérivé de tetrazine - sillustré à la Figure 7, simulation des spectres rpe (ligne rouge) indique l’interaction de l’électron non apparié avec quatre atomes d’azote de s- tetrazine.

Figure 1 : électrochimiques et spectroélectrochimiques cellules utilisés pour les mesures. La figure présente la configuration système des cellules électrochimiques/spectroélectrochimiques à l’aide de voltampérométrie cyclique, UV-visible et proche infrarouge (UV-Vis-NIR) et les électrons des mesures spectroélectrochimiques résonance paramagnétique (RPE). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 2 : voltampérométrie cyclique (CV) de COPO1 composé correctement mesurées (a) et le CV avec un instable de référence potentiels dibenzothiophène-S, S-dioxyde avec ferrocène (b)52. La figure montre deux voltampérogrammes cycliques. (a) présente correctement enregistré CV et (b) montre un voltamogramme enregistrée à l’aide d’une électrode de référence avec aucun potentiel stable. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 3 : voltampérométrie cyclique (CV) du composé COPO1 dans un large éventail de potentiels de. Évaluation des potentiels pour les calculs de EA et de la propriété intellectuelle de la COPO1 apparition composés52. EA = −2.90 eV et IP = 5,35 eV. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 4. Raies spectrales RPE six de manganèse standard. Le signal paramagnétique de manganèse utilisé pour l’étalonnage de l’évolution du signal. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 5 : voltamétrie cyclique (CV) des composés NtVTh. Voltampérogrammes cycliques de 15 mM NtVTh à 0,1 M Bu4NBF4/CH3CN et par rapport à la norme de ferrocène présentant le processus de dégradation sur l’électrode de travail. Vitesse de balayage 0,05 V/s : (un) a été prise dans l’intervalle −1, 4 V à 0,8 V et (b) dans l’intervalle −1, 4 V à 0,9 V. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 6 : Ultraviolet-visible et proche infrarouge (UV-Vis-NIR) spectroélectrochimie du dérivé de poly (OiPrThEE). Spectres UV-Vis-NIR présentant l’évolution de la bande d’absorption à travers la génération des porteurs de charge sur la structure du polymère. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 7 : analyse spectroélectrochimiques EPR du dérivé tetrazine. (a) structure du s- tetrazine dérivé ; (b) les spectres rpe enregistrés pendant la réduction électrochimique du stetrazine - dérivé (spectre simulé noir ligne ligne expérimentale et rouge). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 8 : EPR spectroélectrochimiques polaron signal du polymère conjugué. Spectres rpe (EPR) enregistrées au cours de la première étape de l’oxydation du polymère conjugué (spectres rpe polaron espèces). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 9 : voltampérométrie cyclique (CV) du ferrocène et decamethylferrocene. Comparaison de deux normes électrochimiques comme pure et mélange montrant l’évolution du potentiel. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
Discussion
Techniques électrochimiques et spectroélectrochimiques n’avoir aucune restriction ; On peut analyser tant à l’état solide et des solutions liquides dans un large éventail de conditions de température et autres avec ces techniques. La chose importante dans tous ces cas, c’est que les composés/matériaux sont analysées sous le potentiel appliqué, reproduisant les conditions du monde réel pour l’utilisation des dispositifs de l’électronique organique. La seule différence est que, en électrochimie, la formation de porteurs de charge, est observée.
Les méthodes présentées ici montrent l’utilité de l’analyse des transporteurs chargés générée en composés organiques qui sont en corrélation avec leur applicabilité dans l’électronique organique. En outre, les techniques électrochimiques et spectroélectrochimiques sont moins cher et moins exigeante que celle des méthodes typiques utilisées dans l’analyse de porteur de charge, mais il y a des étapes critiques et modifications au protocole qui sont nécessaires selon le résultats obtenus.
Au cours de la caractérisation électrochimique, toujours commencer avec une concentration particulière. Si l'on compare un ensemble des composés, puis tous les matériaux ont besoin d’avoir la même concentration molaire. Le mieux est de commencer par la concentration de 1 mM et 50 mV/s scan taux comme il est indiqué dans le protocole de cette étude, mais il est bon de connaître la concentration de l’échantillon sur le comportement électrochimique observé. Toujours essayer de mesurer au moins trois balayages. Les deux premiers scans sont habituellement différents parce que les conditions initiales (équilibre) sont différentes. La deuxième et la troisième analyse devrait être le même. Si les scans de deuxième et troisième sont les mêmes, il n’y a probablement aucune réactions secondaires observées dans ce système (Figure 2 a). Dans un processus d’oxydation, un nouveau record à un potentiel inférieur s’affiche indiquant que le matériau conducteur a été déposé sur le WE18,19,24,25,29,30 , 31 , 32. Si la hauteur de la crête inférieure augmente dans les analyses successives, le polymère conjugué était probablement déposé18,19,24,25,29 , 30 , 31 , 32. si tous les courants diminuent dans les analyses successives, la conductrice produit de dégradation a été déposé sur l’électrode. Si l'on observe un pic très faible avant l’oxydation principale ou le pic de réduction (en particulier des polymères), alors c’est probablement responsable piégeage processus19,23,31,34. Si l'on observe un pic de dedoping très forte d’oxydation ou de réduction, alors c’est probablement causé par la décomposition des structures cristallines sur une électrode formée à travers le processus électrocristallisation durant l’oxydation35.
