

Summary
Qui presentiamo un metodo per misurare direttamente il RNA di trasferimento carica livelli da purificato Escherichia coli RNA così come un modo per confrontare i livelli relativi di RNA di trasferimento, o qualsiasi altri RNA breve, attraverso diversi campioni basati sull'aggiunta di spike-a cellule che esprimono un gene di riferimento.
Abstract
RNA transfer (tRNA) è una parte essenziale dell'apparato traduzionale in qualsiasi organismo. tRNAs legano e trasferire gli aminoacidi al ribosoma traduzione. I livelli relativi di tRNAs differenti ed il rapporto di aminoacylated tRNA per tRNA totale, conosciuto come il livello di carico, sono fattori importanti nel determinare l'accuratezza e la velocità di traduzione. Di conseguenza, l'abbondanza e livelli di carica di tRNAs sono variabili importanti per misurare quando si studia la sintesi delle proteine, ad esempio in varie condizioni di stress. Qui, descriviamo un metodo per la raccolta del tRNA e misurando direttamente l'abbondanza relativa e il livello di carico assoluto di determinate specie di tRNA in Escherichia coli. Il tRNA è raccolto in modo tale che il legame labile tra il tRNA e l'aminoacido è conservato. il RNA è quindi sottoposto a elettroforesi su gel e Northern blotting, che si traduce nella separazione dei tRNAs cariche e scariche. I livelli di specifici tRNAs nei diversi campioni possono essere paragonati a causa dell'aggiunta di spike-a celle per la normalizzazione. Prima Purificazione RNA, aggiungiamo 5% delle cellule di Escherichia coli che overproduce la rara di tRNAselC a ciascun campione. La quantità di specie di interesse in un campione di tRNA è poi normalizzata alla quantità di tRNAselC nello stesso campione. Aggiunta di spike-in cellule prima Purificazione RNA ha il vantaggio rispetto alla aggiunta di purificato RNA spike-in che è anche responsabile di eventuali differenze di efficienza di lisi delle cellule fra i campioni.
Introduction
Di seguito, presentiamo un metodo per quantificare specifico tRNAs e misurare la loro carica livelli mediante Northern blotting. Il metodo si basa su una tecnica sviluppata da Varshney et al. 1. raccolta delle cellule in acido tricloroacetico (TCA) e mantenere i campioni a 0 ° C durante la purificazione di RNA, il legame estere tra il tRNA e l'aminoacido è conservata2,3,4. Aminoacylated tRNAs si distinguono dalle loro controparti nonacylated gel elettroforesi e macchiare nordico, dovuto ad una mobilità in diminuzione del aminoacylated tRNA nel gel, causato dalla covalentemente aminoacido5. Inoltre, vi presentiamo un protocollo per la normalizzazione della quantità di tRNA con aggiunta di spike-in cellule che overexpressing il raramente usato tRNAselC 4.
tRNAs sono alcune delle molecole più abbondanti nella cellula batterica e una parte vitale dell'apparato di traduzione. tRNAs legano gli amminoacidi e trasferirli i ribosomi traduzioni. L'associazione di aminoacidi per tRNAs (aminoacylation o ricarica) è facilitato dalla sintetasi del tRNA di aminoacyl. L'abbondanza relativa di diversi tRNAs carica è importante per assicurare la fedeltà della sintesi proteica, poiché sottorappresentanza delle cognate carica che tRNA per un codone determinato RNA messaggero (mRNA) aumenta la probabilità che un tRNA vicino cognate sarà erroneamente consegnare suo aminoacido a catena del polipeptide crescente sul ribosoma6. L'importanza della carica tRNA si riflette nella vasta risposta della cella e. coli a un drastico calo dei livelli di carica di un tRNA; la risposta rigorosa. Durante la risposta rigorosa la sintesi di tRNA, RNA ribosomiale (rRNA) e maggior parte dei mRNAs è abbassata a favore di trascrizione di mRNA specifici connessi con la sopravvivenza dell'amminoacido biosintesi e stress, e il tasso di crescita delle cellule è abbassato drasticamente7 . Inoltre, il recente lavoro da noi e gli altri ha mostrato che e. coli degrada attivamente la maggior parte dei suoi tRNA in risposta alle sollecitazioni che limitano traduzione4,8, suggerendo che la regolazione dei livelli di tRNA può essere importante per far fronte a tali sollecitazioni. Così, misurazioni affidabili delle abbondanze di tRNA e livelli di ricarica sarà uno strumento importante per comprendere pienamente le risposte batteriche di sforzo.
E. coli è comunemente usato per esprimere la proteina ricombinante e a causa delle differenze nell'uso di codone tra specie, espressione non ottimale è un problema spesso affrontato9. Questo può essere aggirato da espressione di tRNAs aggiuntive necessarie per tradurre il mRNA ricombinante10. Misurazioni di tRNA livelli in tali ceppi di ricarica potrebbero guidare gli sforzi sulla risoluzione dei problemi e ottimizzare l'espressione della proteina.
Questo metodo consente anche la rilevazione di "orifizio"; una molecola di tRNA aminoacylated con un aminoacido non-affine. Aminoacylation di aminoacidi diversi può causare un tRNA per la migrazione con velocità leggermente diverse attraverso i gel di poliacrilammide1,11. In alcuni casi, il metodo utilizzabile anche per distinguere diversi modifica modelli altrimenti identici tRNAs3.
Un altro istituita procedura biochimica per l'indagine di tRNA livelli di carica è periodato ossidazione. Il metodo si basa sull'osservazione che periodato aminoacylated tRNA è protetto dall'ossidazione e uncharged tRNA non è. Periodato dopo trattamento di ossidazione della ricarica del tRNA viene utilizzata per stimare i livelli di carica del RNA raccolto. Tuttavia, la ricarica dei diversi tRNAs ha dimostrata di essere colpiti dal trattamento fornendo così qualche inesattezza12. Il metodo qui presentato misure carico direttamente da RNA purificato, escludendo quindi eventuali pregiudizi da reazioni chimiche o enzimatiche. Una limitazione di questo metodo è che solo una specie di tRNA viene rilevato in un momento, così anche se la stessa Northern blot può essere spogliato e reprobed per più tRNAs, è che richiede tempo e un po ' laborioso per raccogliere dati su molti tRNAs.
