Agarose Microchambers for Long-term Calcium Imaging of Caenorhabditis elegans
Summary
Imaging behavior and neural activity over long time scales without immobilization of the animal is a prerequisite to understand behavior. Agarose microfluidic chambers imaging (AMI) can be used to image neural activity and behavior for all life stages of Caenorhabditis elegans.
Abstract
Behavior is controlled by the nervous system. Calcium imaging is a straightforward method in the transparent nematode Caenorhabditis elegans to measure the activity of neurons during various behaviors. To correlate neural activity with behavior, the animal should not be immobilized but should be able to move. Many behavioral changes occur during long time scales and require recording over many hours of behavior. This also makes it necessary to culture the worms in the presence of food. How can worms be cultured and their neural activity imaged over long time scales? Agarose Microchamber Imaging (AMI) was previously developed to culture and observe small larvae and has now been adapted to study all life stages from early L1 until the adult stage of C. elegans. AMI can be performed on various life stages of C. elegans. Long-term calcium imaging is achieved without immobilizing the animals by using short externally triggered exposures combined with an electron multiplying charge-coupled device (EMCCD) camera recording. Zooming out or scanning can scale up this method to image up to 40 worms in parallel. Thus, a method is described to image behavior and neural activity over long time scales in all life stages of C. elegans.
Introduction
Caenorhabditis elegans has been established as a model system to study behavior1. Due to its amenability to genetics, the molecular and cellular mechanisms underlying behavior can be studied. Many behaviors occur on long time scales. Two principal approaches can be used to observe motile animals over long time scales. The first approach is following the animal during movement using an automated stage or camera2-8 and the second is to restrict the movement to a range that is at least as small as the field of view of the camera9-14. Both methods have their advantages. Tracking allows following an animal over long spatial ranges but limits the number of animals that can be discerned in one experiment. Restricting the movement of the animals allows scaling up the observation to many individuals at the same time by using arrays of restricted compartments.
Because the nematode is transparent, live fluorescent imaging can be performed non-invasively15. Calcium imaging provides a functional readout for the activity of excitable cells and is established for C. elegans16-20. Calcium enters the cell via channels in the plasma membrane that open upon depolarization. Thus, calcium acts as a proxy for neural activity. Calcium sensors can be grouped in two major classes, ratiometric and non-ratiometric sensors. Both classes employ conformational changes of calcium-binding proteins induced upon binding of calcium. Ratiometric sensors contain two fluorescent proteins. When the lower-wavelength fluorescent protein is excited, a part of the light energy is transmitted to the higher wavelength fluorescent protein as a function of their distance in a process called Fluorescence Resonance Energy Transfer or Förster Resonance Energy Transfer (FRET)21. Non-ratiometric sensors are based on circularly permuted GFP and employ de-quenching of the fluorophore caused by calcium binding22. Each class has its advantages. While ratiometric sensors are less sensitive to movement or expression artifacts, non-ratiometric sensors typically have a higher dynamic range. Both ratiometric and non-ratiometric sensors have been useful to study the activity of excitable cells in C. elegans16-20,23,24.
When doing long-term fluorescence imaging, the experimenter will have to deal with several potential challenges: 1. Disturbance of behavior through excitation light: Worms are sensitive to short-wavelength fluorescence excitation light and avoid light from the violet to blue range that is used for calcium imaging25,26. Worms respond with either a backward or a forward escape response25,26. Thus, the amount of light needs to be controlled. 2. Bleaching of the fluorescent sensor protein: Often, bleaching of the fluorescent protein hampers long-term imaging. Typically, however, light intensities that are needed to observe bleaching are higher than the light intensities that cause disturbing effects on the behavior of the animals. Thus, bleaching is only a theoretical problem in this type of calcium imaging. 3. Worms inside the microchambers are not fixed and move constantly during wake behavior and images may appear blurred if the worm is not immobilized. All these challenges can be solved using extremely short exposure times with low light intensity. This can be realized by using a highly sensitive EMCCD camera with short exposure times and external triggering of a light emitting diode (LED). To expose the worms to the illumination light as briefly as possible (only during the exposure time of the camera) the LED is externally triggered using a transistor-transistor logic (TTL) signal that the camera emits during exposure resulting in an illumination of the worm for precisely the time the camera chip is exposed. This also means that the EMCCD chip will be dark during data readout, which is optimal for readout performance of this chip.
Previously, agarose microchambers have been developed for long-term fluorescence imaging of C. elegans larvae9. Here, it is described how agarose micro-chambers can be used for long-term calcium imaging for any life stage of C. elegans, how calcium imaging can be performed, and how this method can be upscaled to assay many individual worms in parallel.
Protocol
1. Instruments, Culture Media, and Dishes
- Use a microscope that is capable of keeping the sample in focus and that is equipped with an automatic stage. Build a custom-made lid heater or purchase a commercial solution. Set up an LED-EMCCD camera system in which the exposure of the camera triggers LED illumination through a TTL signal using manufacturer's instructions.
