

Summary
Herein we describe simple methods for the preparation of vesicles, the encapsulation of transcription and translation machinery, and the monitoring of protein production. The resulting cell-free systems can be used as a starting point from which to build increasingly complex cellular mimics.
Abstract
As interest shifts from individual molecules to systems of molecules, an increasing number of laboratories have sought to build from the bottom up cellular mimics that better represent the complexity of cellular life. To date there are a number of paths that could be taken to build compartmentalized cellular mimics, including the exploitation of water-in-oil emulsions, microfluidic devices, and vesicles. Each of the available options has specific advantages and disadvantages. For example, water-in-oil emulsions give high encapsulation efficiency but do not mimic well the permeability barrier of living cells. The primary advantage of the methods described herein is that they are all easy and cheap to implement. Transcription-translation machinery is encapsulated inside of phospholipid vesicles through a process that exploits common instrumentation, such as a centrifugal evaporator and an extruder. Reactions are monitored by fluorescence spectroscopy. The protocols can be adapted for recombinant protein expression, the construction of cellular mimics, the exploration of the minimum requirements for cellular life, or the assembly of genetic circuitry.
Introduction
Cell-free, in vitro transcription-translation reactions and the generation of vesicles from synthetic lipids are nothing new. However, combining the two into a cellular mimic is significantly more challenging1-6. E. coli cell extracts with or without T7 RNA polymerase can be used as a source of transcription-translation machinery7,8. Cell extracts benefit from the presence of additional cellular components that can facilitate protein expression and folding. Alternatively, a mix of individually purified RNA and protein molecules, i.e. the PURE system9, can be used to mediate intravesicular protein synthesis4,10-14. The PURE system allows for the construction of fully defined cellular mimics and does not suffer from the nuclease activity found in cell extracts. Practically, this means that much less DNA template is required, thereby facilitating processes with low encapsulation efficiency11. Although less frequently used, cellular mimics can be built with cell extracts derived from eukaryotic cells15. Thus far, genetically encoded encapsulated cascades and cellular mimics that sense the environment have been reported16-18.
The simplest way to monitor transcription-translation reactions is to measure the fluorescence or luminescence of genetically encoded elements. Typically, firefly luciferase19 or GFP are used, although in vitro reactions are frequently measured by radiolabeling. Fluorescence detection additionally allows for the monitoring of populations of vesicles20,21 through cytometry based methods, thereby offering some insight into the stochastic nature of biological-like processes. These monitoring methods have been used to define a small set of design rules and a library of parts from which to build from, including a collection of fluorescent proteins that are compatible with in vitro transcription-translation22, the influence of genetic organization on expression22, the activity of sigma factors16, and the efficiency of transcriptional terminators23. Nevertheless, there remains much that needs to be done to increase the capability of building predictable in vitro, genetically encoded devices.
There are many methods available to make vesicles. The most common methods depend upon the generation of a thin lipid film on a glass surface followed by resuspension in aqueous solution24. If the aqueous solution contains transcription-translation machinery, for example, then a fraction of the vesicles formed would contain the necessary components for protein production. However, the encapsulation efficiency of such methods is low, meaning that only a small percentage of vesicles are active. Many of the alternative methods characterized by much higher encapsulation efficiency exploit the conversion of water-in-oil emulsion droplets to vesicles. While it is likely that such methods will be commonplace in the future, currently these methods suffer from the need of specialized equipment and give vesicles with altered membrane compositions25. A distinct advantage of water-in-oil to vesicle methods is the potential to control membrane lamellarity. The method described herein is based on the thin lipid film protocol described by the Yomo laboratory11 with slight modifications including an additional homogenization step. This method is easy, cheap, and gives robust vesicles well suited for the encapsulation of transcription-translation machinery.
Protocol
1. Preparing the DNA Template
- Purify plasmid from a standard laboratory strain of E. coli, such as E. coli DH5a or Nova Blue with a commercial kit. Alternatively, a linear PCR product can be similarly purified with a commercial kit. Elute the DNA with H2O only.
- Phenol-chloroform extract the DNA solution26.