Un modo affidabile di normalizzazione dei campioni a vicenda è fondamentale negli studi dove l'obiettivo è quello di confrontare i livelli relativi di una molecola attraverso diversi campioni. Qui introduciamo una procedura di normalizzazione per RNA dove una piccola aliquota di Escherichia coli cellule che overexpressing il tRNA raraselC è aggiunto come spike-in per tutti i campioni sperimentali prima Purificazione RNA. Questo metodo è utile non solo quando si esaminano i tRNAs ma per quantificazione relativa di ogni specie di RNA quando la messa a punto sperimentale è tale che nessun RNA endogeno può essere attendibile per essere presenti alla stessa concentrazione cellulare in tutti i campioni. Ad esempio, il livello di una "pulizia" RNA ribosomiale RNA-come spesso viene utilizzato come un riferimento endogeno per confrontare le relative quantità di RNA un altro tra i diversi campioni13. Ma questo è di scarsa utilità se la concentrazione cellulare del RNA riferimento varia tra i campioni, come può essere il caso per il RNA ribosomiale (rRNA) se il campionamento avviene durante un sforzo risposta15,16,17 o durante entrata in fase stazionaria17. L'aggiunta di spike-in cellule di campioni sperimentali prima Purificazione RNA fornisce un modo solido e preciso di normalizzazione indipendenti del setup di campionamento. Un altro modo di standardizzazione è l'aggiunta di uno o più RNA di spike ai campioni sperimentali dopo la purificazione di RNA. Tuttavia, questo metodo non tiene conto delle eventuali differenze di recupero di RNA tra campioni.
Utilizzando cellule di Escherichia coli che iperesprimere il riferimento RNA come spike-in celle (Vedi Figura 1) in un esperimento su Escherichia coli ha lo svantaggio che risulta inoltre di una piccola quantità di esogeno Escherichia coli totale RNA per la campioni. Correggiamo per questa aggiunta analizzando un campione contenente solo spike-in celle in parallelo con i campioni sperimentali (Vedi il Northern blot nel Figura 2, lane etichettato "selC"). Il protocollo presentato è stato sviluppato per e. coli K-12 ma è probabile che sia applicabile per la maggior parte delle specie batterica.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. preparazione delle colture batteriche.
- Crescere tutte le culture in matracci di Erlenmeyer con un rapporto di volume matraccio di cultura al massimo 1/6. In una tipica configurazione sperimentale, crescono le culture a 37,0 ° C e scuotendo a 160 giri/min in morpholinepropanesulfonic acido (MOPS) minimo medio 18 completati con la fonte di carbonio preferito, ad esempio 0,2% glucosio, per almeno 10 generazioni successive prima raccolta RNA.
- Il giorno prima della raccolta del RNA, diluire una cultura sovrasviluppata del ceppo desiderato in MOPS medium minimo in modo che raggiungerà la desiderata OD 436 al momento desiderato il giorno seguente. Calcolare il volume di cultura sovrasviluppata da usare per l'inoculazione con la formula:
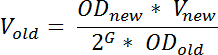
Nota: qui, OD nuovo denota la desiderata OD 436 dopo l'incubazione, V nuovo denota il volume della cultura diluito, OD vecchio denota la OD 436 della cultura sovrasviluppata diluito da, G indica il numero di generazioni la cultura crescerà, dalla tempo di diluizione (t 1) il tempo è necessario il giorno dopo (t 2) e può essere calcolata come: tempo di differenza (in minuti) tra t 1 e t 2 diviso per il tempo di generazione (in min). Per garantire la crescita allo steady-state al momento del campionamento, G dovrebbe essere ≥ 10; V vecchio denota il volume della cultura fuori-coltivato che dovrebbe essere trasferito al medium fresco. Questa equazione è un'approssimazione che non tiene conto per qualsiasi fase di ritardo; un fenomeno spesso osservato dopo diluizione di una cultura fuori-coltivata nei mezzi freschi.- Condizioni di crescita variano a seconda dello scopo dell'esperimento, ma per limitare cellula--cellula variazione nei livelli di tRNA e per incrementare la riproducibilità dei risultati, (consigliato) crescono le culture di stato prima del campionamento stazionario 19.
2. Preparazione delle cellule di spike
Nota: il ceppo di e. coli MAS1074, che esprime il gene selC codifica tRNA selC sotto il controllo di un isopropilico β-D-1-thiogalactopyranoside (IPTG)- promotore inducibile è utilizzato come il ceppo di spike-a.
- Diluire una cultura sovrasviluppata di MAS1074 a un OD 436 di 0.05 in MOPS minimal supplementato con 100 µ g/mL ampicillina e 0,2% di glucosio. Far crescere la cultura a 37 ° C in agitazione a 160 giri/min.
Nota: Vedere la discussione per l'uso di un ceppo di riferimento alternativi. - OD a 436 ~ 0.1, aggiungere IPTG ad una concentrazione finale di 1 mM per indurre l'espressione di tRNA selC. Far crescere la cultura a OD 436 ~0.5 (~ 3-4 h di crescita) e spostare il matraccio di cultura in un bagno di ghiaccio. Mantenere la cultura di spike-in cella su ghiaccio fino a quando tutti i campioni per la purificazione di RNA sono state raccolte.
3. Raccolta di campioni sperimentali.
- Pre-calda (37 ° C), uno capped beuta da 100 mL (o simili) contenente 4 mL 10% (p/v) l'acido tricloroacetico (TCA) per ogni campione collocandolo in un bagno di acqua riscaldata.
Nota: attenzione, TCA si decompone al riscaldamento, producendo fumi tossici e corrosivi, compreso acido cloridrico e cloroformio. La soluzione in acqua è un acido forte; reagisce violentemente con basi ed è corrosivo per molti metalli. Corretta precauzione dovrebbe essere presa quando usando questo liquido. - Immediatamente prima della raccolta delle cellule, misurare e registrare la OD 436 della cultura.
- Per raccogliere le cellule, trasferire 4 mL di coltura in un pallone pre-riscaldato contenente TCA.