Toujours vérifier le comportement de l’essai composé avant, pendant et après les sommets de l’oxydo-réduction. Cela signifie qu’au moins trois balayages de CV doivent être enregistrés : avec le plus faible potentiel de vertex inférieure ou supérieure ou supérieur (dans l’affaire oxydation), respectivement, puis le potentiel du maximum du pic, sommet inférieur ou supérieur potentiel valeur exactement sur le pic maximal et avec Sommet des potentiels plus élevés (oxydation) et inférieur (réduction) du potentiel de la valeur maximale de crête. Le processus observé peuvent varier et parfois deux processus peuvent être observées sous une crête théoriquement. Toujours comparer les voltampérogrammes cycliques recueillies de l’électrolyte (étape 2.6), le ferrocène (étape 2,9), le composé (étape 2.13) et le ferrocène avec composé (étape 2.19) ; Il y a plusieurs questions à prendre en compte.
Comparez toujours les signaux de CV de l’électrolyte et la substance, à tester si tous les signaux de l’électrolyte est visible sur le voltamogramme cyclique du composé mesuré, puis l’électrolyte doit être changé car sa fenêtre électrochimique est trop faible, ou la électrolyte est contaminée. Si le signal (couple redox) du ferrocène (étape 2,9) et le ferrocène avec composé (étape 2.19) sont à la même position, puis tout est exécutée correctement. Si les sommets sont décalés entre eux, puis vérifiez le RE et recommencez la mesure. Si le signal (oxydation, réduction ou couple rédox potentiels) de la substance avec le ferrocène ajouté (étape 2.19) d’essai est à un potentiel plus élevé que celui de la pure composé (étape 2.13), puis examiner les valeurs (oxydation, réduction ou redox couple potentiel) de le voltamogramme cyclique du composé pur. Le changement est causé par le montant le plus élevé du ferrocène dans la solution. Lorsqu’on observe les deux procédés d’oxydation, le premier processus (oxydation ou réduction) qui est toujours sur le WE susceptibles d’affecter la surface active ; Cela peut provoquer une augmentation du potentiel d’oxydation concernant le deuxième processus (Figure 9).
Disclosures
Les auteurs n’ont rien à divulguer.
Acknowledgments
Les auteurs remercient l’appui financier du projet « Excilight » « Donneur-accepteur Light Emitting Exciplexes comme matériaux pour facile-à-tailleur foudre OLED ultra efficace » (H2020-ACEM-ITN-2015/674990) financé par Marie Skłodowska-Curie Actions dans le cadre du programme de recherche et d’innovations « Horizon-2020 ».
Materials
| Name | Company | Catalog Number | Comments |
| Potentiostat | Metrohm | Autolab PGSTAT100 | |
| EPR | JEOL | JES-FA200 | |
| UV-Vis detector | Oceanoptics | QE6500 | |
| NIR detector | Oceanoptics | NIRQuest | |
| Dichloromethane (DCM) | Sigma-Aldrich | 106048 | |
| Tetrabutylammonium tetrafluoroborate (Bu4NBF4) | Sigma-Aldrich | 86896 | |
| 2-propanol, 99.9% | Sigma-Aldrich | 675431 | |
| Acetone, 99.9% | Sigma-Aldrich | 439126 | |
| Ultrasonic Bath | Elma | S30H | |
| Tetrahydrofuran >99.9% | Sigma-Aldrich | 401757 | |
| ferrocene >98% | Sigma-Aldrich | F408 | |
| decamethylferrocene >97% | Sigma-Aldrich | 378542 |
References
- O'Brien, D. F., Baldo, M. A., Thompson, M. E., Forrest, S. R. Improved energy transfer in electrophosphorescent devices. Applied Physics Letters. 74, 442-444 (1999).
- Chin, B. D., et al. Effects of interlayers on phosphorescent blue organic light-emitting diodes. Applied Physics Letters. 86, 133505-133507 (2005).
- Cardon, C. M., Li, W., Kaifer, A. E., Stockdale, D., Bazan, G. C. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bang, A. -M., et al. A novel random terpolymer for high-efficiency bulk-heterojunction polymer solar cells. RSC Advances. 7, 1975-1980 (2017).