Note: See Discussion for details. - Polydimethylsiloxan (PDMS) stamps:
- Fabricate PDMS stamps in a microfluidics facility or have the PDMS stamps produced by a commercial foundry. To use a commercial foundry, send an autocad file to the company and specify the depth (15 µm, for example) of the device. After delivery from the foundry, cut the PDMS chip into its 16 stamps using a scalpel.
- Bond each individual stamp to a glass slide using air plasma. For bonding, expose both the PDMS stamp and the glass slide to air plasma for about 1 min (using the highest plasma settings at 0.5 mbar). Ensure that the surfaces for bonding face upwards during plasma treatment. Then, place the PDMS stamp onto the glass slide.
- Take a 3.5 cm bottom dish and cut out a square area of 18 x 18 mm from the center of the bottom of the dish using a vertical milling machine and sharp rotary cutters. Use low rotation speed of the cutter and slow feeding to prevent the plastic from melting due to heat produced by friction. Prepare several dishes at once.
Note: The bottom dish can be reused many times after the experiment if it is cleaned after soaking in pure ethanol O/N. - Agarose:
- Dissolve 3 g high-melting point agarose in 100 ml S-Basal (5.85 g NaCl, 1 g K2HPO4, 6 g KH2PO4, 1 ml cholesterol (5 mg/ml in ethanol), H2O to 1 L, sterilize by autoclaving) by boiling. Make aliquots of the dissolved agarose in 2 ml Eppendorf tubes and store. Also, prepare a batch of low-melting point agarose exactly the same way as the high-melting point agarose.
- Prior to use, place 3 aliquots of high-melting point agarose onto a heating block at 95 – 98 °C. Prior to use, place one aliquot of low-melting point agarose onto a heating block at 95 – 98 °C until it has melted and then onto a heating block at 30 – 35 °C.
2. Selection of Animals
- Grow worms at a low density on seeded NGM plates to obtain clean animals. Make sure that there is plenty of food and only few animals on the plate.
- For imaging L1 larvae, transfer about 30 eggs containing embryos at the pretzel stage onto a fresh seeded NGM plate. For transferring later larval stages or adult C. elegans, transfer about 30 worms onto a fresh seeded NGM plate.
3. Preparation of Agarose Microchambers
- Preparation of the dish:
- Take a plastic dish with a square opening of 18 x 18 mm at the bottom and place it upside down with the opening facing upwards. Close the opening by placing a piece of double-sided sticky tape of 20 x 20 mm onto the opening. Turn the dish around so that the sticky tape is on the bottom and place the dish onto a hard surface. Cut the opening free using a scalpel.
- Using a P1000 pipette, fill 2 ml of 3% high-melting point agarose in S-Basal into the dish. Place the agarose onto the area that surrounds the opening and let solidify. Wait until the agarose is solid.
- Turn around the dish and peel off the protective film that covers the double-sided sticky tape so that the sticky side will remain on the dish and will be exposed.
Note: As a result, a fine ring of sticky tape will surround the outside of the opening. The agarose serves as a moisture reservoir that will later surround the sample.
- Casting of microchambers:
- Expose the PDMS surface for molding with air plasma for 20 – 60 sec.
Note: This plasma treatment renders the PDMS surface hydrophilic, which prevents the trapping of air bubbles and produces sharper prints. - Construct two spacers of equal height by stacking 5 – 9 glass slides. Place, in parallel to their long sides, the first spacer stack, then a single glass slide, then again a spacer stack. Place the glass slide that contains the PDMS stamp orthogonally across the spacers. Adjust the height of the spacers so that there is a space of about 1.5 mm between the molding surface of the PDMS stamp and the single glass slide.
- Place a drop of hot liquid high-melting point agarose onto the single glass slide near the PDMS stamp and quickly slide the PDMS stamp vertically into the liquid agarose. Let the agarose solidify. Check that it gets an opaque appearance, which usually takes about 2 min. Pull off the stamp vertically with one move.
Note: It is convenient to glue the spacer slides together with double-sided sticky tape. The vertical movement of the stamp prevents air bubbles from getting trapped in the agarose.
- Expose the PDMS surface for molding with air plasma for 20 – 60 sec.
- Transfer the eggs or worms together with OP50 bacteria onto the agarose using a fine platinum wire pick. Distribute one egg or one worm per chamber together with food using an eyelash. Fill around 30 eggs onto one agarose pad.
- Cut the agarose slab containing the filled microchambers into a square of about 15 x 15 mm so that it fits nicely into the opening of the dish. Pick up the square agarose slab with forceps and place it upside down onto a glass coverslip of 20 x 20 mm. Once dropped, do not lift it up again or slide it around because this may cause the bacteria and worms to be pushed out of their chambers.
- Assembly of the dish:
- Place the glass coverslip onto the opening of the plastic dish. Gently press down the glass coverslip onto the ring made of double-sided sticky tape. Take care not to break the glass.