- Mescolare il campione agitando a mano il matraccio per 5 s. miscelazione con TCA immediatamente Disattiva attività enzimatiche tutte e quindi conserva i livelli di carica dei tRNAs.
Nota: Se più campioni devono essere raccolti, di mantenere campioni raccolti in TCA su ghiaccio fino a quando tutti i campioni sono stati raccolti.
4. Aggiunta di spike-a cellule
Nota: preparazione delle cellule di spike è descritta nel passaggio 2.
- Quando tutti i campioni per la preparazione di RNA sono stati raccolti, aggiungere 5% spike-in celle (basate su OD) di ciascun campione.
Nota: Il volume di picco-in cella cultura necessaria è calcolato con la formula:
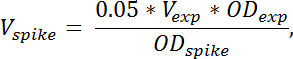
dove V picco indica il volume di picco-in cella cultura necessaria, V Exp indica il volume di coltura raccolto cellulare sperimentale (senza TCA), OD exp denota la OD 436 della cultura sperimentale delle cellule al momento della vendemmia in TCA. OD a spillo denota la OD 436 della cultura spike-in cella. Se campioni multipli vengono raccolte a diversi ODs, il volume delle cellule di spike aggiunto deve essere regolato secondo la formula di cui sopra per ciascun campione. - Quantificare il contributo dalle cellule spike-in per le specie di RNA di interesse, preparare un campione contenente solo spike-in celle da 4 mL di miscelazione spike-in celle con 4 mL TCA. Preparare il RNA da questo campione nello stesso modo per quanto riguarda gli altri campioni.
- Facoltativamente, prendere un altro matraccio contenente solo il campione sperimentale, senza spike-nelle cellule e includerlo per quantificare il contributo del campione sperimentale per il tRNA selC livelli; i livelli endogeni di tRNA selC in Escherichia coli K-12 sono minimo trascurabile.
5. Preparazione di RNA
Nota: protocollo di preparazione di RNA The descritto qui è essenzialmente quella descritta dallo Varshney et al. 1; Per evitare deacilazione dei tRNAs aminoacylated durante la purificazione è importante conservare i campioni a pH acido e a 0 ° C durante tutto il corso della purificazione. Utilizzare tubi/boccette pulito sterile per tenere i campioni e le soluzioni e preparare tutte le soluzioni con ultrapura (18,2 MΩ·cm resistività) acqua.
- Pour 8 mL di ogni campione in una provetta da centrifuga ghiaccio-refrigerati. Centrifugare i campioni per 10 min a 9.500 x g a 4 ° C. completamente rimuovere surnatante e risospendere in acetato di sodio 0,3 mL freddo 0,3 M, pH 4,5 e 10 mM EDTA.
- Trasferire ciascun campione in una provetta da microcentrifuga da 1,5 mL freddo e aggiungere fenolo 0,3 mL equilibrata con lo stesso buffer.
Nota: attenzione, fenolo produce fumi tossici ed è altamente corrosivo per la pelle. - Vortex ciascun campione 10x per 15 s con almeno 1 min tra ogni vortice. Tra Vortex mantenere i campioni a 0 ° C. utilizzare le pause frequenti per garantire che i campioni rimangono freddi.
- Centrifugare i campioni per 15 min a 20.000 x g a 4 ° C. trasferimento la fase acquosa (fase superiore) di nuovo per microcentrifuga da 1,5 mL provette. Aggiungere come prima 0,3 mL freddo fenolo e vortice 4x15 s.
- Centrifuga per 10 min a 20.000 x g a 4 ° C. trasferimento la fase di acqua per microcentrifuga da 1,5 mL nuovo, freddo tubi contenenti 2,5 volte il volume di etanolo al 96% (~ 750 µ l). Precipiti il RNA incubando le provette per 1 h a-20 ° C, o a -80 ° C.
- Centrifugare i campioni per 30 min a 20.000 x g a 4 ° C. eliminare il supernatante e attentamente aggiungere 1 mL di etanolo 70% freddo senza disturbare il pellet di RNA. Capovolgere delicatamente una volta per lavare l'interno dei tubi con etanolo.
Nota: Il pellet può essere difficile da vedere in questa fase. - Centrifuga per 10 min a 20.000 x g a 4 ° C. completamente rimuovere il supernatante e asciugare all'aria il pellet fino a quando non viene lasciato senza etanolo. Risospendere il pellet vigorosamente nel Vortex in 20 µ l freddo 10 mM sodio acetato pH 4,5 e 1 mM EDTA.
Nota: Non asciugare troppo il pellet come diventerà difficile risospendere. - Facoltativamente, valutare la concentrazione di RNA misurando l'assorbanza a 260 nm.
- Facoltativamente, valutare la qualità del RNA purificato da di elettroforesi su gel di agarosio 20.
- Esempi di store a -80 ° C.
Nota: Il protocollo può essere messo in pausa qui.
Nota: per distinguere la banda di aminoacylated tRNA dal suo omologo deacilata il Northern blot, un deacilata aliquota è preparato chimicamente da trattamento alcalino. Maggior parte dei casi, è sufficiente deacylate un singolo campione per ogni macchia nordica.
Campione- disgelo il RNA su ghiaccio. Trasferire un'aliquota di 4 µ l di campione di RNA ad un nuovo tubo. Aggiungere 46 µ l Tris-HCl pH 9.0. Incubare per 2 ore a 37 ° C.
- Aggiungere 15 µ l di acetato di sodio 0,3 M a pH 4,5 e successivamente aggiungere 125 µ l di etanolo al 96%. Precipitare di RNA a-20 ° C per 1 h.
- Centrifuga per 30 min a 20.000 x g a 4 ° C. completamente rimuovere il surnatante e risospendere in 4 µ l freddo 10 mM sodio acetato pH 4,5 e 1 mM EDTA.
Nota: Il protocollo può essere messo in pausa qui, se il campione è conservato a -80 ° C.
7. Gel elettroforesi e macchiare nordico
- Mix 4 µ l di campione (non trattati o chimicamente deacilata) con 6 µ l tampone di caricamento (0.1 M sodio acetato (pH 5.0), 8m urea, 0,05% di blu di bromofenolo e 0.05% xilene cianolo).
