

Summary
Dieses Video zeigt eine Methode zu sezieren und Kultur kommissuralen Neuronen aus E13 Ratte Rückenmark. Dissoziiert kommissuralen Neuronen sind nützlich, um die zellulären und molekularen Mechanismen der Axon-Wachstum und Anleitung zu studieren.
Abstract
Kommissuralen Neuronen wurden häufig verwendet, um die zugrunde liegenden Mechanismen Axon Führung während der embryonalen Entwicklung des Rückenmarks zu untersuchen. Die Zellkörper dieser Neurone sind in der dorsalen Rückenmark und ihre Axone folgen stereotype Trajektorien während der embryonalen Entwicklung. Kommissuralen Axone zunächst ventral Projekt auf die Bodenplatte. Nach dem Überqueren der Mittellinie, diese Axone anterior und Projekt in Richtung des Gehirns. Jeder dieser Schritte wird durch die Wirkung von mehreren Führung Cues geregelt. Kulturen stark in kommissuralen Neuronen angereichert sind ideal für viele Experimente Bewältigung der Mechanismen der Axon Wegfindung, einschließlich Drehen Assays, Immunchemie und Biochemie geeignet. Hier beschreiben wir eine Methode zu sezieren und Kultur kommissuralen Neuronen aus E13 Ratte Rückenmark. Zuerst wird das Rückenmark isoliert und dorsalen Streifen sind herauspräpariert. Die dorsale Gewebe wird dann in eine Zellsuspension durch Trypsinierung und mechanische Störungen dissoziiert. Neuronen sind auf Poly-L-Lysin-beschichtete Deckgläschen oder Gewebe-Kulturschalen plattiert. Nach 30 Stunden
Protocol
1. Dissection der embryonalen Ratte Rückenmark
Allgemeine Empfehlungen
Keep L-15-Medium auf Eis und ändern sich häufig das Medium, in dem Sezieren Gericht zu halten Embryonen cool. Dies schont Gewebe Integrität. Alle Schritte werden mit zwei Paaren von Dumont Nr. 5 Pinzette durchgeführt sofern nicht anders angegeben. Um Verunreinigungen zu vermeiden, sprühen alle Werkzeuge und Arbeitsflächen mit 70% Ethanol und halten Dissektion Medium Flasche geschlossen. Zur Übertragung von Embryonen zwischen Gerichten, verwenden Sie einen Ausschnitt aus Kunststoff Pipette oder einem perforierten Löffel. Es ist wichtig, nicht auf das Rückenmark (nicking, Stretching) Schäden an den erfolgreichen Abschluss der Präparation.
Vorbereitung
- Kalte L-15-Medium
- 50 ml L-15 + 10% Hitze-inaktiviertem Pferdeserum (HiHS). Keep on ice.
Rückenmark Dissektion
- Euthanize ein E13 Schwangerschaft inszeniert Ratte (E0 = ersten Tag nach Decktag) mit einem CO 2-Kammer nach den Richtlinien des Instituts.
- Spray 70% Ethanol auf dem Bauch. Pinch und ziehen Sie die Haut der unteren Bauchgegend mit einer Pinzette und schneiden mit chirurgischer Schere. Wiederholen, um durch die Muskel-und Peritonealdialyse Schichten geschnitten, um die Bauchhöhle gelangen. Erstellen Sie eine V-Form Schnitt durch Schneiden von Gewebe an den Seiten des Bauches, bis zum Brustkorb. Anheben und wieder das Gewebe der Bauchhöhle aussetzen.
- Die Gebärmutter ist an drei Standorten angebracht: an der unteren Mitte Bauch und an den beiden oberen seitlichen Ecken. Heben Sie die Gebärmutter durch Greifen auf Gewebe zwischen embryonalen Säcken. Schneiden Sie das Bindegewebe in die Gebärmutter und in eine Petrischale mit L-15 auf Eis gefüllt zu entfernen.