- Determine the DNA concentration and purity, e.g. by UV absorbance or other suitable methods. It is important to use highly pure DNA for efficient transcription-translation.
2. Preparing the Thin Lipid Film
- Weigh the dry lipid powder and dissolve in solvent. For 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), stock solutions are prepared in chloroform at a concentration of 40 mg/ml.
Note: Organic solvents must always be handled with glass pipettes and bottles. In other words, plastic must never be used. Store stock solutions in air tight, solvent resistant amber glass bottles at -20 °C, preferably under argon. - Aliquot 12 μmol POPC (220 μl of 40 mg/ml stock solution) into a 5 ml round bottom flask.
- Evaporate the solvent with a rotary evaporator (Figure 1) to generate the thin lipid film.
- Securely attach the round bottom flask to the distillation tube with a circular clip and start the rotation of the flask.
- Initiate the circulation of water through the condenser coil. Using a heat bath is not necessary, because chloroform has a low boiling point of 61.2 °C at normal atmospheric pressure.
- Ensure that the system is open to the atmosphere by checking that the stopcock is open. Turn on the vacuum pump and slowly close the stopcock to apply the vacuum to the system.
- A thin lipid film is deposited on the walls of the round bottom flask during rotary evaporation forming an opaque film that is visible by eye. Let the rotary evaporation continue for 0.5-2 hr. To stop the process, slowly release the vacuum pressure, stop the rotation, and remove the round bottom flask.
3. Lipid Resuspension and Vesicle Homogenization
- Add 1 ml 18.2 MΩ H2O to the thin lipid film directly in the round bottom flask. Vigorously vortex the solution at maximum speed, approximately 3,200 rpm, until the lipid film detaches from the glass, which is observable by eye.
- Transfer the lipid dispersion into a 2 ml microcentrifuge tube.
- Set-up a ring stand to hold a homogenizer with a 5 mm tip. Place a microcentrifuge stand below the homogenizer to securely hold the lipid dispersion. Rinse the dispersing element of the homogenizer by immersing the tip in 18.2 MΩ H2O and running for a few seconds.
- Place the dispersing element directly into the lipid dispersion ensuring that the tip does not touch the bottom of the microcentrifuge tube. Homogenize for 1 min at power level 4 or 14,000 rpm.
4. Vesicle Extrusion and Lyophilization
- Assemble the parts of the mini-extruder (Figure 2), which consists of an outer casing and a retainer nut that houses two Teflon internal membrane supports, two O-rings, and one Teflon bearing.
- Place the two O-rings into the grooves of the internal membrane supports. Prewet two filters and one 400 nm membrane. The filters will be placed against the Teflon inside of the O-rings with the membrane between them.
- Place the membrane supports into the extruder outer casing with the membrane surrounded by two filters in between the O-rings. Place the Teflon bearing inside the casing and attach the retainer nut and tighten by hand.
- Rinse two syringes and fill one with 18.2 MΩ water. Insert the syringe needles into the small holes in the Teflon on either end of the extruder assembly. The needles should slide in easily; do not force the needles. Secure the extruder with the syringes into the extruder housing and fasten.
- Pass the water through the extruder by slowly pushing the water out of one syringe and into the other. This represents one passage. Repeat for a total of three passages ensuring that there are no leaks present. Remove the syringe of water and dispose of the water.
- Fill one syringe with the sample, attach to the extruder, and slowly pass the vesicle solution through the membrane as described above in step 4.3. Repeat 10x for a total of 11 passages. As the extrusion process proceeds, the sample will become less turbid and easier to push across the membrane. A sudden decrease in resistance, however, usually indicates a rupturing of the membrane.
- Transfer the extruded vesicle solution and make 40 μl aliquots of the vesicles into microcentrifuge tubes. Flash freeze each aliquot in either dry ice or in liquid nitrogen.
- Lyophilize each aliquot with a centrifugal evaporator overnight at 30 °C. Store the lyophilized empty vesicles at -20 °C.