Nota: Ricordarsi di includere il campione contenente solo spike-in celle, il chimicamente deacilata campione (e il campione senza spike-in celle, se uno è stato preparato). - Caricare i campioni su un 0.4 mm spessore 6,5% gel di poliacrilammide (19:1 acrylam - ide:bisacrylamide) contenenti urea 8 M in tampone di acetato di sodio 0.1 M (pH 5.0). Utilizzare gel lunga 60 cm per la corretta separazione delle specie di tRNA.
- Separare il RNA mediante elettroforesi in gel di 10 V/cm a 4 ° C, fino a quando il blu di bromofenolo raggiunge il fondo del gel (~ 20 h).
- Zona di provenienza del colorante di xilene cianolo e 20cm giù verso il blu di bromofenolo colorante è usato per macchiare (la distanza tra i due coloranti dovrebbe essere 25-30 cm). Attentamente separare le lastre di due vetro, in modo che il gel rimane su uno di essi. Un sottile pezzo di carta da filtro è tagliato nella misura dell'area desiderata macchiante e posizionato sopra la parte del gel che verrà utilizzato per macchiare. Le parti del gel che non sono coperti da carta da filtro sono tagliate e scartate. utilizzare la carta da filtro per sollevare delicatamente il pezzo di gel dalla lastra di vetro.
Nota: Il grande cuscinetto in gel è molto fragile quindi maneggiare con cura. - Una membrana di nylon positivamente caricato (ad esempio Hybond N + membrana) è collocata sulla superficie del gel e trasferiti a 20 V per 90 min con 40 mM Tris-acetato (pH 8,1), 2 mM EDTA come buffer di trasferimento è.
- Crosslink RNA alla membrana tramite luce UV (0,12 J/cm 2).
Nota: La membrana ora può essere conservata a temperatura ambiente e il protocollo può essere messo in pausa qui.
8. Progettazione e verifica delle sonde
Nota: un'attenta progettazione e verifica delle sonde di Northern blot è fondamentale per ottenere risultati affidabili ed evitare indesiderato cross-ibridazione con altre specie di RNA.
- Uso breve oligonucleotides sintetici come sonde. Progettare la sequenza di sonda in un modo che è complementare alla parte più singolare della sequenza del tRNA di interesse - questo è spesso il ciclo anticodone.
Nota: Un elenco di sequenze di tRNA stimate da vari organismi può essere trovato nel tRNA Genomic Database (GtRNAdb) 21. - Regolare la lunghezza della sequenza per ottenere una prevista fusione temperatura di 55-65 ° C.
- BLAST (base locale Alignment Search Tool) la vostra sequenza contro la sequenza del genoma del ceppo batterico utilizzato nell'esperimento, per verificare che la sequenza è unica per il tRNA selezionato, come descritto in 22.
Nota: La tabella 1 elenca suggerito sonda sequenze per diversi tRNAs differenti di e. coli K12. Se la sonda è specifica per il tRNA di interesse, solo due bande sono visibili sulla macchia nordica (cariche e scariche tRNA) e solamente una banda è visibile nella corsia con il campione di deacilata (uncharged tRNA). Se più di due bande sono presenti o se ulteriore convalida della specificità della sonda è desiderato, vedere discussione.
9. L'ibridazione della macchia nordica
Nota: nei passaggi seguenti, la membrana è in sequenza ibridata con sonde complementari per i tRNAs specifico desiderato, tra cui una sonda specifica per il riferimento tRNA selC.
- Posizionare la membrana in un tubo di ibridazione e aggiungere 6 mL di soluzione di ibridazione (0,9 M di NaCl, 0.05 M NaH 2 PO 4 (pH 7,7), 5 mM EDTA, 0,5% (p/v) SDS, 100 mg/mL di tranciati e denaturazione del DNA di sperma di aringhe e 5 x Denhardt ' soluzione s (100 x Denhardt ' soluzione s = 2% di albumina di siero bovino, polivinilpirrolidone 2% 40 e 2% Ficoll)).
- Pre-ibridare la membrana nel buffer per 1 h a sonda di 42 ° C. aggiungere 30 pmol radioattivo oligo-DNA 5 girevole ' - fine - etichettati con 32 P (preparato come descritto da Sambrook et al. 23)
Nota: attenzione, le emissioni di beta ad alta energia da 32 P possono presentare un sostanziale della pelle e degli occhi dose pericolo. Corretta precauzione dovrebbe essere presa quando usando 32 P. radioattivo rifiuti deve essere smaltito secondo i protocolli di sicurezza istituzionali adeguati. - Incubare la sonda con la membrana durante la notte rotante a 42 ° C. Rimuovi la sonda utilizzando una pipetta monouso da 10 mL.
Nota: Se conservati in un congelatore la sonda può essere riutilizzata. - Nel tubo di ibridazione, lavare la membrana poco in 50 mL di 0,3 M di NaCl, 30mm citrato di sodio, 0,1% (p/v) SDS a temperatura ambiente.
Nota: Questo primo lavaggio contiene molto non associato sonda e deve essere gestito come altamente radioattivo. - Ripetere passo 9.4 una volta, quindi spostare la membrana in un contenitore in plastica piatta o vassoio con un coperchio. Coprire la membrana con la soluzione di lavaggio (0,3 M di NaCl, 30mm citrato di sodio, 0,1% (p/v) SDS) e Incubare 30 min a temperatura ambiente.
- Modificare la soluzione di lavaggio e continuare il lavaggio fino a quando un soddisfacente rapporto segnale-rumore è stato ottenuto (spesso 3-4 cambi di soluzione di lavaggio). Utilizzare un tubo Geiger-Müller per rilevare come il rapporto segnale rumore cambia misurando la radiazione da un'area vuota della membrana e confrontarlo con l'area dove la sonda ha ibridato specificamente al tRNA.
- Al fine di essere in grado di rimuovere la sonda dalla membrana dopo l'esposizione, garantire che (importante) la membrana non si asciughi prima della procedura di "stripping". Posizionare la membrana in un sacchetto di plastica sottile o un involucro e fissarlo con plastica avvolgere e sigillarlo per essere a tenuta d'aria di nastro o saldatura. Posizionare la membrana sigillata in una schermata di phosphorimaging.
