


Summary
Riktlinjer för datorbaserad strukturella och funktionella karakterisering av protein med hjälp av I-TASSER pipeline beskrivs. Från frågan protein sekvens, är 3D-modeller skapas med hjälp av flera gängning väglinjer och iterativ konstruktionsmontering simuleringar. Funktionella slutsatser som därefter dras baserade på matcher till proteiner med känd struktur och funktioner.
Abstract
Genomsekvenseringsprojekt projekt har chiffrerat miljontals protein sekvens, som kräver kunskap om deras struktur och funktion för att förbättra förståelsen för deras biologiska roll. Även experimentella metoder kan ge detaljerad information för en liten bråkdel av dessa proteiner är datormodellering behövs för majoriteten av proteinmolekyler som experimentellt är uncharacterized. I-TASSER server är en on-line arbetsbänk för högupplöst modellering av proteiners struktur och funktion. Givet ett protein sekvens, innehåller en typisk effekt från I-TASSER server sekundär struktur förutsägelse, förutspådde lösningsmedel tillgängligheten av varje rester, homolog mall proteiner detekteras genom att trä-och väglinjer struktur, upp till fem fullängds tertiär strukturella modeller, och struktur-baserade funktionella anteckningar för enzym klassificering, Gene Ontology termer och protein-ligand bindningsställen. Alla prognoser är märkta med ett självförtroende värdering somberättar hur noggranna förutsägelser utan att känna till experimentella data. För att underlätta särskilda önskemål av slutanvändare ger servern kanalerna att acceptera användardefinierade mellan rester avstånd och kontakt kartor att interaktivt ändra I-TASSER modellering, utan också tillåter användare att ange några proteiner som mall, eller att utesluta någon mall proteiner under simuleringarna struktur montering. Den strukturella informationen kan samlas in av användare baserat på experimentella bevis eller biologiska insikter i syfte att förbättra kvaliteten på I-TASSER förutsägelser. Servern bedömdes som de bästa programmen för proteiners struktur och förutsägelser funktion i den senaste hela gemenskapen Casp experiment. Det finns för närvarande> 20.000 registrerade forskare från över 100 länder som använder online-I-TASSER server.
Protocol
Metod översikt
Efter sekvensen till struktur-till-funktion paradigmet, innebär I-TASSER förfarande 1-4 för struktur och funktion modellering fyra på varandra följande steg: (a) mall identifiering av LOMETS 5, (b) fragment struktur återmontering av replika- utbyte Monte Carlo-simuleringar 6, (c) atomär nivå struktur förfining med REMO 7 och FG-MD 8, och (d) struktur-baserad funktion tolkningar med hjälp av kofaktor 9.
Mall identifiering: För en fråga sekvens som lämnats av användaren, är sekvensen first träs genom ett representativt preliminära struktur bibliotek genom en lokalt installerad LOMETS meta-threading server. Threading är en sekvens-struktur anpassning förfarande som används för att identifiera mall proteiner som kan ha liknande struktur eller innehåller liknande strukturella motiv som frågan protein. För att öka täckningen av homologa Templåt upptäckter kombinerar LOMETS flera state-of-the-art algoritmer som täcker olika gäng metoder. Eftersom olika gäng program har olika poängsystem och känslighet anpassning, är kvaliteten på den genererade trådning anpassningar från varje gäng program utvärderas av normaliserade Z-score, som definieras som: 
där Z-score är poängen i standardavvikelse enheter i förhållande till den statistiska medelvärdet av alla anpassningar som genereras av programmet, och Z 0 är ett program-specifik Z-score cutoff bestäms utifrån den storskaliga gäng benchmark-tester 5 att skilja "bra "och" dåliga "mallar. En mall med ett högt Z-score innebär att den övre mallar har en anpassning poäng betydligt högre än de flesta andra mallar, som oftast innebär att justeringen motsvarar en bra modell. Om de flesta av de bästa trådning mallar har hiGH normaliserade Z-värdena är noggrannhet i det slutliga I-TASSER modell vanligtvis hög. Men om proteinet är stort och täckning gängning anpassningar är begränsad till en liten region i frågan protein, inte en hög normaliserade Z-score inte nödvändigtvis betyda en stor modell noggrannhet för full längd modell. Topp två gäng väglinjer från varje gäng program samlas in och används för nästa steg i strukturen montering.
Iterativ simulering struktur montering: Efter gängning förfarande, frågan sekvensen delas upp i trä anpassad och icke-justerade regioner. Kontinuerlig fragment i gängning anpassning är censurerade från mallar och används direkt för strukturen montering, medan den icke-justerade slingan regioner byggs av ab initio modellering. Strukturen montering proceduren utförs på ett galler system som styrs av repliken utbyte Monte Carlo-simuleringar 6. I-TASSER kraftfält inkluderar vätgas-boENDE interaktioner 10, kunskapsbaserade statistiska gäller energi kommer från kända proteinstrukturer i det preliminära budgetförslaget 11, sekvens-baserade kontakt förutsägelser från SVMSEQ 12, och rumsliga begränsningar som samlats in från LOMETS 5 gäng mallar. Den konformationsanalys lockbete genereras i låg temperatur repliker under simuleringarna är grupperade i SPICKER 13 för att identifiera strukturer av låg fri energi stater. Cluster centroids av de kluster erhålls som genomsnittet av de 3D-koordinater för alla klustrade strukturella vettar och används för den slutliga modellen generationen. Simuleringen och klustring procedur upprepas två gånger för att ta bort steriska sammandrabbningar och ytterligare förfina den globala topologi.