- Tybrandt, K., Kollipara, S. B., Berggren, M. Organic electrochemical transistors for signal amplification in fast scan cyclic voltammetry. Sensors and Actuators B: Chemical. 195, 651-656 (2014).
- Schaur, S., Stadler, P., Meana-Esteban, B., Neugebauer, H., Sariciftci, N. S. Electrochemical doping for lowering contact barriers in organic field effect transistors. Organic Electronics. 13, 1296-1301 (2012).
- Mullen, K., Scherf, U. Organic Light Emitting Devices: Synthesis, Properties and Applications. , Wiley-VCH. Weinheim. (2006).
- Monk, P. M. S., Mortimer, R. J., Rosseinsky, D. R. Electrochromism and Electrochromic Devices. , Cambridge University Press. Cambridge. (2007).
- Skotheim, T. A., Elsenbaumer, R. L., Reynolds, J. R. Handbook of Conducting Polymers. , Marcel Dekker. New York. (1998).
- Data, P., et al. Kesterite Inorganic-Organic Heterojunction for Solution Processable Solar Cells. Electrochimica Acta. 201, 78-85 (2016).
- Data, P., Pander, P., Okazaki, M., Takeda, Y., Minakata, S., Monkman, A. P. Dibenzo[a,j]phenazine-Cored Donor-Acceptor-Donor Compounds as Green-to-Red/NIR Thermally Activated Delayed Fluorescence Organic Light Emitters. Angewandte Chemie International Edition. 55 (19), 5739-5744 (2016).
- Jankus, V., et al. Highly Efficient TADF OLEDs: How the Emitter-Host Interaction Controls Both the Excited State Species and Electrical Properties of the Devices to Achieve Near 100% Triplet Harvesting and High Efficiency. Advanced Functional Materials. 24 (39), 6178-6186 (2014).
- Data, P., et al. Exciplex Enhancement as a Tool to Increase OLED Device Efficiency. Journal of Physical Chemistry C. 120 (4), 2070-2078 (2016).
- Dias, F. B., et al. The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Advanced Science. , (2016).
- Okazaki, M., et al. Thermally Activated Delayed Fluorescent Phenothiazine-Dibenzo[a,j]phenazine-Phenothiazine Triads Exhibiting Tricolor-Changing Mechanochromic Luminescence. Chemical Science. 8, 2677-2686 (2017).
- Data, P., et al. Efficient p-phenylene based OLEDs with mixed interfacial exciplex emission. Electrochimica Acta. 182, 524-528 (2015).
- Goushi, K., Yoshida, K., Sato, K., Adachi, C. Organic light-emitting diodes employing efficient reverse intersystem crossing for triplet-to-singlet state conversion. Nature Photonics. 6, 253-258 (2012).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of heteroaryl group on electrochemical and spectroscopic properties of conjugated polymers. Electrochimica Acta. 83, 271-282 (2012).
- Data, P., Motyka, M., Lapkowski, M., Suwinski, J., Monkman, A. Spectroelectrochemical Analysis of Charge Carries as a Way of Improving Poly(p-phenylene) Based Electrochromic Windows. Journal of Physical Chemistry C. 119, 20188-20200 (2015).
- Enengl, C., et al. Explaining the Cyclic Voltammetry of a Poly(1,4-phenylene-ethynylene)-alt-poly(1,4-phenylene-vinylene) Copolymer upon Oxidation by using Spectroscopic Techniques. ChemPhysChem. 18, 93-100 (2017).
- Gudeika, D., et al. Hydrazone containing electron-accepting and electron-donaiting moieties. Dyes Pigments. 91, 13-19 (2011).
- Swist, A., Cabaj, J., Soloducho, J., Data, P., Lapkowski, M. Novel acridone-based branched blocks as highly fluorescent materials. Synthetic Metals. 180, 1-8 (2013).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of alkyl chain on electrochemical and spectroscopic properties of polyselenophenes. Electrochimica Acta. 87, 438-449 (2013).
- Laba, K., et al. Electrochemically induced synthesis of poly(2,6-carbazole). Macromolecular Rapid Communications. 36, 1749-1755 (2015).
- Pluczyk, S., et al. Unusual Electrochemical Properties of the Electropolymerized Thin Layer Based on a s-Tetrazine-Triphenylamine Monomer. Journal of Physical Chemistry C. 120, 4382-4391 (2016).
- Blacha-Grzechnik, A., Turczyn, R., Burek, M., Zak, J. In situ Raman spectroscopic studies on potential-induced structural changes in polyaniline thin films synthesized via surface-initiated electropolymerization on covalently modified gold surface. Vibrational Spectroscopy. 71, 30-36 (2014).