Nota: Il tempo di esposizione richiesto sullo schermo di phosphorimaging è determinato dall'attività specifiche della sonda, la quantità di RNA sondato for e l'efficienza di ibridazione della sonda e può variare notevolmente; da pochi minuti ad alcuni giorni. Con esperienza, l'intensità della radiazione rilevata sulla membrana con il tubo Geiger-Müller dà una buona indicazione del tempo di posa richiesto. Per la quantificazione affidabile, è importante che lo schermo non è sovraesposta. - Sullo schermo di phosphorimaging utilizzando un laser scanner per la scansione e salvare il file in " .gel " formato.
Nota: Un alto rapporto segnale-rumore è necessario per la quantificazione affidabile. Sondaggio per un tRNA dovrebbe tradursi in due bande; aminoacylated contenente un tRNA (più lenta migrazione) e uno contenente deacilata tRNA (più veloce migrazione). Vedere la Figura 2.
10. Stripping della membrana
Nota: prima che la membrana può essere sondata per un altro tRNA, la sonda precedente deve essere rimosso prima mettendo a nudo la membrana.
- Calore abbastanza stripping buffer (15 mM NaCl, 1,5 mM di citrato di sodio, 0,1% (p/v) SDS) per coprire la membrana e versare sopra la membrana. Incubare 15 min a temperatura ambiente.
- Ripetere il punto 10.1 fino a nessuna radiazione può essere rilevato con un tubo Geiger-Müller; la membrana può ora essere ri-ibridata seguendo la procedura descritta nella sezione 9.
11. Quantificare il tRNA e livelli di carica
- caricare il file di .gel nell'imaging software (Vedi la Tabella materiali). Tracciare una linea verticale lungo ciascuna delle corsie del campione in modo tale che si estende su gran parte della larghezza della corsia, ma esclude i bordi delle fasce dove il segnale può essere irregolare, come mostrato in Figura 3A.
- Visualize conta da ogni corsia facendo " analisi "-> " creare grafico ". Definire manualmente le due cime che rappresentano la carica e il tRNA uncharged da ogni corsia, come mostrato nella Figura 3B.
- Ottenere conteggi da ogni picco cliccando " analisi "-> " relazione sulla zona di " e registrare l'area sotto ciascuno dei due picchi.
- Quando la macchia è stata sondata per tRNA selC e almeno un tRNA di interesse, normalizzare i livelli del tRNA di interesse per tRNA selC.
- Calcolare la proporzione di tRNA che proviene dalle cellule spike-in ogni corsia e sottrarre dal segnale totale in quanto Vicolo usando l'equazione:
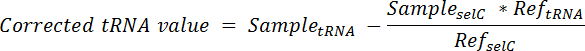
Nota: qui, TRNA di esempio = il segnale ottenuto da una sola corsia di una membrana sondata per il tRNA desiderata; Campione selC = il segnale ottenuto dalla stessa corsia della membrana stessa sondata per tRNA selC; Ref tRNA = il segnale ottenuto dalla corsia contenente RNA solo spike-in cella della membrana stessa sondato per il tRNA desiderata; Ref selC = il segnale ottenuto dalla corsia contenente RNA solo spike-in cella della membrana stessa sondato per tRNA selC. - Per normalizzare, dividere il valore corretto di tRNA da ogni corsia per il segnale di selC tRNA da stessa corsia.
Nota: Il contributo al segnale selC tRNA dalle cellule campionate è trascurabile (vedere la Figura 2, lane etichettato "-selC ").
- Calcolare la proporzione di tRNA che proviene dalle cellule spike-in ogni corsia e sottrarre dal segnale totale in quanto Vicolo usando l'equazione:
- Calcolare il livello per ogni corsia di carica dividendo il segnale del picco che rappresenta la carica tRNA con la somma del segnale da due cime di tRNA (carica + uncharged).
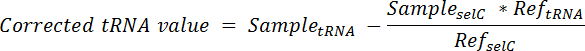
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Utilizzando la procedura descritta qui, l'abbondanza e ricarica i livelli di tre tRNAs sono stati misurati in e. coli K-12 prima e durante l'inedia dell'aminoacido.
Il auxotroph di arginina ceppo NF9154 (thr leu suo argH thi mtl supE44) è stato coltivato per almeno dieci generazioni in MOPS supplementato con 0,4% glicerolo, 50 µ g/mL treonina, leucina, arginina e istidina di 5 µ g/mL a 37 ° C in agitazione al minimo 160 giri/min. Fame di arginina è stato introdotto da filtrazione della cultura e della risospensione in mezzo di MOPS fresco come sopra ma senza arginina. Durante l'inedia di arginina, sintesi proteica è fortemente ridotto, ma continua ad un livello basso utilizzo di arginina rilasciato dal turnover delle proteine esistenti. I campioni sono stati raccolti al momento punti indicati in Figura 4. Dopo che tutti i campioni sono stati raccolti in TCA, 5% di MAS1074 spike-in cellule che overexpressing tRNAselC, è stato aggiunto per ogni campione come illustrato Figura 1. RNA è stato purificato da campioni, separato mediante elettroforesi e cancellato su una membrana come descritto nel protocollo. La membrana è stata sondata per tRNAargVYZQ, tRNAgltTUVW, tRNASuare tRNAselC, come si vede nella Figura 2. Il segnale di radiazione da ogni corsia è stato quantificato per ciascuna delle quattro sonde come illustrato in Figura 3e corretto e normalizzato come descritto nella sezione 11. I risultati sono presentati nella Figura 4. Figura 4A Mostra che i livelli di tutte le specie di tRNA testata diminuiscono rapidamente durante l'inedia dell'aminoacido, come recentemente segnalato4. Figura 4B Mostra che il tRNA ricarica livelli si comportano come previsto: la frazione di carica del tRNA arginina-accettare diminuisce rapidamente dopo la rimozione di arginina dal mezzo di crescita, considerando che i livelli di carica di altri i tRNAs aumentano leggermente al momento inedia perché carica tRNAs si accumulano quando il loro tasso di decharging presso i ribosomi diminuisce a causa della mancanza di substrato per la sintesi di proteina4. Ulteriormente nel periodo di inedia, la frazione di tRNA carica diminuisce ai livelli circa pre-fame a causa della degradazione della maggior parte della piscina tRNA (Figura 4A), che si traduce in un maggior tasso di decharging dei tRNAs rimanenti alle i ribosomi. Al contrario, la frazione carica di tRNAargVYZQ aumenta per almeno due motivi; 1) l'attivazione della risposta rigorosa significa che mRNA, tRNAarg, non a carico diventa il fattore limitante per la traduzione e 2) il pool totale di tRNAarg è ridotto (Figura 4A) così una frazione più grande del tRNA totalearg può essere ricaricata tramite la stessa piccola quantità di arginina. Questo esperimento dimostra come le misurazioni simultanee di abbondanza di tRNA e livelli di carica tRNA fornisce informazioni di alta qualità sulle condizioni per la sintesi proteica all'interno della cellula.