- Die nächsten Schritte sind im Rahmen eines Dissektionsmikroskop getan. Zur Trennung eines Embryos aus extraembryonalen Geweben und embryonalen Membranen, greifen das Gewebe zwischen Gebärmutter Säckchen mit einer Pinzette,. Mit dem anderen Paar, eine Prise mehr transparent Seite des sac, um durch die oberflächlichen Membranen (die dunklere Seite der Plazenta) geschnitten. Squeeze out der Embryo durch sanftes Drücken der Plazenta Seite des sac. Entfernen Sie alle Embryonen und in eine Petrischale mit L-15 auf Eis gefüllt.
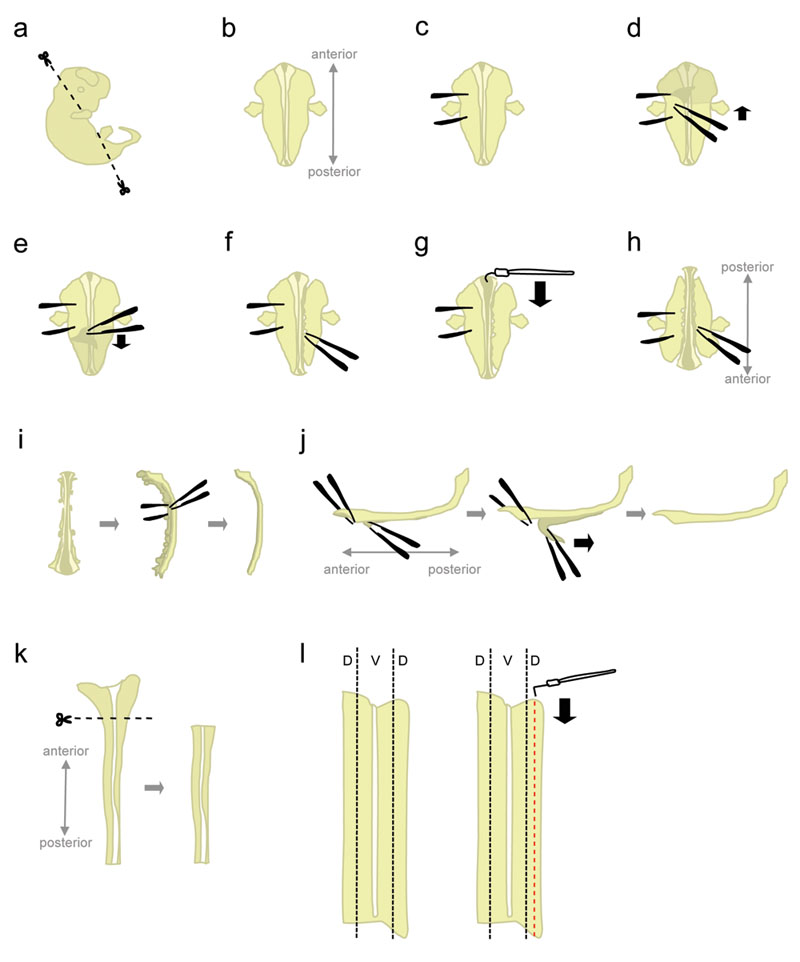
- Setzen Sie einen Embryo in eine 10 cm Petrischale mit eiskaltem L-15 gefüllt. Mit Mikroschere, schneiden Sie den Kopf und hinteren Teile mit Winkeln in Abbildung 1a gezeigt.
Schneiden bei diesen Winkeln wird dazu beitragen, Position der Embryo, wenn gesetzt "ventral face down". - Position des Embryos "ventral face down" (anterior weg zeigt, posterior zeigt auf der Experimentator) (Abb. 1b).
- Halten Sie das Embryo aus einer Seite, mit der Pinzette "festzunageln" des Embryos (Abb. 1c). Bewegen Sie die Zange, da sie das Gewebe zerreißt. Mit der anderen Pinzette, ziehen Sie die Haut über den Rücken des Embryo ab der Region zwischen den "Betrieb" Pinzette (Abb. 1d-e). Dies legt das Rückenmark, die an dieser Stelle, durch Membranen (der Hirnhäute) gewickelt ist.