Atomär nivå modellbygge och förfining: Klustret centroids erhållits efter SPICKER klustring reduceras protein modeller (varje rest representeras av dess C α och sidokedjan masscentrum) och have begränsade biologiska ansökan. Byggandet av full-atommodell från reducerade modellerna görs i två steg. I det första steget är REMO 7 används för att konstruera full atommodeller från C-alfa-spår genom att optimera H-band nätverk. I det andra steget, är REMO full atommodeller vidareutvecklats av FG-MD 14, vilket förbättrar vinklarna ryggraden vridning, längder band och sidokedjan riktlinjer rotamer genom molekylära dynamiska simuleringar, som styrs av de strukturella fragment sökte från preliminära strukturer av TM-align. Den FG-MD raffinerade modeller används som slutlig modeller för tertiärstruktur förutsägelser av I-TASSER.
Kvaliteten på den genererade modeller beräknas utifrån ett förtroende poäng (C-poäng), vilket definieras baseras på Z-score av LOMETS threading anpassningar och konvergens av I-TASSER simuleringar, matematiskt formuleras som: 
där
C-värdering har ett starkt samband med kvaliteten på I-TASSER modeller. Genom att kombinera C-score och protein längd, kan riktigheten i den första I-TASSER modeller skattas med ett medelfel på 0,08 för TM-poäng och 2 A för RMSD 15. I allmänhet modeller med C-score> - är 1,5 förväntas ha en korrekt fold. Här RMSD och TM-poäng är båda kända åtgärder av topologiska likhet mellan modell och infödda struktur. TM-poäng VALUES-serien i [0, 1], där en högre poäng indikerar en bättre struktur match 16,17. Men för lägre rankade modellerna (dvs 2: a -5: e modeller), är korrelationen av C-poäng med TM-poäng och RMSD mycket svagare (~ 0,5), och kan inte användas för tillförlitlig uppskattning av absoluta modell kvalitet.
Är första modell alltid är den bästa modellen i I-TASSER simuleringar? Svaret på denna fråga beror på målet typ. För enkla mål, är den första modellen oftast den bästa modellen och dess C-värdering är vanligtvis mycket högre än resten av modellerna. Men för hårda mål, där trä inte har betydande mall träffar, är den första modellen inte nödvändigtvis den bästa modellen och I-TASSER faktiskt har svårt att välja ut de bästa mallen och modeller. Det rekommenderas därför att analysera alla fem modeller för hårda mål och väljer dem utifrån experimentell information och biologisk kunskap.
Funktion Predictions: I det sista steget, är slutgiltiga 3D-modeller som genererats från FG-MD användas för att förutsäga tre aspekter av proteiners funktion, nämligen: en) Enzyme kommissionen (EC) nummer 18 och (b) Gene Ontology (GO) 19 termer och ( c) bindningsställen för liten molekyl ligander. För alla de tre aspekterna, är funktionella tolkningar genereras med kofaktor, vilket är en ny metod för att förutsäga proteinets funktion bygger på globala och lokala likhet med mallen proteiner i det preliminära budgetförslaget med känd struktur och funktioner. För det första är den globala topologi förutspådde modellerna matchas mot funktionell mall bibliotek med hjälp av strukturell anpassning programmet TM-align 20. Därefter är en uppsättning proteiner som mest liknar målet modellerna väljs från biblioteket baserat på deras globala struktur likhet, och en omfattande lokal sökning utförs för att identifiera struktur och ordning likheten nära den aktiva / bindningsställe regionen. Den resulterande globala och lokala likhet poäng används för att rangordnamall proteiner (funktionell homologer) och överför Noten (EG siffror och Gene Ontology 19 termer) baserat på toppen poänggivande träffar. Likaså är ligand bindningsställe rester och ligandbindande läget härledas baserat på den lokala anpassningen av frågan med känd ligand bindningsställe rester i toppen mallar poäng funktion 9.
Kvaliteten på funktion (EG och GO sikt) förutsägelse i I-TASSER utvärderas baseras på funktionella homologi poäng (FH-poäng), som är ett mått på globala och lokala likhet mellan sökfråga och mallen, och definieras som: 
där C-poäng är en uppskattning av kvaliteten på förutspådde modellen enligt Ekv. (2), TM-poäng mäter globala strukturell likhet mellan modell och proteiner mall, RMSD Ali är RMSD mellan modell och mallen struktur i strukturellt anpassade regionen från TM-align 20, Cov representerar täckning av den strukturella anpassningen (dvs. förhållandet mellan strukturellt anpassade rester dividerat med frågan längd), ID-Ali är sekvensen identitet i TM-align justering. Den beräknade förtroende poäng för EG-nummer prognoser innehåller även en term för utvärdering av verksamma site match (ACM) mellan fråga och mall inom ett definierat lokala regionen, beräknas som: 
där N T betecknar antalet mallen rester som finns inom det lokala området, är N Ali numret på den linje query-mall rester par, är D II C α avståndet mellan I th par anpassade rester, är d 0 = 3.0 A avståndet cutoff, M II BLOSUM poäng mellan ed par anpassade rester. I allmänhet är det FH-poäng i intervallet [0, 5] och ACM värdering är mellan [0, 2], Där högre poäng indikerar mer självsäker funktionell uppdrag. ACM poäng används också för att utvärdera den lokala strukturen och sekvensen likheten nära ligand-bindningsställen som kallas BS-poäng.