Figura 1. Aggiunta di spike-in celle a campioni. In questo esempio, campioni da una singola coltura di Escherichia coli NF915 sono raccolte in una serie temporale, prima e durante l'inedia dell'aminoacido. Viene mostrato uno schema della curva di crescita di NF915, dove i punti blu indicano i punti di raccolta del campione. La y sono scala logaritmica. Campioni della coltura sperimentale sono raccolti, come descritto nella sezione 3. Un disegno schematico della curva di crescita del ceppo spike-in MAS1074 viene anche mostrato, con il punto di raccolta del campione indicato come un punto blu. Il punto della figura è di illustrare che tutti i campioni sperimentali (di e. coli NF915 in questo esempio) ricevano un'aliquota di spike-in cellule della stessa cultura di riferimento. Per ogni campione, 5% delle cellule di spike-a sono aggiunti alle cellule sperimentale, calcolate basato sul diametro esterno delle colture cellulari (Vedi sezione 4). Successivamente, RNA è purificato da tutti i campioni e utilizzato per le macchie nordiche. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 2. Scansione di imager di fosforo di un Northern blot sondato per i tRNAs indicato. La membrana contiene RNA dal ceppo di e. coli NF915 raccolte presso i punti di tempo indicato prima e dopo induzione di fame di arginina (minuti). Inoltre, la membrana contiene due corsie di campioni dove sono stati omessi spike-in celle (-selC DA e - selC), e una corsia contenente RNA dallo spike-in cellule solo (selC). Il campione - selC DA contiene chimicamente deacilata tRNA. Le due bande che rappresentano le specie cariche e scariche di tRNA sono chiaramente separate. Nota il segnale trascurabile dal tRNAselC nei vicoli senza aggiunto spike-in celle. La membrana stessa è stata successivamente spogliata e reprobed per i tRNAs indicato sulla sinistra. L'area circoscritta nella macchia superiore indica la parte della macchia, che è indicata per i successivi approfondimenti di tRNA. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 3. Quantificazione del Northern blot. abbondanza di tRNA è misurata utilizzando il software. (A) A corsia singola da Northern blot visto in Figura 2 sondato per tRNAargVYZQ. La freccia superiore e inferiore indicano la aminoacylated tRNA e il deacilata tRNA, rispettivamente. Le linee rosse che corre parallela con la corsia vengono aggiunti e utilizzate per definire l'area per la quantificazione. (B) quantificazione del segnale all'interno delle due linee rosse rotte visto in (A) raffigurato come un grafo, dove l'asse orizzontale indica la distanza dalla parte superiore della linea in (A) (in pixel), e l'asse verticale indica l'intensità del segnale (in conteggi). I due picchi contenente il segnale che proviene dalla aminoacylated tRNA e il deacilata il tRNA, rispettivamente, sono definite manualmente. I dati che viene esportati per un'ulteriore analisi sono il valore corrispondente alla zona sotto ciascuno dei due picchi. Clicca qui per visualizzare una versione più grande di questa figura.

Nella figura 4. Risultato rappresentativo: quantificazione delle abbondanze di tRNA und livelli di carica durante l'inedia di arginina. Quantificazione delle bande il Northern blot illustrato nella Figura 2. (A), la relativa abbondanza di tRNAgltTUVW (rosso), tRNAargVYZQ (blu) e tRNASuar (verde). Il livello trovato a zero minuti (campioni raccolti appena prima dell'inizio di fame) è impostato su 1. (B) livello dei tre tRNAs stesso come in (A) di carica. Clicca qui per visualizzare una versione più grande di questa figura.
| Specificità della sonda | Sequenza della sonda |
| tRNAargVYZQ | 5'-TCCGACCGCTCGGTTCGTAGC |
| tRNAgluTUVW | 5'-CCTGTTACCGCCGTGAAAGGG |
| tRNAhisR | 5'-CACGACAACTGGAATCACAATCC |
| tRNAleuPQVT | 5'-GTAAGGACACTAACACCTGAAGC |
| tRNAleuU | 5'-TATTGGGCACTACCACCTCAAGG |
| tRNAleuW | 5'-CTTGCGGCGCCAGAACCTAAATC |
| tRNAleuX | 5'-TATTTCTACGGTTGATTTTGAA |
| tRNAleuZ | 5'-AAAATCCCTCGGCGTTCGCGCT |
| tRNAlysQTVWYZ | 5'-TGCGACCAATTGATTAAAAGTCAAC |
| tRNAthrV | 5'-TGGGGACCTCACCCTTACCAA |
| tRNAtyrTV | 5'-TCGAACCTTCGAAGTCGATGA |
| tRNAselC | 5'-ATTTGAAGTCCAGCCGCC |
Tabella 1. Tabella delle sequenze di sonda suggerito per tRNAs selezionato in e. coli K-12.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Questo protocollo descrive come misurare contemporaneamente il livello di carico delle specifiche Escherichia coli tRNAs e confrontare i livelli relativi dei tRNAs in diversi campioni. I punti critici del protocollo sono 1) per gestire i campioni in modo che i livelli di carica tRNA cellulare vengono mantenuti, 2) per normalizzare la quantità di tRNA in modo tale che i livelli relativi di tRNA in diversi campioni possono essere paragonati in modo affidabile e 3) per garantire la sp ecificity delle sonde selezionate per i tRNAs di interesse. Questi punti vengono affrontati separatamente sotto.
purificazione di tRNA
I tRNAs sono purificati da e. coli in modo delicato che è ottimizzato per mantenere i loro livelli di ricarica cellulari. È la nostra esperienza che questo protocollo estrae principalmente RNA inferiore a 150 nt, così tRNA costituisce una grande frazione di RNA purificato. Per questo motivo, non è consigliabile utilizzare questo protocollo per purificare il RNA totale da Escherichia coli. Il protocollo presentato qui dovrebbe anche essere adatto per la rilevazione e quantificazione degli altri RNA come piccolo RNA (sRNA) senza qualsiasi ulteriori adattamenti. Perché il protocollo di estrazione di RNA arricchisce per brevi sequenze di RNA dovrebbe anche trovare uso anche se umile è espresso il sRNA sotto inchiesta.