5. Encapsulating Transcription-translation Machinery
- Mix the components of the transcription-translation reaction and add 20 units of RNase inhibitor. Incubate on ice.
- Add the DNA template. For a control reaction, 250 ng of a plasmid encoding mVenus or a similar fluorescent protein behind a T7 transcriptional promoter and a strong E. coli ribosome binding site is recommended.
- Bring the final volume to 25 μl with RNase-free water.
- Hydrate an aliquot of lyophilized vesicles (from step 4.6) with 10 μl of the reaction assembled in step 5.3. Briefly vortex the mixture until the vesicles are resuspended. This should take less than 30 sec.
- Incubate the reaction on ice for 30 min to allow the vesicles to swell.
- Dilute the vesicle mixture 20-fold to a final volume of 30 μl by adding 1.5 μl of vesicles into 27.0 μl of 50 mM Tris-HCl, 50 mM NaCl, pH 7.4 and 1.5 μl of 20.2 mg/ml Proteinase K. DNase and RNase can also be added at this point as an alternative to proteinase K to degrade extravesicular material.
- Incubate for at least 2.5 hr at 37 °C.
6. Microscopy
- Examine the vesicles and the progress of fluorescent protein production at different time points. The vesicles will have a larger diameter than the 400 nm pore-size of the membrane used for vesicle extrusion.
- Prepare a sample chamber by placing a 20 x 5 mm silicon spacer onto a standard microscope slide. Pipette 10 μl of vesicles into the sample chamber. Place a siliconized glass cover slip over the chamber.
- Observe the vesicles with a 63X oil-dispersion or similar objective by bright field and fluorescence microscopy using the appropriate filter set for the exploited fluorescent protein.
Representative Results
Fluorescence microscopy reveals that fluorescence is only observed inside of the vesicles, because extravesicular material is enzymatically degraded (Figure 3). For the expression of mVenus, intravesicular fluorescence begins to be observed after 1.5 hr at 37 °C and reaches maximum fluorescence intensity within 6 hr. The optimal temperature and incubation time can vary depending upon the specific constructs used. For example, different fluorescent proteins mature quite differently in a temperature dependent manner. In other words, the observation of protein production is not solely dependent upon protein synthesis and folding but also on chromophore formation. Overall protein synthesis can be increased by incorporating membrane protein pores to allow for an influx of depleted components necessary for transcription and translation27.
It is recommended to carry out an analogous transcription-translation reaction in the absence of vesicles to ensure that the exploited genetic construct is functional. This control reaction is more easily monitored by fluorescence spectroscopy rather than microscopy. Figure 4 shows an in vitro transcription-translation reaction of a construct encoding mVenus. Unencapsulated reactions give much higher total fluorescence intensities than similar intravesicular reactions. This is due to encapsulation efficiency and because the total intravesicular volume is much less than the extravesicular volume (i.e. a dilution effect).

Figure 1. The rotary evaporator and vacuum pump. Click here to view larger image.

Figure 2. The housing and parts of the extruder are shown separately. From left to right, syringe, retainer nut, teflon bearing, internal membrane supports with black O-rings facing one another, extruder outer casing, and second syringe. Click here to view larger image.
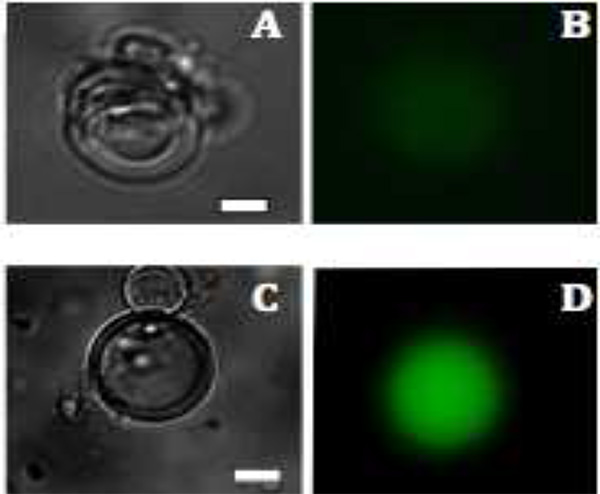
Figure 3. Fluorescence images of mVenus protein production in liposomes. A and C: Bright field images of multilamellar vesicles after 1.5 hr and 2.5 hr. B and D: The production of mVenus is visualized by fluorescence (colored green) after 1.5 and 2.5 hr, respectively. Scale bar is 20 μm. Click here to view larger image.