Besorgen Sie sich die Haut von den Seiten und nicht von oben auf das Rückenmark zu vermeiden nicking des Rückenmarks. - Teilweise lösen das Gewebe auf der rechten Seite des Rückenmarks (Abb. 1f). Beginnend auf der Ebene der Haltezange, poke das Gewebe mit geschlossenen Pinzette so nah wie möglich an das Rückenmark. Dann öffnen Sie langsam die Zange, um das Gewebe zu reißen. Dies sollte zu lösen die Spinalganglien (DRGs) aus dem Rückenmark, und sollte auch stören ventralen Organe. Lassen Sie einige Gewebe an der vorderen und hinteren Enden. Verlassen Gewebe angebracht fügt Gewicht des Embryos verbunden, somit verhindern, dass es aufwärts im nächsten Schritt gezogen. Darüber hinaus ist das Gewebe verwendet zu halten, um den Embryo in späteren Schritten.
- Ausgehend von der vorderen Ende, verwenden Sie die hakenförmigen Wolframnadel auf die Hirnhäute geschnitten und öffnen das Rückenmark entlang der roofplate (Abb. 1g).
- Drehen Sie den Embryo 180 Grad (hinten weg zeigt, anterior zeigt auf der Experimentator) (Abb. 1 Std.).
Dies ermöglicht es dem Forscher, der gleichen Hand benutzen, um das Gewebe von der anderen Seite des Embryos zu lösen. - Halten Sie den Embryo durch Greifen auf die zuvor freistehende Seite, und stören das Gewebe von den übrigen Seiten mit der gleichen Methode (siehe 1.8.). Wenn Sie fertig sind, vollständig lösen das Gewebe auf beiden Seiten des Rückenmarks.
Alternative: Vollständig lösen das Gewebe, das auf der linken Seite, aber lassen Sie einige Gewebe an der vorderen und hinteren Ende auf der rechten Seite angebracht. Dies kann manchmal helfen, die Wirbelsäule in ihrer Position in Schritt 1.12. - Legen Sie das Rückenmark auf die Seite und entfernen Sie die meisten der übrigen mesenchymalen Gewebe und DRGs (Abb. 1i).
- In diesem Schritt werden die Hirnhäute und das Rückenmark zwei "Blätter" des Gewebes aufeinander apposed. Pin nach unten die größeren, anterior-meisten Teil des Rückenmarks, und ziehen Sie ein kurzes Segment der Hirnhäute aus dem Rückenmark cord (Abb. 1j, links). Jetzt halten die beiden getrennten Segmente durch Greifen über mit einer Pinzette, und ziehen Sie die Hirnhäute mit einer glatten, ständig in Bewegung (Abb. 1j).
Ungleiche "Peeling" führt zum Bruch des Rückenmarks und / oder der Hirnhäute Schicht. Es ist normalerweise nicht möglich, den Anteil des Rückenmarks noch Meningen befestigt erholen. - Mit einem Kunststoff-Pipette das isolierte Rückenmark eine Petrischale mit L-15 + 10% HiHS und verlassen auf dem Eis. Typischerweise sind Rückenmark aller Embryonen, die vor Sezieren der dorsalen Teile gesammelt.
Rückenmark Dissektion
- Legen Sie ein Rückenmark in eine Petrischale mit L-15 +10% HiHS, liegen in einem "open-book"-Konfiguration flach. Schneiden Sie den breiteren, vorderen Teil (die auch Teil der Hinterhirn) (Abb. 1K).
- Die dorsale Gewebe wird in den seitlichen-weiten Teilen des Rückenmarks (Abb. 1l, links). Während Pinning das Kabel mit einem geraden Wolframnadel, verwenden Sie die L-förmige Wolframnadel zum Ausschneiden eines Streifens, der 1:5 die Breite der Hälfte des Rückenmarks. (Abb. 1l, rechts). Legen Sie die dorsalen Streifen in einem 15 ml Kunststoffröhrchen mit L-15 + 10% HiHS auf Eis.