- Turn the dish upside down and use a P1000 pipette to fill the gap between the agar slab containing the microchambers and the agarose reservoir with liquid low-melting point agarose cooled to about 30 °C. Wait until the agarose has solidified.
- Seal the dish with a lid. For inverted microscopes use a heated lid. For upright microscopes use a normal lid and seal the dish with Parafilm.
- After finishing the preparation of microchambers, check them under a stereomicroscope. The correct filling is crucial. See the Discussion for details.
4. Calcium Imaging
- Use transgenic strains expressing genetically encoded calcium sensors such as HBR16 (goeIs5[pnmr-1::SL1-GCaMP3.35-SL2::unc-54-3'UTR, unc-119(+)])27.
- Use a compound microscope equipped for wide-field epifluorescence. Connect the TTL output of the EMCCD camera to the TTL input of the LED, so that each time the camera records a frame the sample will be illuminated. Use an exposure time of about 5 msec. Use EM gain in the range of 50 – 300.
- Specify a burst movie running for 24 hr with each worm being imaged every 15 – 30 min first for 20 sec with DIC, then for 20 sec with a GFP fluorescence to record GCaMP, and then take a final image of the mKate2 signal is taken to control for expression levels. Use a frame rate of 2/sec during each burst.
- For visual data inspection, use a false-color map to enhance the visibility of small changes in fluorescence intensity. Plot fluorescent data as ΔF/F, with F being the average baseline value of fluorescence. A detailed description of calcium data analysis can be found in the literature20.
5. Parallel AMI of Multiple Worms
- Place the dish containing the microchambers onto the microscope, focus on the sample and engage the autofocus. Set up a software protocol so that the camera acquires a burst of 40 image frames in 20 sec every half an hour for 24 hr, which will result in 1,920 frames per worm, a reasonable amount of data. Set up the scan so that it visits each worm using the stage. Aim to film about 30 worms in one run.
- Image multiple worms by zooming out, i.e., using a lower magnification. Use a lower magnification to cover several microchambers with the camera chip and film several adjacent microchambers simultaneously. After the end of image acquisition, separate the data for each individual chamber by cropping a region of interest covering one animal.
- To quickly assess mobility data, use frame subtraction28-32.
6. Imaging Different Life Stages of C. elegans
- Use agarose microchambers for all life stages of C. elegans from L1 to adult and including dauers. Use the appropriate chamber dimensions for different life stages that are shown in Table 1.
| Life stage | Chamber size | Chamber depth | Typical recording time | Magnification |
| L1 | 190 x 190 μm | 10 – 15 μm | egg – mid L2 | 400X |
| L2 | 370 x 370 μm | 15 μm | egg – L3 | 200X |
| Dauer larva | 370 x 370 μm | 25 μm | 4 days | 200X |
| L3 | 700 x 700 μm | 45 μm | L2 – L4 | 200X |
| L4 | 700 x 700 μm | 45 μm | young L4 – young adult | 100X |
| Young adult | 700 x 700 μm | 45 μm | 12 – 24 hr | 100X |
Table 1. Chamber sizes for different life stages. Shown are chamber dimensions and magnifications that are useful for various stages optimized for a 8 x 8 mm camera chip.
Representative Results
Agarose microchambers can be applied to any life stage of C. elegans. As can be seen in Figure 1A, larval development and sleep behavior during L1 lethargus can be observed. Shown are chamber sizes that are 190 x 190 µm, 10 µm deep. If longer time scales are required, larger chambers can be used. As can be seen in Figure 1B, using a chamber with larger dimensions (370 x 370 µm, 25 µm deep) allows development of C. elegans from egg until adult. Figure 1C shows an analysis of long-term changes in behavior using frame subtraction of a worm grown in a chamber from egg until adult. Shown are mean intensities after frame subtraction for selected bursts during wake and lethargus. The lower the mean intensity after frame subtraction values is, the lower the mobility of the worm. Shown are selected burst traces from one wake condition and one lethargus condition per larval stage. The observed increase in mean intensity during development is mostly due to increased contrast and size of the animal. Figure 1D shows a dauer larva, an alternative life stage that is engaged during adverse environmental conditions33. Chamber size is 370 x 370 µm, 15 µm deep. Note that the dauer larva was placed into the chamber without bacterial food, which would cause exit from the dauer larval stage. Figure 1E shows an adult worm that has already laid many eggs into the chamber. Chamber dimensions used for the adult were 700 x 700 µm, 45 µm deep. Finally, Figure 1F shows an adult hermaphrodite and a male mating inside an adult chamber.
Calcium imaging in motile worms is possible with GCaMP calcium sensors. Figures 2A,B show calcium imaging of the command interneuron AVA for an L1 larva27. Figures 2C,D show calcium activity for the same type of neuron in an adult animal.
Scaling up long-term imaging is possible by scanning and by zooming out. Figure 3A shows the image quality of simultaneous imaging of 30 worms on one camera chip with a 5 megapixel camera. Figure 3B shows the image quality of 4 worms calcium imaged simultaneously.