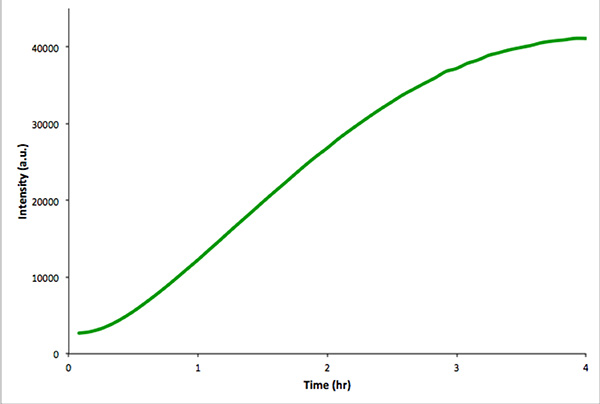
Figure 4. In vitro control reaction of nonencapsulated in vitro transcription and translation of mVenus. Fluorescence intensity was measured every 5 min over 4 hr. The data were acquired with a Real-Time PCR instrument. Click here to view larger image.
Discussion
Although cell-free synthetic biology is still in its infancy, advances have laid a foundation from which increasingly complex cell-like systems can be made. The reconstitution of transcription-translation machinery from fully defined components9 inside of vesicles28 was particularly significant in facilitating later efforts in constructing environmentally responsive artificial cells17,18. Similarly, artificial cell studies have been used to probe evolutionary processes4,29,30, the mechanistic details of RNA and protein synthesis22,31, the influences of metabolic load32,33, and the assembly of viral particles34. Importantly, enough knowledge now exists that basic cellular function can be reconstituted inside of vesicles in the laboratory following these previous reports and the protocols described herein.
In addition to being easy, the described encapsulation procedure has several benefits. For example, many empty, lyophilized vesicle aliquots can be made in advance and stored at -20 °C for later use. The protocol does not subject biological molecules to organic solvents, drastic temperature changes, or long periods of dialysis. We expect that the gentleness of the procedure will facilitate the incorporation of additional components as needed. We also have not observed adverse effects to changing the lipid composition of the membrane on encapsulation or transcription-translation efficiency. Therefore, lipids more amenable to the incorporation of membrane proteins, specific morphologies, or visualization could conceivably be exploited.
The major limitation of the described method is that the resulting vesicles are not homogenous in size or lamellarity. For many applications, these difficulties do not interfere with the interpretation of data. However, if needed, additional steps can be incorporated to narrow the size distribution and decrease the layers of membranes, such as further rounds of extrusion after encapsulation, freeze-thawing, or dialysis35. Undoubtedly better methods that circumvent these and other problems will be developed. Until then, we find the protocol described here to be well suited for the construction of cellular mimics.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors acknowledge the Armenise-Harvard Foundation, the Marie-Curie Trentino COFUND (ACS), the Autonomous Province of Trento (Ecomm), and CIBIO for funding.