Dorsal Neuralrohr Abschnitte ~ 12 E13 Embryonen ~ 3,5 bis 4.000.000 Zellen für die Beschichtung nach der Dissoziation ergeben. Weitere Zellen können durch eine grobe Zerlegung der dorsalen Rückenmarks (Schneiden breiteren dorsalen Streifen) erhalten werden, aber kommissuralen Neuron Reinheit reduziert werden.
2. Kommissuralen Neuron Kultur
Allgemeine Empfehlungen
Alle Schritte sollten unter sterilen Bedingungen in einer Gewebekultur Haube sofern nicht anders angegeben durchgeführt werden. Verwenden Sie frisches Medium und frisch aufgetaut Ergänzungen und Reagenzien. Die Dissoziation und Verreiben Schritte sind in Ca 2 + / Mg 2 +-freiem HBSS zu minimieren Ca 2 + / Mg 2 +-abhängige Adhäsion durchgeführt.
Vorbereitung
- beschichteten Deckgläsern (Einsatz deutscher Desag Glas) oder Zellkulturplatten (siehe Beschichtungsverfahren unten).
- warme Neurobasal Plating Medien (siehe unten), in Gewebekultur-Schalen und CO 2 äquilibriert in der Gewebekultur-Inkubator für mindestens 1,5 h vor dem Ausplattieren.
- 2,5% Trypsin in der 37 ° C im Wasserbad
- 2 Flaschen HBSS Ca 2 + / Mg 2 +-freien, einer bei 4 ° C, einer bei 37 ° C.
- zwei Feuer-poliertem Glas Pasteurpipetten eins mit einem Durchmesser der Hälfte der üblichen Größe, und eine mit einem Durchmesser, der geringfügig kleiner als die Hälfte. Gebrauch sterilisiert Pasteur Pipetten. Um Feuer-polish der Pipetten, mit einem Bunsenbrenner (oben in der hellblauen Flamme) an der Spitze schmilzt ein leichter Rückgang zu dem Durchmesser. Da dieser Schritt außerhalb der Gewebekultur Haube durchgeführt wird, sprühen die Pipetten mit 70% Ethanol, bevor unter der Gewebekultur Kapuze. Für eine effizientere Wiederherstellung von Neuronen, Mantel des Pasteur-Pipetten mit serumhaltigen vor Beginn der Dissoziation oder kurz vor der Verreibung Schritt (fill Pipetten mit Medien und halten für 30 Sekunden). Dies wird das Anhaften von Zellen der Innenseite der Pasteurpipette während der Verreibung zu verhindern.
- sterile milliQ Wasser zum Waschen PLL-beschichteten Schalen oder Deckgläser
- 12,5% MgSO 4-Lösung in HBSS
Poly-L-Lysin-Beschichtung
Bei Verwendung von Deckgläsern, Acid-Wash für 24 Stunden und sterilisieren vor dem Beschichten (siehe Kaech und Banker, 2006). Verwenden deutschen Desag Deckgläschen.
Zur Beschichtung mit Poly-L-Lysin auf Deckgläschen oder Kunststoff Gewebekulturschalen:
- unter einer Gewebekultur Kapuze, Oberflächen mit einer kleinen Kuppel des 100 ug / ml PLL-Lösung für 1,75-2 Stunden.
- waschen zweimal mit milliQ Wasser, mindestens 5 Minuten pro Waschgang (kann während der Zelle Dissoziationsstufe unten durchgeführt werden).
- Geschäft in Wasser bis zur Verwendung. Lassen Sie sich nicht die PLL-beschichtete Oberfläche trocken.