1. Inlämning av protein sekvens
- Besök I-TASSER webbsida på http://zhanglab.ccmb.med.umich.edu/I-TASSER att börja med uppbyggnad och funktion modellering experiment.
- Kopiera och klistra in den aminosyrasekvens i de medföljande form eller direkt ladda upp den från din dator genom att klicka på "Bläddra"-knappen. I-TASSER servern accepterar för närvarande sekvenser med upp till 1500 rester. Proteiner är längre än 1500 restprodukter är oftast flera domän proteiner, och rekommenderas att delas upp i enskilda domäner innan de lämnar in till I-TASSER.
- Ge din e-postadress (obligatoriskt) och ett namn för jobbet (tillval).
- Användare kan välja att ange yttre inter-residue kontakt / distans begränsningar, add-in ytterligare en mall eller utesluta vissa mall proteiner under uppbyggnad modellering processen. Läs mer om hur du använder dessa alternativ i "Discussion"-avsnittet.
- För att lämna sekvensen, klicka på "Kör I-TASSER"-knappen. Webbläsaren kommer att hänvisas till en bekräftelse sida som visar användaren specificerade uppgifter, jobb identifiering (Job-ID-nummer) och en länk till en webbsida där resultaten kommer att deponeras efter avslutad jobb. Användare kan bokmärke denna länk eller anteckna numret jobbet identifiering för framtida referens.
2. Tillgång till resultat
- Kontrollera status för dina lämnats jobb genom att besöka I-TASSER kö sidan på http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . Klicka på fliken Sök och använder numret Job ID eller frågan sekvensen för att söka din in jobb.
- Efter struktur och funktion MOdeling är klar kommer ett meddelande via e-post som innehåller bild av den förväntade strukturer och en web-länk skickas till dig. Klicka på denna länk eller öppna länken bokmärkt i steg 1,5 för att visa och ladda ner resultaten.
3. Sekundär struktur och vätska förutsägelser tillgänglighet
- Kontrollera Fasta formaterade frågan ordningsföljd som visas överst på resultatsidan. Om ytterligare återhållsamhet / mall angavs under sekvens underkastelse, en länk till den webbsida som visar användardefinierade information kan också ses (Figur 1A).
- Undersök sekundär struktur förutsägelse visas som: Alpha Helix (H), beta-strängen (S) eller spole (C) och förtroende poäng förutsägelse (0 = låg, 9 = hög) för varje rester. Leta efter region med långa sträckor av regelbundna sekundär struktur (H eller S) förutsägelser, att uppskatta core-regionen i protein. Strukturella klass av proteiner kan också analyseras utifrån fördelning av sekundära strukturer element. Alså länge regioner spole delar i proteinet brukar tyda ostrukturerade / oordnad regioner.
- Visa förutspådde lösningsmedlet tillgänglighet (Figur 1C) att se till begravdes och vätska utsatta regionerna i frågan. Värden för förutspådde lösningsmedel tillgängligheten varierar från 0 (begravd rester) till 9 (exponerade rester). Region innehåller mestadels begravd restprodukter kan användas för att beskriva kärnan regionen i protein, medan regioner med lösningsmedel exponerade och hydrofila rester är potentiella hydrering / funktionella webbplatser.
4. Tertiärstruktur förutsägelser
- Bläddra nedåt för att visa den förväntade tertiära strukturer frågan protein, som visas i en interaktiv Jmol applet (figur 2). Vänsterklicka på appleten att ändra utseendet på displayen struktur, zooma in specifik region, välja ut vissa rester typer i den förutsagda modellen eller beräkna mellan rester avstånd.
- Analysera modeller för förekomsten av långa ostrukturerade regioner. Dessa regions motsvarar oftast störda regioner i protein eller visar brist på mall anpassning. Dessa regioner har i allmänhet låg modellering noggrannhet och ta bort dessa regioner under modellering från N & C-terminus regionen kommer att förbättra modelleringen noggrannhet.
- Ladda ner det preliminära budgetförslaget formaterade struktur filer av modellen genom att klicka på "Ladda ner Model" länkar. Du kan öppna dessa filer i en molekylär visualiseringsprogram (t.ex. Pymol, Rasmol etc.) för vidare analys av de strukturella egenskaper.
- Analysera förtroendet poäng (C-poäng) av struktur modellering för att uppskatta kvaliteten på förutspådde strukturer. C-poäng (Eq. 2) värden är vanligtvis i intervallet [-5, 2], där en högre poäng avspeglar en modell av bättre kvalitet. Den beräknade TM-poäng och RMSD av första modellen visas som "Beräknad noggrannhet Modell 1". För långa proteiner, rekommenderas att utvärdera modellen kvalitet baserat på TM-värdering, som TM-poäng är mer känslig för topologiska förändringar än RMSD. < li> Klicka på "mer om C-poäng" för att analysera C-poäng, kluster storlek och kluster täthet av alla modeller. Beräknad TM-poäng och RMSD presenteras endast för första I-TASSER modell, eftersom C-poäng lägre rankade modeller som inte är starkt korrelerad med TM-poäng eller RMSD. Kvaliteten på lägre rankade modellerna kan delvis bedömas utifrån sina kluster densitet och kluster storlek i förhållande till den första modellen, där modeller från större kluster och högre densitet är i genomsnitt närmare infödda struktur.