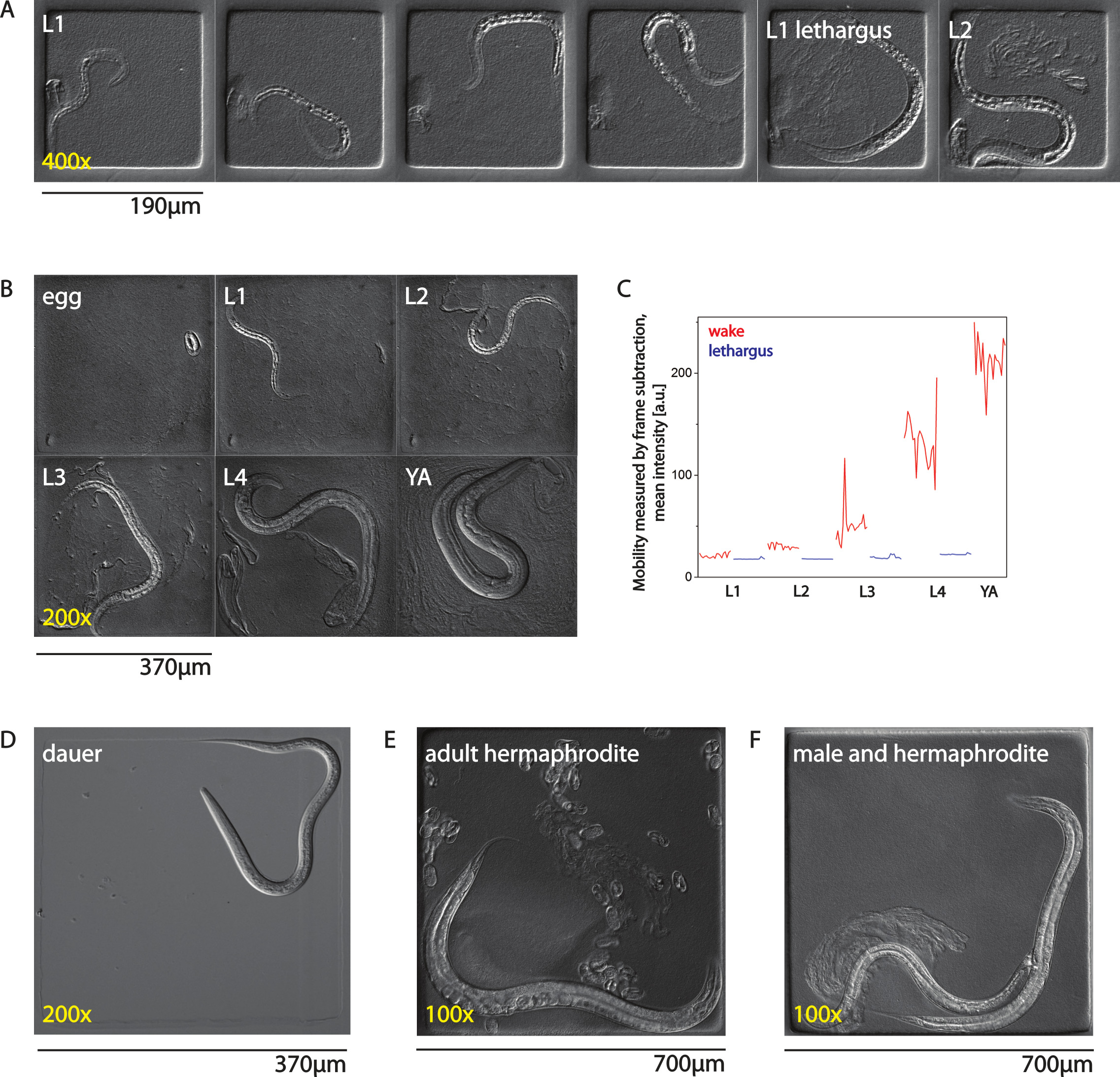
Figure 1. Adaptation of AMI to all life stages of C. elegans. (A) Larva imaged from early L1 until early L2 stage in 190 x 190 µm chambers. (B) Development from egg until adult in a 370 x 370 µm chamber. (C) Mobility of a worm assessed using frame subtraction. Shown is the mean intensity of all pictures of the image after frame subtraction for one selected burst movie for each larval stage for wake and lethargus behavior. (D) A dauer larva in the absence of food in a 370 x 370 µm chamber. (E) Adult hermaphrodite with many eggs laid into a 700 x 700 µm chamber. (F) Mating of a male and a hermaphrodite inside a 700 x 700 µm chamber. YA means young adult. Please click here to view a larger version of this figure.