Normalizzazione di spike-a cellule
Per confrontare i livelli del tRNA attraverso diversi campioni, è necessario normalizzare i livelli di tRNA ai livelli di una trascrizione che è presente nello stesso numero medio per la cellula in tutti i campioni, al fine di correggere variazioni di campione a campione in RNA recupero e gel di caricamento. Per questo scopo, abbiamo spike i campioni con il 5% delle cellule di riferimento che iperesprimono il tRNA raraselC prima Purificazione RNA. Il metodo di spike è preferibile rispetto all'utilizzo di un RNA endogeno come riferimento, poiché ne permette il confronto dei livelli di tRNA attraverso circostanze dove nessun RNA endogeni è note con certezza di essere trovato alla stessa concentrazione cellulare. Utilizzando cellule di Escherichia coli come lo spike-in ha lo svantaggio che il 5% supplementare Escherichia coli RNA viene aggiunto a tutti i campioni sperimentali, dove contribuisce al segnale del RNA di interesse. Questa polarizzazione è rappresentata da compreso un campione contenente solo le celle spike-in su ogni macchia nordica e correzione dei valori di RNA come descritto al punto 11.4.1. L'espressione di tRNAselC in cellule wild type è così basso rispetto ad altri tRNA (Vedi Figura 2) che esso non fa una differenza significativa nella quantificazione e quindi può essere trascurata. Tuttavia, nel ceppo di riferimento MAS1074, dove selC è espresso da un promotore di7 T su un plasmide alta copia, si stima che almeno 1000 volte più tRNAselC è prodotto in presenza di 1 mM IPTG rispetto a quello prodotto da wildtype E. coli K-12 in qualsiasi condizione. MAS1074 cessa di crescere dopo due generazioni di induzione di IPTG completo. A quel punto, le differenze di cultura all'induzione di tRNAselC sono piccole, e non una preoccupazione per questo protocollo come tutte le aliquote di spike-a celle per un singolo esperimento sono prese da una stessa cultura indotta di MAS1074. Come alternativa alla MAS1074, le cellule di un organismo cui RNA sarà non cross-reagiscono con la sonda di ibridazione possono essere utilizzate come spike-in celle. Sulfolobus solfataricus cellule sono state utilizzate con successo come spike-in per esperimenti con Escherichia coli tRNA4. Tuttavia, spike-in cellule di Escherichia coli sono probabili riflettere più accuratamente il grado di lisi delle cellule delle cellule sperimentali, come è incerto se S. solfataricus lisi con la stessa efficienza e. coli. Inoltre, S. solfataricus in crescita è più noioso che crescono a 85 ° C con un tempo di generazione di ~ 16 h.
Garantire la specificità di tRNAs selezionato
Se sia aminoacylated e deacilata tRNA è stata correttamente purificati due bande devono essere rilevati sulla membrana del Nord. La distanza tra le due bande sarà diverso a seconda il tRNA specifico sotto inchiesta ed è una conseguenza combinata della differenza di dimensioni, proprietà polari e conformazione che un particolare amminoacido causa quando si lega una particolare tRNA5 . Ad esempio, la distanza tra l'amminocilato e il deacilata tRNA è maggiore per tRNAargVYZQ è per tRNAgltTUVW, come si vede nella Figura 2. In conseguenza le grandi dimensioni del gel utilizzato qui per separare il RNA, la risoluzione è abbastanza alta per distinguere tRNA caricato da uncharged, ma anche di distinguere la maggior parte dei tRNAs diversi gli uni dagli altri a causa delle loro differenze in lunghezza, sequenza composizione e modifica modelli. Anche isoaccepting tRNAs può essere distinto come illustrato per la leucina accettando tRNAs2. Tuttavia, nota che in questo studio non era possibile distinguere tRNAleuPQV da tRNAleuT, come la differenza nella sequenza è solo uno G alla sostituzione di T.
Se più di due bande appaiono sulla membrana del Nord potrebbe essere una conseguenza del limite di rilevabilità della sonda con altro RNAs. Aumentare il rigore della fase di lavaggio (passo 9,5) aumentando la temperatura in genere può risolvere questo. 30 min di lavaggio a 55 ° C è di solito sufficiente. Se aumento il rigore di lavaggio non rimuove cross-ibridazione, la soluzione può essere di progettare una nuova sonda che è complementare a una parte diversa (spesso parzialmente sovrapposta) della sequenza tRNA. Bande supplementari possono anche essere riporto da una sonda precedente, a seguito di insufficiente stripping della membrana. Per evitare questo problema, è necessario fare un controllo striscia esponendo la membrana prima disondaggio. Se bande appaiono sull'esplorazione di controllo la membrana potrebbe essere necessario ulteriori stripping. È solitamente possibile ri-sonda una membrana almeno 8 - 10 volte, ma dopo parecchie sonde può essere difficile da rimuovere completamente. Se questo è il caso, la membrana sono memorizzabili per pochi mesi prima disondaggio come 32P decadimenti veloci.
Un'ulteriore verifica della specificità sonda possa essere ottenuta eseguendo un Northern blot con cellule raccolte in un insieme di condizioni in cui la trascrizione di interesse è previsto per essere colpiti in modo prevedibile. Ad esempio, dell'espressione genica del tRNA da un plasmide o inedia dell'aminoacido per l'aminoacido cognata, dovrebbe comportare un livello di maggiore espressione relativa o un livello di carico inferiore, rispettivamente, del tRNA di interesse.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori non hanno nulla a rivelare.