- Lågt C-score prognoser indikerar vanligtvis en låg noggrannhet förutsägelse. I de flesta sådana fall saknar frågan proteinet en bra mall i biblioteket och har en storlek bortom utbud av ab initio modellering (dvs> 120 rester). I dessa fall kan användare söka efter ytterligare rumsliga begränsningar och använda dem för att förbättra I-TASSER modellering (se diskussion avsnitt). Det är också uppmuntras att lämna in sekvenser till vår QUARK server (QUARK / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) för en rent från början modellering om proteinet storlek ligger under 200 rester.
5. LOMETS mål mall anpassning
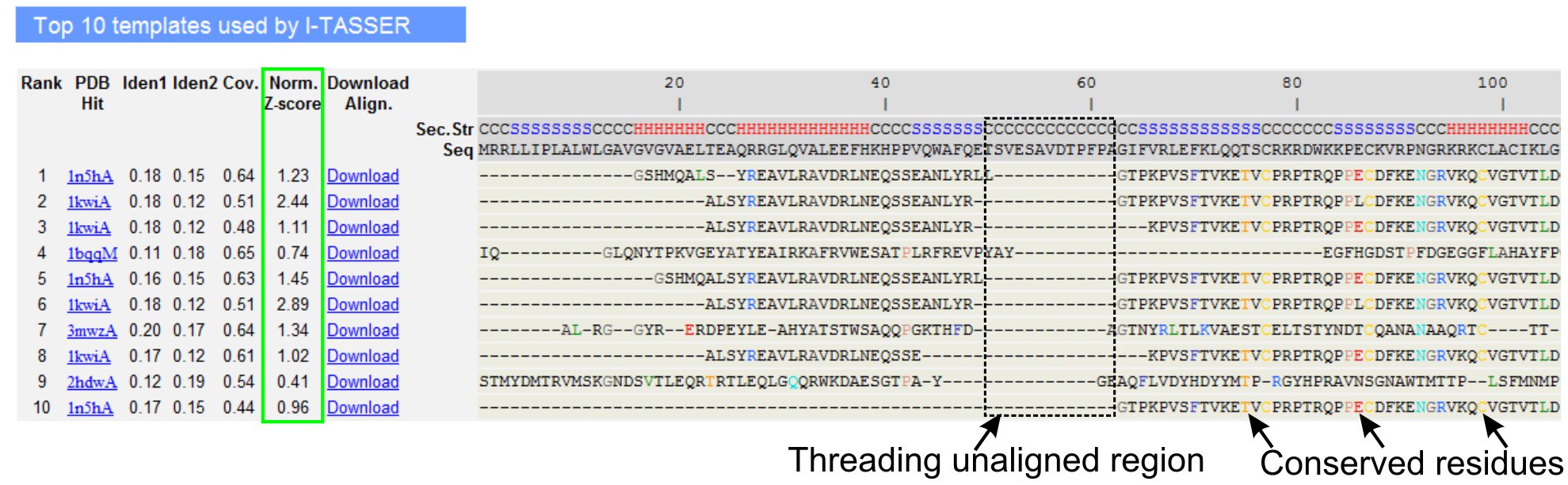
- Bläddra ner för att analysera de tio bästa gäng mallar av frågan protein, som identifierats av LOMETS gäng program (Figur 3). Visa normaliserade Z-score (Eq. 1), som visas i "norm. Z-score "kolumnen för att analysera kvaliteten på trådning väglinjer. Anpassningar med en normaliserad Z-score> 1 avspeglar en säker anpassning och troligen har samma vik som frågan protein.
- Analysera sekvensen identitet i den gängning alliansfria regionen (kolumn "Iden. 1") och för hela kedjan (kolumn "Iden. 2 ') för att bedöma homologi mellan frågan och de proteiner som mall. Hög sekvens identitet är en indikator på evolutionära släktskap mellan frågan och proteiner mall.
- Visa gängning linje rester som visas i färg för att visuellt identifiera nackdelarerved överskott / motiv i frågan och de proteiner som mall. En högre sekvens identitet i gängning linje-regionen, jämfört med hel-kedjan anpassning visar också förekomsten av bevarade strukturella motiv / domäner i frågan.
- Bedöm täckning av trådning anpassning genom att titta på "Cov." kolumn och inspekterar anpassning. Om täckningen av de bästa anpassningarna är låg och begränsad till endast en liten region i frågan protein eller frånvarande under en längre del av frågan sekvens, då frågan protein innehåller oftast mer än en domän och det rekommenderas att dela sekvensen och modell domäner individuellt (Figur 3).