Zur Verringerung der PLL Verschwendung für Deckgläser Beschichtung, legen Sie die Deckgläser in sterile bakteriellen Petrischalen. Coat und waschen Sie die Deckgläschen, indem eine Kuppel von Flüssigkeit auf dem Deckgläschen. Die bakterielle Petrischalen sind hydrophob, so sollte die Flüssigkeit auf die Deckgläser bleiben. Übertragen Sie die Deckgläser zu Gewebekulturschalen kurz vor dem Ausplattieren der Zellen.
Für Neuronen auf Kunststoff-Gewebe Kulturschalen plattiert, Haftung in der Regel höher ist, könnte somit Neuriten Dehnung vermindert werden.
Dissoziation und Plattieren
- Stellen Sie sicher, dass die dorsale Streifen haben bis auf den Boden der Röhre nieder. Entfernen Sie die meisten der L-15 +10% HiHS mit einer Pasteurpipette. Schnell Waschen der dorsalen Neuralrohr Streifen einmal durch Zugabe von 3 ml kaltem (4 ° C) HBSS.
- Lassen Sie die dorsalen Neuralrohr Streifen für 2 Minuten absitzen, dann entfernen Sie die HBSS mit einer Pasteurpipette.
- Fügen Sie warmes (37 ° C) HBSS auf ein Volumen von 4,7 ml. THenne add 0,3 ml 2,5% Trypsin, um eine endgültige Konzentration von 0,15% Trypsin geben.
- Bei 37 ° C im Wasserbad für 7 min. Mix einmal sanft auf halbem Wege durch die Inkubation.
- Zugabe von 30 ul DNAse (25 000 U / mL) für eine Endkonzentration von 150 U / mL. Add 60 ul MgSO 4 und kurz mischen, für eine endgültige Konzentration von 0,15%.
Für Gewebe aus groben Dissektionen, gegen 1 Minute bei 37 ° C im Wasserbad.
Zu diesem Zeitpunkt sollte der dorsalen Neuralrohr Abschnitte fragmentiert. Wenn der dorsalen Neuralrohr Abschnitte nicht zu fragmentieren begonnen haben, bedeutet das normalerweise, dass die 2,5% Trypsin Lager alt ist, und neue Lager sollte aufgetaut werden, oder das Waschen mit kaltem HBSS nicht effektiv entfernen HiHS aus der Probe. - Zentrifugieren Sie die Gewebestücke bei 200 g für 4 min.
- Entfernen Sie den Überstand mit einer Pasteurpipette verlassen ~ 50-100 ul der Flüssigkeit am Boden des Röhrchens.
- Flick das Rohr vorsichtig, um das Pellet zu lösen, dann waschen Sie die Zellen durch Zugabe von 5 ml warmem HBSS. Lassen Sie regeln bei Raumtemperatur für 2 min.
- Zentrifugation bei 200 g für 5 min.
- Entfernen Sie den Überstand mit einer Pasteurpipette verlassen ~ 50-100 ul der Flüssigkeit am Boden des Röhrchens.
- Flick das Rohr vorsichtig, um das Pellet zu lösen und teilweise resuspendieren. Dann fügen Sie 2 ml warmem HBSS.
- Mit dem kleinen (Halb-Durchmesser) feuerpolierten Glas Pasteur Pipette auf die Zellen durch Pipettieren langsam up-and-down 4-6 mal distanzieren. Vermeiden Sie Blasen und Pipette die Flüssigkeit gegen die Seite des Rohres. Nicht zu verreiben.
- Verwenden Sie den kleinsten Feuer-poliertem Glas Pasteurpipette weiter distanzieren die Zellen durch Pipettieren langsam up-and-down 3-4 mal. Vermeiden Sie Blasen und Pipette die Flüssigkeit gegen die Seite des Rohres. Nicht zu verreiben.
Für Gewebe aus groben Dissektionen, fügen Sie ein extra 1 ml warmem HBSS auf das Rohr am Ende der Dissoziation.