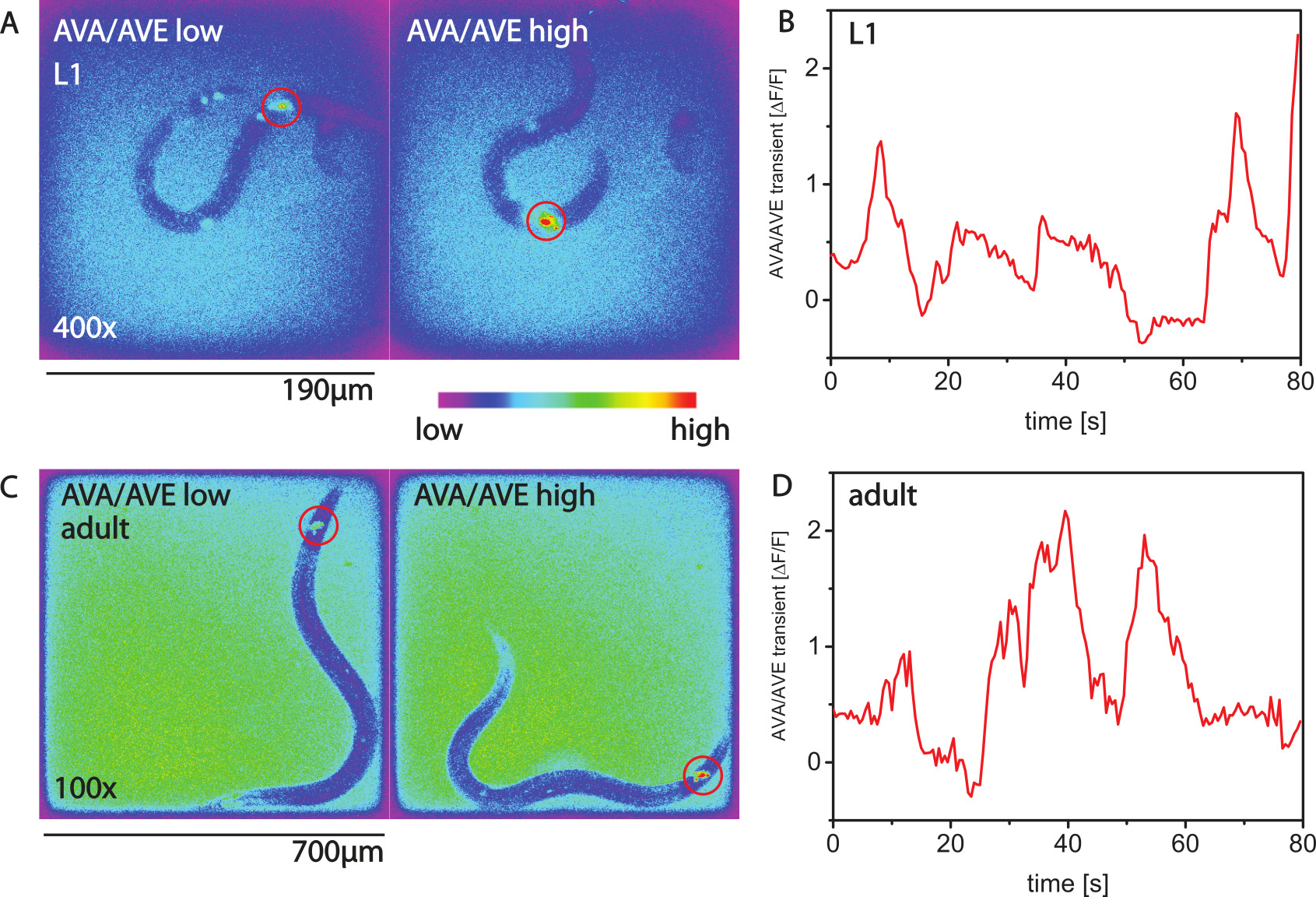
Figure 2. Calcium imaging with AMI. (A) Calcium imaging with GCaMP3 in the command interneuron AVA of L1 larvae using the nmr-1 promoter. Shown are two false-color images. In the left image, the activity of AVA is low and the worm is not making a backwards movement. In the right image the activity of AVA is high, and the worm is moving backwards. (B) Calcium transients over time for an L1 worm. (C,D) Calcium transients in an adult animal. Please click here to view a larger version of this figure.

Figure 3. Imaging multiple worms in parallel by zooming out. (A) Simultaneous imaging of 30 L1 worms per frame using 190 x 190 µm chambers and a 100X magnification (using a 10X objective) and a 16.6 x 14 mm camera chip. (B) Simultaneous imaging of four L3 worms per frame using 370 x 370 µm chambers and 100X magnification and a region of interest of a 16.6 x 14 mm camera chip. Please click here to view a larger version of this figure.