Materials
| Name | Company | Catalog Number | Comments |
| Quick Spin Mini-prep kit | Qiagen | 27104 | |
| Spectrometer | NanoDrop 1000 | NDB767ND | |
| POPC | Avanti Polar Lipids | 770557 | MW 760 g/mol Transition Temp -2 °C CAS# 26853-31-6 |
| Ethanol | Sigma Aldrich | 459836 | Anhydrous, >99.5% |
| Phenol-choloroform-isoamyl alcohol 25:24:1, for molecular biology use | Sigma Aldrich | P3803-100mL | Saturated with 10 mM Tris, pH 8.0, 1 mM EDTA |
| Chloroform | Biotech Grade Fluka | 496189-1L | Contain ethanol at 0.5-1.0% v/v as stabilizer |
| Brown amber glass bottles | VWR | 89043-518 | 55X 48 mm |
| Rotary evaporator | Buchi Rotovapor R-210/Sigma | Z563846EU-1EA | With jack and water bath, 29/32 joint 240 V |
| Analog vortex mixer | VWR | 945300 | Speed 1,000-3,200 rpm |
| Homogenizer | IKA T10 Basic Ultra-Turbax | 3420000 | |
| Mini-extruder | Avanti Polar Lipids | 610020 | |
| Extruder filters | Whatman | 610014 | drain disc 10 mm |
| Extruder polycarbonate membrane 400 nm | Whatman | 61007 | nuclepore polycarbonate |
| Speed vacuum | Labconco | 7970011 | Centritrap DNA concentrator |
| PURExpress kit | New England Biolabs | NRM #E6800S | |
| RNAse inhibitor (40,000 U/ml) | New England Biolabs | #M0307S | |
| Proteinase K (20.2 mg/ml) | Fermentas | #EO0491 | |
| Microscope | Zeiss Observer Z1with a AxioCam MRm camera | ||
| RealTime | CFX96 Real time PCR Detection System (Biorad) | ||
| Silicon press to seal -Molecular Probe | Life Technologies | P18174 | Resistant from -25-30 °C |
| Siliconized glass circle cover slides | Hampton Research | HR3-231 | Diameter= 22 mm |
| ImageJ | NIH |
References
- Forster, A. C., Church, G. M. Towards synthesis of a minimal cell. Mol. Syst. Biol. 2, 1-10 (2006).
- Noireaux, V., Maeda, Y. T., Libchaber, A. Development of an artificial cell, from self-organization to computation and self-reproduction. Proc. Natl. Acad. Sci. U.S.A. 108, 3473-3480 (2011).
- Harris, D. C., Jewett, M. C. Cell-free biology: Exploiting the interface between synthetic biology and synthetic chemistry. Curr. Opin. Biotech. 23, (2012).
- Nishikawa, T., Sunami, T., Matsuura, T., Yomo, T. Directed Evolution of Proteins through In Vitro Protein Synthesis in Liposomes. J. Nucleic Acids. 2012, 1-11 (2012).
- Forlin, M., Lentini, R., Mansy, S. S. Cellular imitations. Curr. Opin. Chem. Biol. 16, 586-592 (2012).
- Chiarabelli, C., Stano, P., Anella, F., Carrara, P., Luisi, P. L. Approaches to chemical synthetic biology. FEBS Lett. 586, 2138-2145 (2012).
- Noireaux, V., Shin, J. Efficient cell-free expression with the endogenous E. Coli RNA polymerase and sigma factor 70. J. Biol. Eng. 4, 1-9 (2010).
- Fujiwara, K., Nomura, S. -iM. Condensation of an Additive-Free Cell Extract to Mimic the Conditions of Live Cells. PLoS ONE. 8, e54155 (2013).
- Shimizu, Y., et al. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 19, 751-755 (2001).
- Hosoda, K., et al. Quantitative Study of the Structure of Multilamellar Giant Liposomes As a Container of Protein Synthesis Reaction. Langmuir. 24, 13540-13548 (2008).
- Sunami, T., Matsuura, T., Suzuki, H., Yomo, T. Synthesis of Functional Protiens Within Liposomes. Methods Mol. Biol. 607, 243-256 (2010).
- Murtas, G., Kuruma, Y., Bianchini, P., Diaspro, A., Luisi, P. L. Protein synthesis in liposomes with a minimal set of enzymes. Biochem. Biophys. Res. Commun. 363, 12-17 (2007).
- Pereira de Souza, T., Stano, P., Luisi, P. L. The Minimal Size of Liposome-Based Model Cells Brings about a Remarkably Enhanced Entrapment and Protein Synthesis. ChemBioChem. 10, 1056-1063 (2009).
- Caschera, F., et al. Programmed Vesicle Fusion Triggers Gene Expression. Langmuir. 27, 13082-13090 (2011).