- Ladda ner det preliminära budgetförslaget formaterad sekvens-struktur anpassning filer genom att klicka på "Ladda ner Rikta" länkar. Dessa anpassning Filen kan öppnas i en molekylär visualisering program som anges på material sektionen, och kan även användas för att lägga till ytterligare begränsningar under uppbyggnad modellering (steg 1,4).
6.Strukturella analoger i det preliminära budgetförslaget
- Visa nästa tabell (Figur 4) av resultatet sidan för att avgöra de tio bästa strukturella analoger av den förväntade första modellen, som identifierats av den strukturella anpassningen programmet TM-align 20. En TM-poäng> 0,5 indikerar att de upptäckta analoga och modell har en liknande topologi och kan användas för att bestämma den strukturella klassen / protein familj frågan protein 16, medan de med TM-score <0,3 innebär en slumpmässig struktur likhet.
- Analysera sekvensen identitet och RMSD i strukturellt anpassade regionen visas i "iDEN ett" och "RMSD en" kolumnerna för att bedöma bevarandet av rumsliga motiv i modellen och de strukturella analog. Inspektera färgade och anpassade rester par i justeringen att identifiera dessa strukturellt bevarade rester och motiv.
- Klicka på det preliminära budgetförslaget som visas i "preliminära Hit kolumnen att besöka RCSB hemsida och läsa mer om deras strukturella klassificering (Scop, CATH och PFAM) och funktionella uppgifter (EG-nummer, tillhörande GO termer och bunden ligand).
7. Funktion förutsägelse med hjälp kofaktor
- Bläddra nedåt i resultatsidan för att analysera funktionella tolkningar för frågan protein. Protein funktioner är uppräknade i tre sammanhang tabeller, visa: Enzyme kommissionen (EC) nummer, Gene Ontology (GO) termer och ligandbindande webbplatser.
- Visa "TM-poäng", "RMSD en", "iDEN ett" och "Cov." kolumnerna i varje tabell för att analysera parametrar för globala struktur likheter och bevarande av rumsliga mönster mellan modell och identifierade funktionella homologer (mallar).
8. Enzym kommissionen antalet prognos
- Se de fem potentiella enzym homologer av fråga protein som visas i "Predicted EG-nummer" tabellen (Figur 5). Den konfidensnivå på EG-nummer förutsägelse med hjälp av dessa mallar visas i "EG-Score" kolumnen. Baserat på benchmarking analys 23, funktionella likheten (första 3 siffrorna i EG-nummer) mellan frågan och mall protein tillförlitligt kan tolkas med hjälp av EG-poäng> 1,1.
- Leta efter konsensus funktion (EG-nummer) bland de mallar som har liknande gånger (dvs TM-poäng> 0,5) som frågan protein. Om flera mallar har samma EG-nummer och EG-poäng> 1,1 är konfidensnivån för förutsägelse mycket hög. Men om EG-Score är hög men det finns en brist på samsyn bland de identifierade träffar, då förutsägelse blir mindre tillförlitliga och användare rekommenderas att rådfråga GO sikt förutsägelser.
- Klicka på länken som finns på EG-nummer för att besöka ExPASy Enzyme databasen och analysera funktion, däribland reaktion som katalyseras, co-faktor krav och metaboliska väg, av mallen protein i detalj.
9. Gene Ontology (GO) sikt förutsägelser
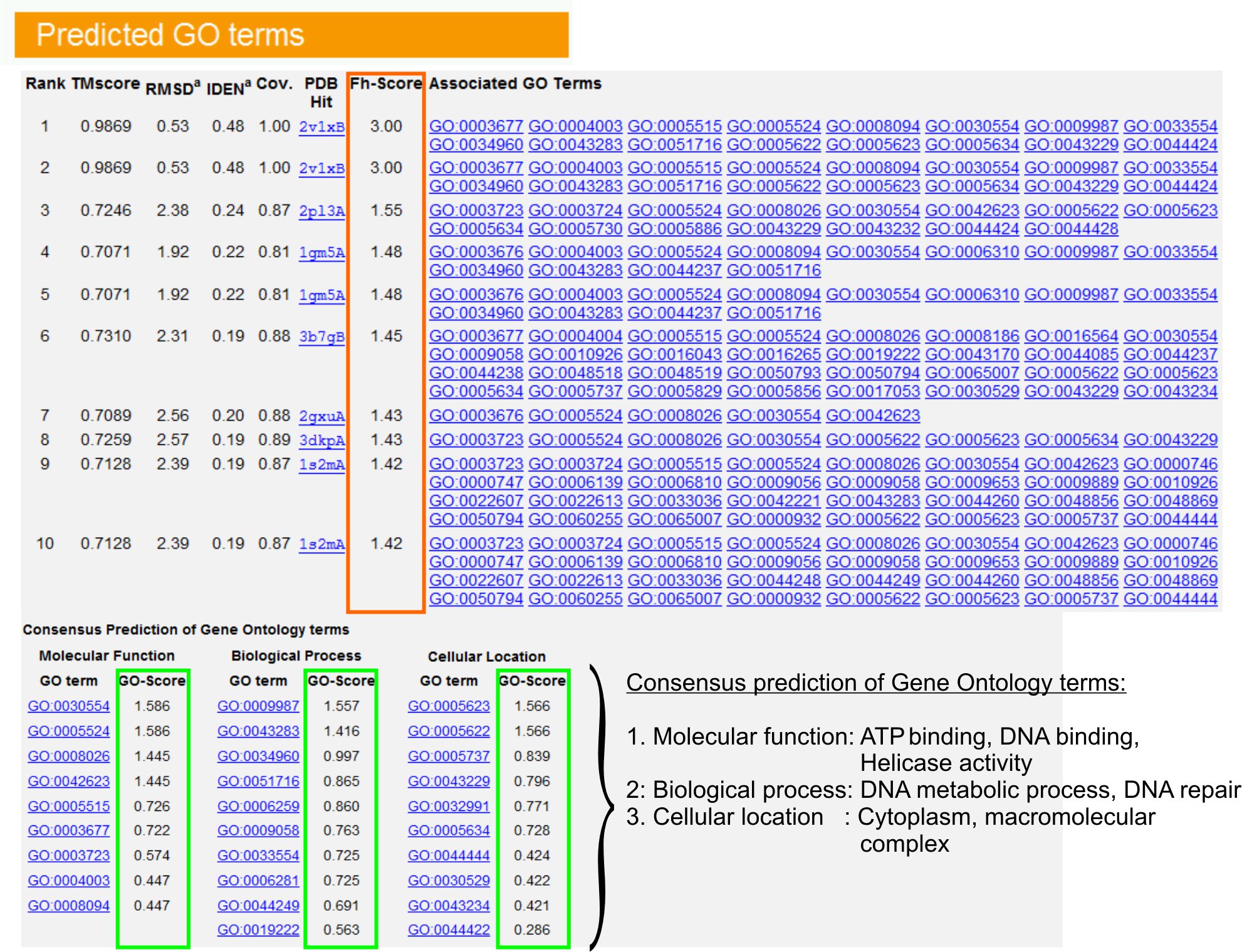