Discussion
Hardware
The focus of a microscope will typically drift during long-term image acquisition. For compound microscopes, focus-keeping systems can be purchased from the major microscope manufacturers. In case a compound microscope with focus control is too pricy, a simple alternative would be to use a stereomicroscope. Compound microscopes allow the use of objectives with high numerical aperture (NA) and can be easily automated. 40X oil objectives are well suited. Water immersion lenses are not ideal because of evaporation during long-term imaging. Differential interference contrast (DIC) generates a nice contrast that helps to follow morphological and behavioral changes, but simple bright field imaging can also be used. For DIC or bright field imaging, use red light by placing a red light filter into the dia-illumination path of the microscope. For scanning of several microchambers an automated stage is necessary. Best performance is achieved when a stage is used that has nonlinear acceleration and deceleration and can be set to low scan speed to prevent disturbing the animals during scanning. We have not observed behavioral responses or calcium increase in mechanosensitive neurons (ALM and PLM) during scanning, suggesting that slow scanning indeed does not activate the mechanosensitive system of the worm (data not shown). Commercial LED systems can be used. Several companies offer ready-to-use solutions that include the LEDs at different wavelengths. The LED should have the option of externally triggering the LED with a TTL signal. A highly sensitive camera is needed for calcium imaging of moving animals. EMCCD cameras are the most sensitive cameras on the market. The camera needs to have a TTL output during exposure (also called “fire” output). A lid heater is required to prevent condensation on the lid if using an inverted microscope. On an upright microscope, the dish will be placed so that the lid will be at the bottom and thus condensation is prevented and no lid heating is required. The lid should tightly close the dish to prevent evaporation of water during long-term imaging.
PDMS stamps
The surface of the PDMS stamp that contains the structure for molding the agarose is faced away from the glass slide, which supports the PDMS stamp. A list with companies that offer custom microfluidic chips can be found on Wikipedia (http://en.wikipedia.org/wiki/List_of_microfluidics_related_companies).
Filling the nematodes into their chambers
This is the most critical step in the protocol and the following points should be considered: A) The moistness of the agarose is crucial for transferring the worms and for imaging. If the agarose is too moist, i.e., there is a liquid covering an area of several microchambers, it will not be possible to distribute bacterial food and worms in a controlled manner because the liquid will make the bacteria flow away. If the agarose is too dry then the bacteria and worms may not get off the pick easily which means that increased force needs to be used to drop the worms and bacteria which easily causes damage to the agarose. When filling in a lot of worms the agarose may dry up. In the worst case the agarose will be so dry in the end that the chambers will collapse. If the agarose is too dry it can be rehydrated by placing a small drop (about 2 µl) of S-Basal onto the side of the chip where there are no worms. Then dip the platinum wire pick into the liquid and even pull some of the liquid into the area where the chambers are filled with worms. B) The amount of food is critical for successful long-term imaging. If there is not enough food, the worms may run out of it. If there is excessive food the cavity of the chamber will not behave like a liquid but rather like a solid and will allow worms to escape from the chamber by pushing off the bacteria. Aim to obtain a bacterial suspension that fills the entire chamber. The worms are in constant physical contact with the agar and glass surface along most of their length during the experiment and the crawling behavior appears to be similar to movement on a plate and dissimilar to thrashing in liquid. C) Mechanical damaging of the agarose can ruin the experiment. A fine pick is essential. It should not have sharp corners. A soft eyelash attached to a pipet tip can be used to move bacteria or worms into the chambers instead of using a platinum pick. In most cases, however, overdried agarose is the reason for damage. The pick or eyelash should, ideally, only barely touch the agarose itself, and the water film on the agarose surface should pull off worms and bacteria. When sealing the chambers, again, the moisture of the agarose is essential. There should not be any free liquid on the agarose surface because this may wash away bacteria and worms during sealing. Large bubbles may be removed by gently lifting a corner of the agar slab. Small bubbles that are smaller than the chamber often get trapped inside the chambers. These bubbles are not a problem and will disappear by absorption. After assembling the dish, check again that the agarose is not too dry or too wet. The chambers should be nicely sealed and there should not be any flow of liquid between chambers. If the agarose is too wet, the chambers will not seal properly. Worms may escape or their food will be washed away. If the sample is too moist, simple open the lid and let the agarose dry for a minute or two. If the agarose is too dry, the chambers may collapse and the worms will escape.
Scaling up by zooming out
For fluorescence imaging, zooming out is limited by the low amount of light obtained at lower magnifications. Also, EMCCD cameras are optimized for sensitivity and often have a relatively low resolution. However, scaling up imaging four-fold is well possible. For instance, four chambers of 190 µm x 190 µm can fit onto one frame when using 140X magnification (achieved by using a 20X Objective and a 0.7X camera mount) and a 512 x 512 pixel, 8 x 8 mm camera. A high-resolution camera with a large chip (such as sCMOS cameras, 16.6 mm x 14 mm chip, 2560 x 2560 pixels = 5.5 Megapixel) optimizes scaling up of DIC and bright field imaging. For instance, up to 30 L1 worms in 190 µm x 190 µm chambers can fit onto each frame of this camera when using a 100x magnification (see Figure 3). In principle, zooming out and scanning can also be combined to obtain even greater numbers of animals. Most neurons stay quite well in focus, so that only one focal plane needs to be imaged. If neurons are found to move out of the focus, a z scan using a piezo drive can be taken at each time point.
Adaptation to different behaviors
This protocol gives a good idea of the behavior across long time scales. Obviously, the timing of the bursts needs to be adapted to different behaviors and life stages.