Wenn Dissoziation der Zellen, ist es nicht notwendig, alle Zellklumpen und Aggregate zu distanzieren. Stoppen Pipettieren up-and-down oder wechseln Sie zu einer Pasteurpipette mit einem kleineren Durchmesser, wenn Sie keine weitere Abnahme in der Größe der Zellaggregate mit weiteren Pipettieren zu sehen. - Lassen Sie alle verbleibenden Gewebestücke in die Röhre niederzulassen für 1 min. Es ist nicht nötig, die Zellen in ein neues Röhrchen übertragen.
- Nehmen Sie 20 ul Zellsuspension und mit 5 ul der Trypanblau. Zählen von Zellen auf einem Hämocytometer.
Neuronen sollte ≥ 95% lebensfähig durch Trypanblau-Ausschluss. - Platte der Neuronen in Neurobasal Plating Medien (siehe Rezepte unten).
- Empfohlene plating Dichten für den Erhalt von isolierten Neuronen (Low-Density-Kulturen von Neuronen Verklumpen oder einander überlappend zu vermeiden):
- 120 000 - 180 000 Zellen / Well für eine 6-Well-Platte
- 60 000 - 75 000 Zellen / Well für eine 12-Well-Platte
- 16-18 Stunden später, ändern Sie die Medien Neurobasal Growth Media (siehe Rezepte unten)
Verwenden Sie nicht eine Vakuumpumpe, die Medien von der Kulturschale absaugen beim Wechseln der Medien; vorsichtig mit einer Pipette. Dadurch wird vermieden, verdrängen die Neuronen.
Repräsentative Ergebnisse:
Vier Stunden nach dem Ausplattieren sollte Neuronen, die Poly-L-Lysin (PLL)-beschichteten Fläche geklebt haben. Unter Phasenkontrast-Beleuchtung, eingehalten werden Zellkörper in der Regel relativ flach und oval (Abb. 2a). Zellen, die nicht gut sind eingehalten erscheinen als Kugeln, die etwas bewegen, wenn die Schale sehr schonend ist auf der Seite erschlossen. Viele Faktoren können möglicherweise behindern die Adhäsion von Zellen (siehe Diskussion).
Nach 30 Stunden in vitro, haben die meisten Neuronen ein Axon mit einem sichtbaren Wachstum Kegel (Abb. 2c, d) verlängert. Wenn arme axonale Wachstum beobachtet wird, ob die Neurobasal Nährmedien mit frischem Medium und Ergänzungen vorgenommen wurde. Neuronen bleiben für mindestens 6 Tage in diesen Bedingungen gesund. Dieses Verfahren hat sich als zuverlässig liefern Präparate stark in kommissuralen Neuronen angereichert mit ~ 90% der Neuronen, DCC (Yam et al. 2009). Die Breite des dorsalen Rückenmarks Streifen, der für die Vorbereitung der Zellsuspension verwendet wird, wird Einfluss auf die Reinheit der Kultur, mit größerer Reinheit als dünner Streifen verwendet werden. Ein Anwendungsbeispiel ist in Abbildung 3 (Immunfluoreszenz) gezeigt. Siehe den Artikel von Yam et al. (2009) für weitere Beispiele.

Abbildung 1. Schematics des Rückenmarks Dissektion Schritte. D = Dorsal, V = Ventrale. Klicken Sie hier, um eine größere Abbildung zu sehen.

Abbildung 2. Repräsentatives Ergebnis von isolierten kommissuralenNeuronen auf einer PLL-beschichteten Deckglas beschichtet. a, b) 4 Stunden nach dem Ausplattieren wurden Neuronen an der Oberfläche festhält. Bar = 20 um. c, d) 30 Stunden nach dem Ausplattieren wurden die meisten Neuronen ein Axon mit einem sichtbaren Wachstum Kegel verlängert. Bar = 20 um.

Abbildung 3. A kommissuralen Neuron immungefärbt für Gamma-Tubulin (grün), mit F-Aktin durch Phalloidin (F-Aktin, rot) und die im Kern mit DAPI (blau) markiert.
