Primerforlængelse 1 er en molekylær metode til bestemmelse af 5 'enderne af specifikke RNA-molekyler op til én base opløsning. Fordelen ved at andre metoder såsom 5'-RACE (rapid amplifikation af cDNA-ender) er den hurtige behandlingstid og evnen til let at analysere en blanding af forskellige længder af RNA-molekyler.
Denne metode fungerer ved at underkaste RNA-molekyler til at vende transkriptionsreaktioner anvendelse af specifikke fluorescerende primere generere cDNA-fragmenter af visse længder. Disse cDNA-molekyler er sideløbende traditionelle Sanger sekventeringsreaktioner 2 på denaturerende polyacrylamidgeler og kan påvises ved deres fluorescens som følge af anvendelsen af fluorescens-mærkede primere. Længderne af de cDNA-fragmenter vurderes derefter ved sammenligning med den sekventeringsstige, tillader kortlægning af 5'-RNA-ender.
Traditionelt er primerforlængelsesreaktioner anvendes i forbindelsemed radioaktive isotoper til påvisning af cDNA-molekyler på X-ray film. Grundet sundhedsfarer, bortskaffelse af affald spørgsmål og lette håndtering, nyere protokoller udnytte fluorescens til påvisning af primerforlængelsen med automatiserede sequencere, omend deres følsomhed er lidt lavere. Brug af fluorescens-mærkede primere, kan den tilbagevendende procedure radio-mærkning undlades, som fluorescerende primere er stabile i lang tid (mere end et år i vores hænder).
Fremgangsmåden beskrevet her anvender en automatiseret gel sequencer, men med små modifikationer kan kapillære sequencers også anvendes til cDNA-separation og detektion 3. Den parallelle karakter gel analyse gør det muligt at detektere selv en lille mængde RNA spaltning eller forarbejdning. En anden fordel er den høje opløsning af denne fremgangsmåde, som terminal spaltning eller forarbejdning af endnu en base kan detekteres.
Med hensyn til påvisning af RNA-spaltningen eller forarbejdning, typically to forskellige typer af primerforlængelser skelnes. I et tilfælde er den enzymatiske behandling udføres in vitro ved anvendelse af oprenset RNA og oprenset enzym, mens i det andet tilfælde er behandlingen udføres in vivo, og den resulterende RNA renses. I begge tilfælde RNA underkastes et primerforlængelsesprodukt udføres in vitro, dog afhængigt af kilden til RNA, er fremgangsmåden enten kaldes en in vitro eller in vivo primerforlængelse. I protokollen præsenterer vi her fokuserer vi udelukkende på in vivo primerforlængelse grund af brugervenlighed (ingen oprensede proteiner nødvendigt) og muligheden for at bestemme transkriptionelle udgangspunkter og forarbejdning på samme tid. Men i vitro primerforlængelser i princippet oprettet på samme måde og denne protokol kan tjene som udgangspunkt.
Fremgangsmåden er illustreret her, kan anvendes til mange bakteriearter, så længe de er medgørlige til højforberedelse renhed og højt udbytte af nukleinsyrer.
Forskningen i vores laboratorium fokuserer på den regulerende rækkevidde af toksin-antitoksin (TA) systemer 4,5, et område, hvor primerekstensionsproduktet metode benyttes flittigt. TA-systemer er små genetiske elementer til stede i prokaryote genomer, der består af en stabil og endogent aktivt toksisk protein og en hovedsagelig ustabilt protein eller RNA antitoksin, der modvirker toksicitet 6,7. Toksin-aktivitet er undertiden udøves ved hæmning af replikation, cellevægsyntese eller andre mekanismer, men oftest af RNase-aktivitet 8,9. Typisk er RNase specificitet bestemmes ved at udføre forskellige test, hvoraf den ene er primerekstensionsproduktet metode. Primerforlængelsesreaktioner er velegnede til denne anvendelse, som en blanding af spaltede og fuld længde fragmenter samtidig kan analyseres for at bestemme deres 5'-ender. Ved hjælp af en blanding af in vitro og in vivo primerforlængelser, denspecifikke toksin RNase-spaltning, f.eks sekvens specificitet kan bestemmes 10-13.