Limitations of the technique
Several factors limit the duration of imaging. The most important is the amount of food. Once the food is consumed, larvae stop developing. Thus, in small chambers (190 x 190 µm) worms develop until the L3 stage and then arrest. If longer imaging time is required, larger chambers have to be used. The maximum duration of long-term imaging is in the range of 2.5 – 3 days. If longer imaging is required, the worms need to be recovered and placed into new chambers. When imaging adult worms, another limitation is caused by the offspring of these worms. Adult worms lay eggs from which larvae hatch. These larvae will also stay inside the chamber, consume food, and may disturb the image analysis. If offspring is a problem, a solution is to either use sterile adults or to repeatedly place the worms into fresh chambers. To recover worms, the agarose slab containing the chamber is cut free with a scalpel, is pulled off the coverslip, and is placed onto an NGM plate from which the worms can be recovered. Another limit is the restriction of the worms to relatively small areas. This may be a problem if long-range movement needs to be analyzed. While animals in the chambers can be stimulated mechanically and optogenetically21,27,34,35, the sealed nature of the chamber will make it difficult to apply soluble or volatile stimulants. Biologically important gases such as oxygen or carbon dioxide can diffuse freely in the agar. The large air reservoir in the dish should keep the gas concentrations in the chambers constant over the time needed for experiments. However, it should be kept in mind that the local oxygen concentration in the chamber may resemble more the conditions found in liquid culture than culture on the plate.
Significance with respect to existing methods
Microfluidic devices have greatly advanced behavioral and developmental studied in C. elegans. Often, microfluidic structures are made of PDMS12. Here we describe a protocol for generating microfluidic culture chambers made from agarose. The strength of this technique is the combination of high imaging quality, correlation of behavior with physiological measurements, long-term imaging, and a reasonably high throughput. High image quality is achieved by imaging through the glass coverslip using high NA objectives. As a result, fluorescence imaging such as calcium imaging and confocal imaging of subcellular structures can be performed. Because the animals are not immobilized like in other systems, it allows a correlation of behavior with physiological measurements. Because the animals have ample food, they continue developing allowing long-term imaging. This system can image many worms in one run because animals are restricted to their defined chambers. Thus, this method can be easily scaled up9,27,34-36.
Future applications
So far, this system has been used mainly to study sleep behavior in C. elegans L1 larvae. However, the adaptation to all stages will make it possible to study a wide range of behaviors also in dauers and adults. A wide array of behaviors can be studied with this technique ranging from mating to egg laying.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The Max Planck Society and a Göttingen Graduate School Junior Group stipend to H.B. funded this work.
Materials
| high-melting point agarose | Fisher Bioreagents | BP(E)164-1000 | |
| Top Vision Low Melting Point Agarose | Thermo Scientific | R0801 | |
| LED system | CoolLED | pE-2 | |
| compound microscope | Nikon | TiE | |
| stereomicroscope | Leica | M5 | |
| EMCCD camera | Andor | iXon 897 | now sold as iXon Ultra 897 |
| sCMOS camera | Andor | Neo | |
| plasma cleaner | Harrick | PDC-23G2 | |
| lid heater | MPI workshop | custom made | |
| plastic dish | Falcon | 08-757-100A | |
| z-stage for the microscope | Prior | Nano Scan Z | |
| x-y-stage for the microscope | Prior | Proscan III | |
| PDMS stamp | Abbott Laboratories | custom made | |
| red light filter | Chroma | ET660/50m | |
| 35mm petri dish | Falcon | Falcon Disposable Petri Dishes, Sterile, Corning | |
| double sided sticky tape | Sellotape/Henkel | double sided 12mm x 33 m | alternatively Advance double sided sticky tape from Rubance Adhesives can be used. |
References
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics. 77, 71-94 (1974).
- Husson, S. J., Costa, W. S., Schmitt, C., Gottschalk, A. Keeping track of worm trackers. WormBook. , 1-17 (2012).
- Yemini, E., Jucikas, T., Grundy, L. J., Brown, A. E., Schafer, W. R. A database of Caenorhabditis elegans behavioral phenotypes. Nature Methods. 10, 877-879 (2013).
- Likitlersuang, J., Stephens, G., Palanski, K., Ryu, W. S. C. elegans tracking and behavioral measurement. Journal of Visualized Experiments : JoVE. , e4094 (2012).
- Yemini, E., Kerr, R. A., Schafer, W. R. Tracking movement behavior of multiple worms on food. Cold Spring Harbor Protocols. 2011, 1483-1487 (2011).
- Swierczek, N. A., Giles, A. C., Rankin, C. H., Kerr, R. A. High-throughput behavioral analysis in. C. elegans. Nature Methods. 8, 592-598 (2011).
- Ramot, D., Johnson, B. E., Berry, T. L., Carnell, L., Goodman, M. B. The Parallel Worm Tracker: a platform for measuring average speed and drug-induced paralysis in nematodes. PLoS One. 3, e2208 (2008).