Rezepte und Kommentare
Neurobasal Plating Medien
- Neurobasal
- 10% Hitzeinaktivierte FBS (HiFBS)
- 2 mM L-Glutamin (200 mM Stammlösung)
Optional: Penicillin / Streptomycin Antibiotika (Einsatz die Hälfte der normalen Konzentration)
Neurobasal Growth Medien
- Neurobasal
- B27 (1 / 50 Verdünnung ab Lager lieferbar)
- 2 mM L-Glutamin (200 mM Stammlösung)
- Optional: Penicillin / Streptomycin Antibiotika (Einsatz die Hälfte der normalen Konzentration)
Nachdem die Medien gemacht wird, kann es bei 4 ° C für zwei Wochen aufbewahrt werden. Um die Temperatur und den pH-Wert der Medien vor dem Ausplattieren der Zellen, setzen Medien in Gewebekultur Petrischalen und in einer Gewebekultur-Inkubator für mindestens 1,5 Stunden ruhen.
Neurobasal
Nach einer Flasche Neurobasalmedium geöffnet worden ist, kann es für einen Monat bei 4 ° C aufbewahrt werden in der Dunkelheit. Entsorgen Neurobasal die seit mehr als einem Monat eröffnet wurde sonst das Überleben der Zelle niedriger sein wird.
Hitze-inaktiviertes fötales Rinderserum (HiFBS) oder Pferdeserum (HiHS)
Um Wärme-Inaktivierung FBS oder HS-, Wärme bei 56 ° C im Wasserbad für 30 Minuten. Schwenken Sie die Flasche ca. alle 10 Minuten oder so. (Für Richtigkeit Verwendung einer Flasche von ähnlicher Größe mit Wasser gefüllt. Setzen Sie ein Thermometer in die Wasserflasche, um zu sehen, wenn 56 ° C erreicht ist. Begin Zeitpunkt an dieser Stelle.) Hitzeinaktivierte FBS müssen gegebenenfalls zentrifugiert, um klar ausfällt, und kann aliquotiert und wieder gefroren bei -20 ° C.
L-Glutamin
Immer Tauwetter ein frisches Aliquot von L-Glutamin für jeden Versuch.
B27
Aliquots der B27 kann bei 20 ° C gehalten werden für die langfristige Lagerung oder bei 4 ° C für bis zu 1 Monat.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Wir haben eine Methode zu sezieren und Kultur kommissuralen Neuronen aus embryonalen Ratte Rückenmark beschrieben. Dieses Verfahren wurde routinemäßig in unserem Labor verwendet werden, um zuverlässig vorzubereiten Neuronen, die zellulären und molekularen Mechanismen der Axon Anleitung zu studieren. Für Zellbiologie und Immunchemie Experimenten Dissektion einen Wurf erzeugt genügend Neuronen. Wenn mehr Zellen benötigt werden, können wie in vielen biochemischen Experimenten Dissektion der zwei Würfe erforderlich. Für eine ausgebildete Person kann Dissektion und Dissoziation von ~ 20 Embryonen in weniger als 4 Stunden durchgeführt werden. Längere Zeiträume werden in einem immer schwieriger Präparation von Rückenmark aufgrund von Veränderungen im Gewebe Integrität führen, und kann auch Kompromisse effektive Wiederherstellung lebensfähiger Neuronen.
Bei der Durchführung des Verfahrens zum ersten Mal, Platten-Zellen auf verschiedenen PLL-beschichteten Oberflächen (acid-washed Deckgläser, Gewebekulturschalen) zum Vergleich. Normalerweise sollten die Zellen robust Wert auf PLL-beschichtete Kunststoff-Gewebekulturschalen oder Multi-Well-Platten. Dies kann als eine allgemeine Prüfung für die Lebensfähigkeit der Zellen und Wartung verwendet werden, wenn es Probleme mit der Zelladhäsion und Wachstum auf Deckgläser sind. Der wichtigste Faktor für die schlechte Zelladhäsion an Glas ist die Qualität des Glases und Deckglas Reinigung (Säure-Waschen und Sterilisieren). Dies wird die Zelladhäsion teilweise durch eine Verringerung der PLL-Beschichtung Effizienz beeinflussen. Andere häufige Faktoren gehören schlechte PLL-(Beschichtung zu kurz oder PLL-Lösung zu alt), bakterielle oder Pilzbefall, oder die Nutzung von Medien oder Ergänzungen, die alt oder abgelaufen sind.