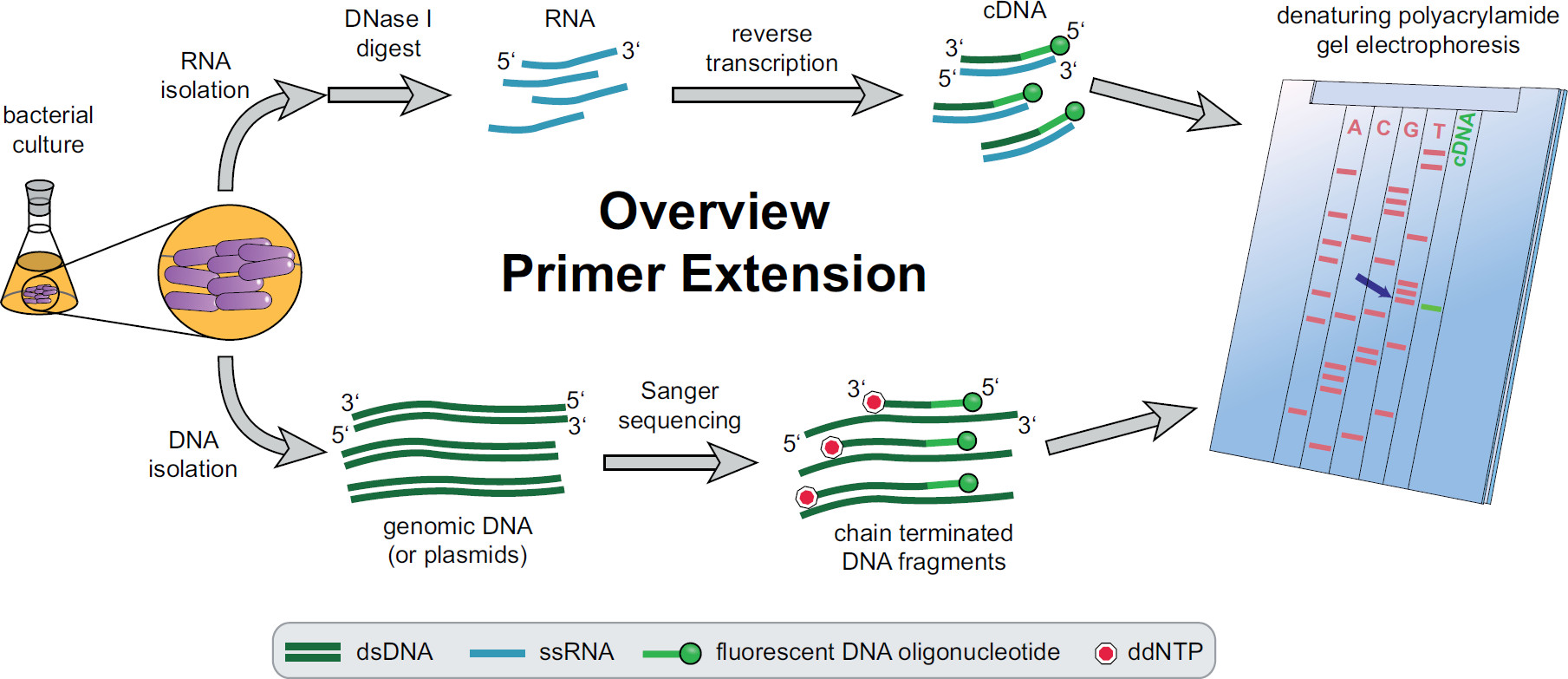
Figur 1. Oversigt over primerforlængelse procedure. Bakteriekulturer inkuberes og behandles i henhold til de eksperimentelle behov. Totalt RNA ekstraheres fra cellerne, der er behandlet med DNase I for at fjerne DNA-spor og underkastet en revers transkription under anvendelse af målspecifikke fluorescerende DNA-primere, der giver cDNA. Genomisk DNA eller plasmider ekstraheres og derefter anvendes til fluorescerende Sanger sekventeringsreaktioner for størrelse sammenligning med cDNA-fragmenter. Primerforlængelsesprodukter køres sideløbende Sanger sekventering produkter på en denaturerende polyacrylamidgel urinstof og analyseret med et automatiseret laser og mikroskop. Den sekventering base, der flugter med cDNA bandet er last base af 5'-cDNA-ende (blå pil). Mere information i Fekete, et al. 3 Klik her for at se en større udgave af dette tal.
Et overblik over hele primerforlængelse procedure kan findes i figur 1. Kort fortalt bakterieceller dyrkes, høstes, cellepelleten lyseret, og RNA ekstraheret. Oprenset RNA behandles dernæst med DNase I for at fjerne spor af DNA-molekyler, som kunne fungere som skabeloner for revers transkriptase. Specifikke fluorescerende primere tilsættes til RNA hybridiseret til regionen af interesse og efterfølgende revers transkriberet, resulterer i enkeltstrenget komplementært DNA (cDNA). En sekventeringsstige er skabt af traditionelle Sanger-sekventering anvender fluorescerende primere og separeret på en denaturerende polyacrylamidgel ved siden af primerekstensionsproduktet cDNA-fragmenter. Den resulterendegel analyseres ved at sammenligne de fluorescerende bånd, som muliggør identifikation af 5 'enderne af interesse. Transkriptionelle udgangspunkter og forarbejdning sites derefter vurderes individuelt i sekvens sammenligninger.