- Larsch, J., Ventimiglia, D., Bargmann, C. I., Albrecht, D. R. High-throughput imaging of neuronal activity in Caenorhabditis elegans. Proceedings of the National Academy of Sciences U S A. 110, E4266-E4273 (2013).
- Bringmann, H. Agarose hydrogel microcompartments for imaging sleep- and wake-like behavior and nervous system development in Caenorhabditis elegans larvae. Journal of Neuroscience Methods. 201, 78-88 (2011).
- Yu, C. C., Raizen, D. M., Fang-Yen, C. Multi-well imaging of development and behavior in Caenorhabditis elegans. Journal of Neuroscience Methods. 223, 35-39 (2014).
- Luke, C. J., Niehaus, J. Z., O’Reilly, L. P., Watkins, S. C. Non-microfluidic methods for imaging live. C. elegans. Methods. 68, 542-547 (2014).
- San-Miguel, A., Lu, H. Microfluidics as a tool for C. elegans research. WormBook. , 1-19 (2013).
- Shi, W., Qin, J., Ye, N., Lin, B. Droplet-based microfluidic system for individual Caenorhabditis elegans assay. Lab Chip. 8, 1432-1435 (2008).
- Krajniak, J., Lu, H. Long-term high-resolution imaging and culture of C. elegans in chip-gel hybrid microfluidic device for developmental studies. Lab Chip. 10, 1862-1868 (2010).
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science. 263, 802-805 (1994).
- Kerr, R., et al. Optical imaging of calcium transients in neurons and pharyngeal muscle of. C. elegans. Neuron. 26, 583-594 (2000).
- Suzuki, H., et al. In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron. 39, 1005-1017 (2003).
- Hilliard, M. A., et al. In vivo imaging of C. elegans ASH neurons: cellular response and adaptation to chemical repellents. EMBO Journal. 24, 63-72 (2005).
- Frokjaer-Jensen, C., et al. Effects of voltage-gated calcium channel subunit genes on calcium influx in cultured C. elegans mechanosensory neurons. Journal of Neurobiology. 66, 1125-1139 (2006).
- Kerr, R. A. Imaging the activity of neurons and muscles. WormBook. , 1-13 (2006).
- Miyawaki, A., Griesbeck, O., Heim, R., Tsien, R. Y. Dynamic and quantitative Ca2+ measurements using improved cameleons. Proceedings of the National Academy of Sciences U S A. 96, 2135-2140 (1999).
- Nakai, J., Ohkura, M., Imoto, K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nature Biotechnology. 19, 137-141 (2001).
- Akerboom, J., et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Frontiers in Molecular Neuroscience. 6 (2), (2013).
- Tian, L., et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nature Methods. 6, 875-881 (2009).
- Edwards, S. L., et al. A novel molecular solution for ultraviolet light detection in Caenorhabditis elegans. PLoS Biology. 6, e198 (2008).
- Ward, A., Liu, J., Feng, Z., Xu, X. Z. Light-sensitive neurons and channels mediate phototaxis in C. elegans. Nat Neurosci. 11, 916-922 (2008).
- Turek, M., Lewandrowski, I., Bringmann, H. An AP2 transcription factor is required for a sleep-active neuron to induce sleep-like quiescence in. C. elegans. Current Biology. 23, 2215-2223 (2013).
- Singh, K., et al. C. elegans Notch Signaling Regulates Adult Chemosensory Response and Larval Molting Quiescence. Current Biology. 21, 825-834 (2011).
- Raizen, D. M., et al. Lethargus is a Caenorhabditis elegans sleep-like state. Nature. 451, 569-572 (2008).
- Iwanir, S., et al. The microarchitecture of C. elegans behavior during lethargus: homeostatic bout dynamics, a typical body posture, and regulation by a central neuron. Sleep. 36, 385-395 (2013).
- Nagy, S., Raizen, D. M., Biron, D. Measurements of behavioral quiescence in Caenorhabditis elegans. Methods. , (2014).
- Nagy, S., et al. A longitudinal study of Caenorhabditis elegans larvae reveals a novel locomotion switch, regulated by Galphas signaling. eLife. 2, e00782 (2013).
- Cassada, R. C., Russell, R. L. The dauerlarva, a post-embryonic developmental variant of the nematode Caenorhabditis elegans. Developmental Biology. 46, 326-342 (1975).
- Schwarz, J., Lewandrowski, I., Bringmann, H. Reduced activity of a sensory neuron during a sleep-like state in Caenorhabditis elegans. Current Biology. 21, R983-R984 (2011).
- Schwarz, J., Spies, J. P., Bringmann, H. Reduced muscle contraction and a relaxed posture during sleep-like Lethargus. Worm. 1, 12-14 (2012).
- Schwarz, J., Bringmann, H. Reduced sleep-like quiescence in both hyperactive and hypoactive mutants of the Galphaq Gene egl-30 during lethargus in Caenorhabditis elegans. PLoS One. 8, e75853 (2013).