- Visa "förutspådde GO termer" tabell (Fig.ure 6) för att identifiera de tio homologer av fråga protein i det preliminära budgetförslaget biblioteket, kommenterad med Gene Ontology (GO) termer. Varje protein är vanligtvis förknippas med flera GO termer, som beskriver dess molekylära funktioner (MF), biologiska processer (BP) och cellulära komponenter (CC). Klicka på varje termin för att besöka Amigo webbplatsen och analysera dess definition och härstamning.
- Analysera FH-poäng (Functional homologi poäng) kolumn för att få tillgång till funktionella likheten mellan sökfrågan och proteiner mall och uppskatta konfidensnivå överföra funktionella anteckningar från dessa proteiner. I vår jämförande studie 23, skulle 50% av de infödda GO termer korrekt identifieras de identifierade första mallen med en FH-poäng cutoff på 0,8, med en total noggrannhet på 56%.
- Visa "Consensus prognoser om GO termer" bord att analysera bifall av funktion mellan mallarna. Dessa gemensamma funktioner används för att förutsäga GO termer (MF, BP och CC) av fråganprotein och bedöma konfidensnivå (GO-poäng) av GO sikt förutsägelser. Grundad på benchmarking tester 23, är den bästa falskt positiva och falskt negativa resultat erhålls för förutsägelser med GO-poäng cutoff = 0,5, med minskande täckning av förutsägelse på djupare ontologi nivåer.
10. Protein-ligand bindningsställe förutsägelser
- Bläddra ned till botten av sidan för att se de tio bästa ligand bindningsställe prognoser för frågan protein. Förutspådde bindningsställen rangordnas utifrån antalet förutspådde ligand konformationer som delar gemensamma bindande fickan. Den bästa identifierade bindningsställe redan visas i Jmol applet. Klicka på knapparna för att analysera andra förutsägelser och visualisera rester ligand interagerande.
- Analysera BS-poäng kolumn utvärdera lokala likhet mellan modell och mallens bindningsställe. Baserat på riktmärket 9, BS-poäng> 1,1 indikerar hög sekvens och struktur similarity nära förutspådde bindningsstället i modell och känd bindningsställe i mallen.
- Ladda ner det preliminära budgetförslaget formaterad struktur-fil av komplexet genom att klicka på "Download" länken. Användare kan öppna dessa filer i en molekylär visualisering program och interaktivt visa förutspådde bindningsstället och ligand-protein interaktioner på sin lokala dator.
11. Representativa resultat

Figur 1 Ett utdrag av I-TASSER resultatsidan med (A) Fasta formaterad fråga sekvens;. (B) förutspådde sekundär struktur och tillhörande poäng förtroende, och (C) förutspådde lösningsmedel tillgänglighet rester. Analyseras kärna regionen och potentiella hydrering plats i frågan är markerade i cyan och röda rektanglar, respektive.

Figur 2.

Figur 3. Ett exempel på I-TASSER resultat sida som visar de tio bästa identifierat gäng mallar och anpassningar av LOMETS 5 trådning program. Kvaliteten på gängning anpassningar utvärderas baserat på normaliserade Z-score (markerad i grönt), där ett värde> 1 avspeglar en säker linje. Alliansfria rester i den mall som är identiska med motsvarande fråga rester markeras med färg för att indikera förekomst av bevarade rester / motiv, medan en brist på anpassning i de flesta av topp mallar indikerar närvaro av flera domäner i frågan protein och den icke-justerade återstoden motsvarar regioner domän länkaren. Klicka här för att se fullstor version av figur 3.