Wir haben kommissuralen Neuron Kulturen vorbereitet nach diesem Verfahren in verschiedene Typen von Experimenten, einschließlich Immunchemie, Biochemie und Drehen Assays (Yam et al. 2009) verwendet. Bemerkenswert ist, behalten kommissuralen Neuronen auf PLL-beschichteten Glas überzogen die Fähigkeit zur Führung Signale reagieren, dh, sie werden ihre Wuchsrichtung in Reaktion auf eine angelegte chemotropische Faktor, wie Sonic Hedgehog ändern, die zuvor gezeigt, dass ein Axon Führung Cue werden ( Charron et al, 2003; Okada et al, 2006; Yam et al 2009; Fabre et al, 2010).. Daher ist dieses ein leistungsfähiges System, um die Wirkung von Axon Führung Cues in-vitro-Studie und ermöglicht Experimente, die nicht in vivo möglich.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Diese Arbeit wurde durch Zuschüsse der Canadian Institutes of Health Research (CIHR), der Peter Lougheed Medical Research Foundation, die McGill-Programm in Neuroengineering unterstützt, dem Fonds de Recherche en Santé du Québec (FRSQ) und der Canada Foundation for Innovation (CFI ). Sébastien D. Langlois wurde von einem Master-Training Award aus dem Fonds unterstützten de la recherche en santé du Québec (FRSQ) und durch einen Frederick Banting und Charles Best Canada Graduate Stipendien Master Award der Canadian Institutes of Health Research (CIHR). Wir sind dankbar, dass Jessica MT Pham für die Unterstützung bei Zahlen.
Materials
| Name | Company | Catalog Number | Comments |
| Neurobasal medium, liquid | Invitrogen | 21103-049 | See Recipes and Comments |
| B27 supplement 50X | Invitrogen | 17504 | See Recipes and Comments |
| Poly-L-lysine 0.01% solution | Sigma-Aldrich | P4707 | |
| L-15 medium, powder | Invitrogen | 41300-070 | |
| Trypsin 2.5% (10X) | Invitrogen | 15090-046 | |
| DNAse I, 25000 U/mL | Worthington Biochemical | ||
| MgSO4 | Sigma-Aldrich | M2643 | |
| HBSS, Ca2+/Mg2+-free | Invitrogen | 14170-112 | |
| L-glutamine 200mM, liquid | Invitrogen | 25030-081 | See Recipes and Comments |
Dissection of embryonic rat dorsal spinal cord (see also Table I)
Commissural neuron culture (see also Table I)
|
|||
References
- Charron, F., Stein, E., Jeong, J., McMahon, A. P., Tessier-Lavigne, M. The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell. , 113-1111 (2003).
- Fabre, P., Shimogori, T., Charron, F. Segregation of ipsilateral retinal ganglion cell axons at the optic chiasm requires the Shh Receptor Boc. Journal of Neuroscience. 30, 266-275 (2010).
- Helms, A. W., Johnson, J. E. Progenitors of dorsal commissural interneurons are defined by MATH1 expression. Development. 125, 919-928 (1998).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1, 2406-2415 (2006).
- Okada, A., Charron, F., Morin, S., Shin, D. S., Wong, K., Fabre, P. J., Tessier-Lavigne, M., McConnell, S. K. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature. 444, 369-373 (2006).
- Yam, P. T., Langlois, S. D., Morin, S., Charron, F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 62, 349-362 (2009).

{kind=link}