Acknowledgments
Gli autori ringraziano Marit Warrer per l'eccellente assistenza tecnica. Questo lavoro è stato supportato dal Consiglio danese per la ricerca indipendente | Scienze naturali [1323-00343B] e la Fondazione di ricerca nazionale danese [DNRF120].
Materials
| Name | Company | Catalog Number | Comments |
| Urea | Merck | 57-13-6 | Purity >99% |
| Polyacrylamide | Serva | 79-06-7 | |
| Bis (N,N'-Methylene-bis-acrylamide) | BIO-RAD | 161-0201 | |
| Trichloroacetic acid (TCA) | Sigma | 76-03-9 | |
| Phenol | Merck | 108-95-2 | |
| IPTG | |||
| Glucose | |||
| Sodium Acetate | |||
| EDTA | |||
| Ethanol | |||
| Tris | |||
| Hydrochloric acid | |||
| Sodium Chloride | |||
| NaH2PO4 | |||
| Sodium Citrate | |||
| SDS | |||
| Herring Sperm DNA | Sigma | 100403-24-5 | |
| BSA | |||
| Polivinylpyrrolidone | |||
| Ficoll | |||
| P32 γATP | Perkin Elmer | ||
| DNA oligos (probes) | TAG Copenhagen | ||
| Hybond N+ membrane | GE Healthcare | RPN203B | |
| Crosslinker | |||
| Electroblotter | BIO-RAD | ||
| Typhoon FLA 7000 Scanner | GE Healthcare | 28955809 | |
| Spectrophotometer | |||
| Hydridization oven | |||
| Geiger-Müller tube | |||
| Phosphor imager screen | GE Healthcare | ||
| Hybridization tube | |||
| Culture Flasks | |||
| 1.5 ml microcentrifuge tubes | |||
| 20 ml centrifuge tubes | |||
| Name | Company | Catalog Number | Comments |
| Software | |||
| ImageQuant | GE Healthcare | ||
| Excel | Microsoft |
References
- Varshney, U., Lee, C. -P., RajBhandary, U. L. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem. 266 (36), 24712-24718 (1991).
- Sorensen, M. A. Charging levels of four tRNA species in Escherichia coli Rel(+) and Rel(-) strains during amino acid starvation: a simple model for the effect of ppGpp on translational accuracy. J Mol Biol. 307 (3), 785-798 (2001).
- Krüger, M. K., Sørensen, M. A.
- Svenningsen, S. L., Kongstad, M., Stenum, T. S., Muñoz-Gómez, A. J., Sørensen, M. A. Transfer RNA is highly unstable during early amino acid starvation in Escherichia coli. Nucleic Acids Res. 45 (2), 793-804 (2017).
- Enriquez, J. A., Attardi, G. Evidence for aminoacylation-induced conformational changes in human mitochondrial tRNAs. Proc Natl Acad Sci U S A. 93 (16), 8300-8305 (1996).
- Parker, J. Errors and alternatives in reading the universal genetic code. Microbiol Rev. 53 (3), 273-298 (1989).
- Traxler, M. F., et al. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol. 68 (5), 1128-1148 (2008).
- Zhong, J., et al. Transfer RNAs Mediate the Rapid Adaptation of Escherichia coli to Oxidative Stress. PLoS Genet. 11 (6), e1005302 (2015).
- Kane, J. F. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr Opin Biotechnol. 6 (5), 494-500 (1995).
- Baca, A. M., Hol, W. G. Overcoming codon bias: a method for high-level overexpression of Plasmodium and other AT-rich parasite genes in Escherichia coli. Int J Parasitol. 30 (2), 113-118 (2000).
- Li, S., Pelka, H., Schulman, L. The anticodon and discriminator base are important for aminoacylation of Escherichia coli tRNA (Asn). J Biol Chem. 268 (24), 18335-18339 (1993).
- Rizzino, A. A., Freundlich, M. Estimation of in vivo aminoacylation by periodate oxidation: tRNA alterations and iodate inhibition. Anal Biochem. 66 (2), 446-449 (1975).
- Dittmar, K. A., Sorensen, M. A., Elf, J., Ehrenberg, M., Pan, T. Selective charging of tRNA isoacceptors induced by amino-acid starvation. EMBO Rep. 6 (2), 151-157 (2005).
- Zhou, K., et al. Novel reference genes for quantifying transcriptional responses of Escherichia coli to protein overexpression by quantitative PCR. BMC Mol Biol. 12 (1), 18 (2011).
- Jacobson, A., Gillespie, D. Metabolic events occurring during recovery from prolonged glucose starvation in Escherichia coli. J Bacteriol. 95 (3), 1030-1039 (1968).
- McCarthy, B. The effects of magnesium starvation on the ribosome content of Escherichia coli. Biochim Biophys Acta (BBA)-Specialized Section on Nucleic Acids and Related Subjects. 55 (6), 880-889 (1962).
- Zundel, M. A., Basturea, G. N., Deutscher, M. P. Initiation of ribosome degradation during starvation in Escherichia coli. Rna. 15 (5), 977-983 (2009).
- Piir, K., Paier, A., Liiv, A., Tenson, T., Maivali, U. Ribosome degradation in growing bacteria. EMBO Rep. 12 (5), 458-462 (2011).
- Schaechter, M., Maaloe, O., Kjeldgaard, N. O. Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J Gen Microbiol. 19 (3), 592-606 (1958).
- Neidhardt, F. C., Bloch, P. L., Smith, D. F.
- Sambrook, J., Fritsch, E. F., Maniatis, T. Molecular cloning: a laboratory manual. , Cold spring harbor laboratory press. (1989).
- Sorensen, M. A., Jensen, K. F., Pedersen, S. High concentrations of ppGpp decrease the RNA chain growth rate. Implications for protein synthesis and translational fidelity during amino acid starvation in Escherichia coli. J Mol Biol. 236 (2), 441-454 (1994).
- Tian, C., et al. Rapid Curtailing of the Stringent Response by Toxin-Antitoxin Module-Encoded mRNases. J Bacteriol. 198 (14), 1918-1926 (2016).