- Noireaux, V., Bar-Ziv, R., Godefroy, J., Salman, H., Libchaber, A. Toward an artificial cell based on gene expression in vesicles. Phys. Biol. 2, P1-P8 (2005).
- Shin, J., Noireaux, V. An E. coli Cell-Free Expression Toolbox: Application to Synthetic Gene Circuits and Artificial Cells. ACS Synth. Biol. 1, 29-41 (2012).
- Kobori, S., Ichihashi, N., Kazuta, Y., Yomo, T. A controllable gene expression system in liposomes that includes a postive feedback loop. Mol. Syst. Biol. 9, 1282-1285 (2013).
- Martini, L., Mansy, S. S. Cell-like Systems with Riboswitch Controlled Gene Expression. Chem. Commun. 47, 10734-10736 (2011).
- Noireaux, V., Bar-Ziv, R., Libchaber, A. Principles of cell-free genetic circuit assembly. Proc. Natl. Acad. Sci. U.S.A. 100, 12672-12677 (2003).
- Sunami, T., et al. Detection of Association and Fusion of Giant Vesicles Using a Fluorescence-Activated Cell Sorter. Langmuir. 26, 15098-15103 (2010).
- Saito, H., et al. Time-Resolved Tracking of a Minimum Gene Expression System Reconstituted in Giant Liposomes. ChemBioChem. 10, 1640-1643 (2009).
- Lentini, R., et al. Fluorescent Proteins and in Vitro Genetic Organization for Cell-Free Synthetic Biology. ACS Synth. Biol. , (2013).
- Du, L., Villarreal, S., Forster, A. C. Multigene Expression In Vivo: Supremacy of Large Versus Small Terminators for T7 RNA Polymerase. Biotechnol. Bioeng. 109, 1043-1050 (2011).
- Trochilin, V. P., Weissig, V. Liposomes: A Practical Approach. , 2nd edn, Oxford University Press. (2003).
- Walde, P., Cosentino, K., Engel, H., Stano, P. Giant vesicles: preparations and applications. ChemBioChem. 11, 848-865 (2010).
- Sambrook, J., Russell, D. W. Molecular Cloning. , 3rd edn, Cold Spring Harbor Laboratory Press. (2001).
- Noireaux, V., Libchaber, A. A vesicle bioreactor as a step toward an artificial cell assembly. Proc. Natl. Acad. Sci. U.S.A. 101, 17669-17674 (2004).
- Yu, W., et al. Synthesis of Functional Protein in Liposome. J. Biosci. Bioeng. 92, 590-593 (2001).
- Caschera, F., et al. Stable vesicles composed of monocarboxylic or dicarboxylic fatty acids and trimethylammonium amphiphiles. Langmuir. 27, 14078-14090 (2011).
- Pereira de Souza, T., Steiniger, F., Stano, P., Fahr, A., Luisi, P. L. Spontaneous crowding of ribosomes and proteins inside vesicles: a possible mechanism for the origin of cell metabolism. ChemBioChem. 12, 2325-2330 (2011).
- Niederholtmeyer, H., Xu, L., Maerkl, S. J. Real-Time mRNA Measurement during an in Vitro Transcription and Translation Reaction Using Binary Probes. ACS Synth. Biol. 10, (2012).
- Stögbauer, T., Windhager, L., Zimmer, R., Rädler, J. Experiment and mathematical modeling of gene expression dynamics in a cell-free system. Integr. Biol. 4, 494-501 (2012).
- Lazzerini-Ospri, L., Stano, P., Luisi, P., Marangoni, R. Characterization of the emergent properties of a synthetic quasi-cellular system. BMC Bioinformatics. 13, 1-10 (2011).
- Shin, J., Jardine, P., Noireaux, V. Genome Replication, Synthesis, and Assembly of the Bacteriophage T7 in a Single Cell-Free Reaction. ACS Synth. Biol. 1, 408-413 (2012).
- Zhu, T. F., Szostak, J. W. Preparation of Large Monodisperse Vesicles. PLoS ONE. 4, 1-4 (2009).