Figur 4. Ett exempel på resultat sida som visar de tio bästa identifierade strukturella analoger och strukturella anpassningar, som identifierats av TM-align 20 strukturell anpassning programmet. Den rangordning av analoger som visas i är baserat på TM-poäng (markeras i blått) av den strukturella anpassningen. En TM-poäng> 0,5 indikerar att två jämfört strukturer som har en liknande topologi, medan en TM-score <0,3 innebär en likhet mellan två slumpmässiga strukturer. Strukturellt linje rester par är markerade i färg baserat på deras aminosyror egendom, medan den icke-justerade regioner markeras med "-".ove.com/files/ftp_upload/3259/3259fig4large.jpg "> Klicka här för att se fullstor version av figur 4.

Figur 5. Ett exempel på I-TASSER resultat sida som visar identifierade enzym homologer av frågan protein i det preliminära budgetförslaget biblioteket. Den konfidensnivå på EG-nummer förutsägelse är analyseras utifrån EG-score (markerad i grönt), där EG-score> 1,1 indikerar funktionella likheten (samma första 3 siffrorna i EG-nummer) mellan fråga och mall protein.

Figur 6. Ett exempel på I-TASSER resultat sida som visar GO sikt prognoser för frågan protein. Funktionell homologer för frågan protein i Gene Ontology mallbiblioteket rangordnas utifrån deras FH-poäng (i orange rektangel). Gemensamma funktionella egenskaper från dessa top-poänggivande träffar härrör att generera åt den slutliga GO sikt prognoser för frågan protein. Kvaliteten på den förväntade GO termer beräknas baserat på GO-score (visas i grönt), där en GO-poäng> 0,5 indikerar en tillförlitlig förutsägelse. Klicka här för att se fullstor version av figur 6.

Figur 7. Ett exempel på I-TASSER resultat sida som visar de tio bästa protein ligand bindningsställe förutsägelser med kofaktor 9 algoritm. Rangordningen av den förväntade bindningsställen är baserat på antalet förutspådde ligand konformationer som delar gemensamma bindande fickan i frågan. BS-score (markerade i rött) är ett mått på lokal ordning och struktur likheten mellan den förväntade och mallens bindningsställe, och är användbart för att analysera bevarandet av bindningsställe fickor.
les/ftp_upload/3259/3259fig8.jpg "/>
Figur 8. Ett exempel på externa återhållsamhet filer som används för att för att ange rester rester kontakt / begränsningar avstånd.

Figur 9. Exempel på återhållsamhet filer som används för att ange en mall protein till I-TASSER server. Användare kan ange frågan-template anpassning antingen (A) Fasta-format, eller (b) 3D-format.

Figur 10. Ett exempel på fil som används för att utesluta mall under I-TASSER struktur modellering förfarande. Den första kolumnen innehåller det preliminära budgetförslaget ID mallen proteiner som ska uteslutas. Den andra kolumnen används för att ange cutoff sekvensen identitet som kommer att användas för andra liknande mallar i mallen bibliotek.
Discussion
Protokollet presenteras ovan är en allmän riktlinje för struktur och funktion modellering med hjälp av I-TASSER server. Även om detta automatiserad procedur fungerar mycket bra för de flesta av de proteiner, mänskliga ingrepp hjälper ofta att avsevärt förbättra modelleringen noggrannhet, särskilt för de proteiner som saknar nära mallar i det preliminära budgetförslaget biblioteket. Användare kan ingripa vid I-TASSER modellering på följande sätt: (a) uppdelning av multi-domän proteiner, (b) tillhandahålla yttre begränsningar för att förbättra strukturen församlingen, och (c) att ta bort mallar under modellering.
Dela upp multi-domain protein:
Många långa proteinsekvenser innehåller ofta flera domäner uppbundna genom flexibla länkare regioner, vilket gör att deras struktur belysning svårt med både experimentella och beräkningsteknik. Men som domäner är självständigt fällbara enheter och kan utföra olika molekylära funktionen, det ärönskvärt att dela upp långa multi-domän proteiner och modell varje domän separat. Modellering domäner individuellt kommer inte bara att påskynda förutsägelse processen, men ökar också kvaliteten i query-mall anpassning, vilket resulterar i mer tillförlitlig struktur och förutsägelser funktion.
Domain gränser i proteinsekvenser kan förutsägas med hjälp av fritt tillgängliga externa online-program som NCBI CDD 24, PFAM 25 eller InterProScan 26. Dessutom, om LOMETS gäng anpassningar finns tillgängliga för frågan proteinet kan domängränser placeras genom att visuellt identifiera långa sträckor av icke-justerade rester i toppen gäng mallar (se steg 5.4). Dessa icke-justerade regioner motsvarar mestadels till områden domän länkaren. Om flera domäner mallar finns redan i mallen preliminära biblioteket med alla de anpassade frågan domäner, då frågan proteinet kan modelleras som full längd.