- Laba, K., et al. Diquinoline derivatives as materials for potential optoelectronic applications. Journal of Physical Chemistry C. 119, 13129-13137 (2015).
- Enengl, S., et al. Spectroscopic characterization of charge carriers of the organic semiconductor quinacridone compared with pentacene during redox reactions. Journal of Materials Chemistry C. 4, 10265-10278 (2016).
- Data, P., et al. Electrochemically Induced Synthesis of Triphenylamine-based Polyhydrazones. Electrochimica Acta. 230, 10-21 (2017).
- Brzeczek, A., et al. Synthesis and properties of 1,3,5-tricarbazolylbenzenes with star-shaped architecture. Dyes Pigments. 113, 640-648 (2015).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Electrochemistry and spectroelectrochemistry of a novel selenophene-based monomer. Electrochimica Acta. 59, 567-572 (2012).
- Data, P., et al. Electrochemical and spectroelectrochemical comparison of alternated monomers and their copolymers based on carbazole and thiophene derivatives. Electrochimica Acta. 122, 118-129 (2014).
- Data, P., et al. Evidence for Solid State Electrochemical Degradation Within a Small Molecule OLED. Electrochimica Acta. 184, 86-93 (2015).
- Data, P., et al. Unusual properties of electropolymerized 2,7- and 3,6- carbazole derivatives. Electrochimica Acta. 1282, 430-438 (2014).
- Etherington, M. K., et al. Regio- and conformational isomerization critical to design of efficient thermally-activated delayed fluorescence emitters. Nature Communications. 8, 14987 (2017).
- Trasatti, S. The absolute electrode potential: an explanatory note. Pure and Applied Chemistry. 58, 955-966 (1986).
- Cardona, C. M., et al. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bredas, J. -L. Mind the gap! Mater Horiz. 1, 17-19 (2014).
- Gerson, F., Huber, W. Electron Spin Resonance Spectroscopy of Organic Radicals. , (2003).
- Brustolon, M., Giamello, E. Electron Paramagnetic Resonance. A Practitioner's Toolkit. , John Wiley & Sons Inc. New Jersey. (2009).
- Weil, J., Bolton, J. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. , John Wiley & Sons, Inc. (2007).
- Rieger, P. H. Electron Spin Resonance. Analysis and Interpretation. , The Royal Society of Chemistry. (2007).
- Roznyatovskiy, V. V., Gardner, D. M., Eaton, S. W., Wasielewski, M. R. Radical Anions of Trifluoromethylated Perylene and Naphthalene Imide and Diimide Electron Acceptors. Organic Letters. 16, 16-19 (2013).
- Pluczyk, S., Kuznik, W., Lapkowski, M., Reghu, R. R., Grazulevicius, J. V. The Effect of the Linking Topology on the Electrochemical and Spectroelectrochemical Properties of Carbazolyl Substituted Perylene Bisimides. Electrochimica Acta. 135, 487-494 (2014).
- Ledwon, P., et al. A Novel Donor-acceptor Carbazole and Benzothiadiazole Material for Deep Red and Infrared Emitting Applications. Journal of Materials Chemistry C. 4, 2219-2227 (2016).
- Heinze, J., Frontana-Uribe, B. A., Ludwigs, S. Electrochemistry of Conducting Polymers-Persistent Models and New Concepts. Chemical Reviews. 110, 4724-4771 (2010).
- Kurowska, A., Kostyuchenko, A. S., Zassowski, P., Skorka, L., Yurpalov, V. L., Fisyuk, A. S., Pron, A., Domagala, W. Symmetrically Disubstituted Bithiophene Derivatives of Properties. Journal of Physical Chemistry C. 118, 25176-25189 (2014).
- Zotti, G., Schiavon, G., Zecchin, S., Morin, J. F., Leclerc, M. Electrochemical, Conductive, and Magnetic Properties of 2,7-Carbazole-Based Conjugated Polymers. Macromolecules. 35, 2122-2128 (2002).
- Kaim, W., Fiedler, J. Spectroelectrochemistry: The Best of Two Worlds. Chemical Society Reviews. 38, 3373-3382 (2009).
- Duling, D. R. Simulation of Multiple Isotropic Spin-Trap EPR Spectra. Journal of Magnetic Resonance, Series B. 104, 105-110 (1994).
- Benson, C. R., et al. Multiplying the Electron Storage Capacity of a Bis-Tetrazine Pincer Ligand. Dalton Transactions. 43, 6513-6524 (2014).
- Nobuyasu, R. S., et al. Rational Design of TADF Polymers Using a Donor-Acceptor Monomer with Enhanced TADF Effi ciency Induced by the Energy Alignment of Charge Transfer and Local Triplet Excited States. Advanced Optical Materials. 4, 597-607 (2016).