Ge externa begränsningar
A. Ange kontakt / distans begränsningar
Experimentellt kännetecknas mellan rester kontakter / avstånd, t.ex. från NMR ellerförnätning experiment, kan specificeras genom att ladda upp en återhållsamhet fil. Ett exempel på filen visas i Figur 8, där kolumn 1 anger vilken typ av återhållsamhet, dvs "DIST" eller "kontakt". Vid distansavtal fasthållningsanordningar (DIST), kolumnerna 2 och 4 innehåller rester position (i, j), kolumnerna 3 och 5 innehåller atom-typer i återstoden och kolumn 6 anger avståndet mellan de två angivna atomer. För kontakt begränsningar (kontakt), kolumnerna 2 och 3 innehåller de positioner (i, j) av restprodukter som bör vara i kontakt. Avståndet mellan sidokedjor centrum för dessa kontakta rester par avgörs baserat på observerade avstånd i kända strukturer i det preliminära budgetförslaget. I-TASSER kommer att försöka dra dessa atom par nära visst avstånd under simuleringarna strukturen förfining.
B. Ange en mall proteinstruktur
LOMETS gängning program använder en representant preliminära bibliotek för att hitta rimliga veck för frågan protein. Även med hjälp av ett bibliotek representant struktur bidrar till att minska den tid som krävs för att beräkna sekvens-struktur väglinjer, är det möjligt att en bra mall protein missas i biblioteket eller mallen kanske inte har identifierats av LOMETS gäng program, även om det är som finns i biblioteket. I dessa fall bör användaren ange önskat proteinstruktur som mall.
För att ange proteinstruktur som en extra mall kan användare ladda upp antingen ett preliminärt budgetförslag formaterad struktur fil eller ange det preliminära budgetförslaget ID för en deponeras proteinstruktur i det preliminära budgetförslaget bibliotek. I-TASSER kommer att generera frågan-template anpassning med hjälp av UPPBJUDA programmet 23 och kommer att samlas rumsliga begränsningar från såväl angivna användaren mallen och LOMETS mallar för att styra simuleringen struktur montering. Eftersom noggrannheten i LOMETS begränsningar är olika för olika mål, är vikten av LOMETS begränsningar starkare i lätt (homolog) TArgets än i hårda (icke-homologa) mål, som har systematiskt inställda i vår jämförelseindex träning.
Användarna kan även ange sin egen query-mall anpassningar. Servern tar emot anpassning i två format: den Fasta format (Figur 9A) och 3D-format (Figur 9B). Den Fasta format är standard och beskrivs på http://zhanglab. ccmb.med.umich.edu / Fasta / . 3D-formatet liknar den vanliga preliminära budgetförslaget format ( http://www.wwpdb.org/documentation/format32/sect9.html ), men två ytterligare kolumner från mallarna läggs till ATOM poster (se Figur 9B):
Kolumnerna 1-30: Atom (C-alfa bara) och rester namn för frågan sekvensen.
Kolumnerna 31-54: Koordinater för C-alfa atomer i frågan kopierade från motsvarande atomer i mallen.
Kolumnerna 55-59: motsvarande resthalter siffra i mallen baseras på anpassning
Kolumnerna 60-64: motsvarande resthalter namn i mallen
Uteslut mallar proteiner
Proteiner är flexibla molekyler och kan anta flera konformationsanalys stater att ändra deras biologiska aktivitet. Till exempel har strukturer av många proteinkinaser och membranproteiner lösts i både aktiva och inaktiva konformation. Också närvaron eller frånvaron av bundna ligand kan orsaka stora strukturella rörelser. Medan alla konformationsändringar stater i mallen är lika för gängning program, är det önskvärt att modellera frågan med mallar i endast en viss stat. Ett nytt alternativ på servern tillåter användaren att utesluta mall proteiner under uppbyggnad modellering. Denna funktion skulle också möjliggöra för användaren att välja den homologi nivån av mallar som ska användas för modellering. Användare kan utesluta mall proteiner frOm I-TASSER bibliotek genom att:
A. Ange ett cutoff sekvens identitet
Användarna kan använda denna möjlighet att utesluta homologa proteiner från I-TASSER mall bibliotek. Den homologi är inställd bygger på sekvensen identitet cutoff, dvs antal identiska rester mellan frågan och mallen protein dividerat med sekvens längden på frågan sekvensen. Till exempel, om användaren skriver i "70%" i formuläret, alla mallar proteiner som har en sekvens identitet> 70% till frågan proteinet I-kommer att uteslutas från I-TASSER mall bibliotek.
B. utesluta viss mall proteiner
Specifik mall proteiner kan uteslutas från I-TASSER mallbibliotek genom att ladda upp en lista som innehåller preliminära ID av de strukturer som ska undantas. Ett exempel på fil visas i Figur 10. Eftersom samma protein kan existera som flera poster i det preliminära budgetförslaget biblioteket, I-TASSER SEtill floden som standard utesluter den angivna mallar (i kolumn1) samt alla andra mallar från biblioteket som har en identitet> 90% till angiven mallar. Användarna kan även ange en annan identitet cutoff, t.ex. 70%, där alla mallar med identitet> 70% till angiven mall proteiner kommer att uteslutas.
Disclosures
Inga intressekonflikter deklareras.
Acknowledgments
Projektet stöds delvis av Alfred P. Sloan Foundation, NSF Career Award (DBI 1.027.394) och National Institute of General Medical Sciences (GM083107, GM084222).
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H.
- Finn, R. D.
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}