Successful labeling of hemogenic endothelial cells and HSPC from embryonic YS or AGM will yield FACS scatter plots similar to the representative plots presented in Figure 3. Following standard live cell and doublet discrimination by forward and side scatter (not shown), side population (SP) events are visualized in a linear Hoechst Red vs. Hoechst Blue differential plot in the absence of Verapamil as a "shoulder" left-shifted from the majority of (non-SP) events (Figure 3A). When the SP gate is properly drawn, SP cells will represent approximately 1 – 3% of total viable YS cells and 3 – 5% of total viable AGM events. Verapamil treatment should result in >50% inhibition of SP events regardless of tissue source (Figure 3A, top panels). We have previously determined that other populations that appear outside of the SP shoulder but are also blocked by verapamil are Ter119-positive erythroblasts, and are therefore excluded from our SP population5.
Compared to the SP, non-SP cells are identified as the dense cluster of cells adjacent to the SP "shoulder" (Figure 3A). This population contains non-Hemogenic EC which can be distinguished using a CD31-PE vs. CD45-FITC daughter plot (Figure 3B), as CD31+/CD45- events. Non-Hemogenic EC are typically 2 – 5% of non-SP cells from AGM (Figure 3B) or yolk sac (not shown), and high numbers of these cells can be sorted back with relative ease.
For identification of HSPC and Hemogenic EC, daughter gates are drawn from the SP fraction, identifying CD45+ and CD45- cells, where CD45+ cells are typically <20% of total events in both AGM (Figure 3C) or YS (not shown). Hemogenic EC are subsequently identified from CD45- cells in a differential cKit-APC vs. Flk1-PECy7 daughter plot as double-positive events, and typically represent 1 – 3% of CD45- events when isolated from either AGM (Figure 3D) or YS (not shown). HSPC are identified from the CD45+ fraction in a separate cKit-APC vs. Flk1-PECy7 daughter plot as Flk1-/cKit+ cells, and also typically represent approximately 25 – 30% of the small population of CD45+ cells when obtained from either YS (not shown) or AGM (Figure 3E). Thus, both Hemogenic EC and HSPC are exceptionally rare (~0.01% of total cell events), and it is typical for this protocol to return a few hundred of each cell type even when tissue from multiple embryos are pooled. Gates in daughter plots should be established with reference to negative control groups. In order to distinguish between specific and non-specific antibody staining, gates should be initially drawn in reference to both Unstained controls (Figure 3C), as well as samples that have been treated with fluorescently-conjugated isotype-matched (IgG2A or IgG2B) control antibodies (not shown). This latter control has demonstrated that non-specific staining is minimal by this protocol, therefore while we recommend that isotype-matched control antibodies (e.g., IgG2A-PE or IgG2B-FITC) be initially used to optimize FACS sorter settings and determine gate boundaries, we also find that unstained controls are sufficient to verify gate boundaries and experimental quality during routine FACS sorting. If gates are properly drawn, minimal positive scatter should be observed in single- or double-positive gates when recording from unstained or isotype-matched controls.
Hemogenic endothelial cells from the AGM and HSPC undergo hematopoietic differentiation over 14 days of culture in methylcellulose (Figure 4A). The relative proportion of colony types typically observed in these cultures is dependent upon tissue source. Hemogenic endothelial cells isolated from yolk sac tissues at E8.5-E9.5 give rise to BFU-E, CFU-GM, and few CFU-GEMM5, whereas hemogenic endothelial cells isolated from the E10.5 AGM give rise to predominantly CFU-GEMM12, although other lineages are still observed, as shown in Figure 4B (top left panel). HSPC isolated from E10.5 AGM also give rise predominantly to CFU-GEMM upon culture in methylcellulose (Figure 4B, top middle panel), although these cells will also differentiate into other hematopoietic colony types as well. Non-hemogenic endothelial cells (CD31+/CD45- non-SP) have also been plated (Figure 4B, top right panel). These cells demonstrate no growth in hematopoietic culture after 14 days.
Assays of hematopoietic capacity of individual surface marker expressing cell types, including cells with and without expression of hematopoietic and endothelial markers CD31, Flk1, c-Kit, VE-cad, CD41, and CD45 within the SP fraction of the AGM at E10.5 have been performed and demonstrate that multi-lineage colony forming activity in the E10.5 AGM is restricted to CD31+, VE-cadherin+, c-Kit+, CD41+ and CD45+ SP cells12. Interestingly, colony forming activity was noted in both Flk-1+ and Flk-1- SP cells, but only Flk1+c-Kit+CD45- SP cells gave rise to multi-lineage colonies via an endothelial monolayer intermediate characterized by both "cobblestone" endothelial cell morphology (Figure 4B, bottom left panel) and by Dil-AcLDL uptake12. Furthermore, expression of c-Kit is necessary for hematopoietic activity of AGM SP cells12.
While it has been shown that some myeloid progenitors have similar morphologic characteristics when compared to hemogenic endothelial cells, myeloid progenitor cells express CD45 and cannot generate multi-lineage colonies in vitro. HSPC also generate multi-lineage colonies, but although these cells CD45, they lack Flk-1 expression12,23 and give rise to rounded cell clusters without an underlying endothelial monolayer (Figure 4B, bottom right panel). Therefore, the population we define as hemogenic endothelium (or, Flk-1+/c-Kit+/CD45-SP cells) represent endothelial cells with blood forming potential and robust hematoendothelial gene expression, including GATA-1/2, Lmo2, SCL/Tal-1, Runx-1, c-Kit, CD34, CD41, and CD4511 that are distinct from hematopoietic stem and progenitor cells, as well as their non-hemogenic endothelial cell counterparts.
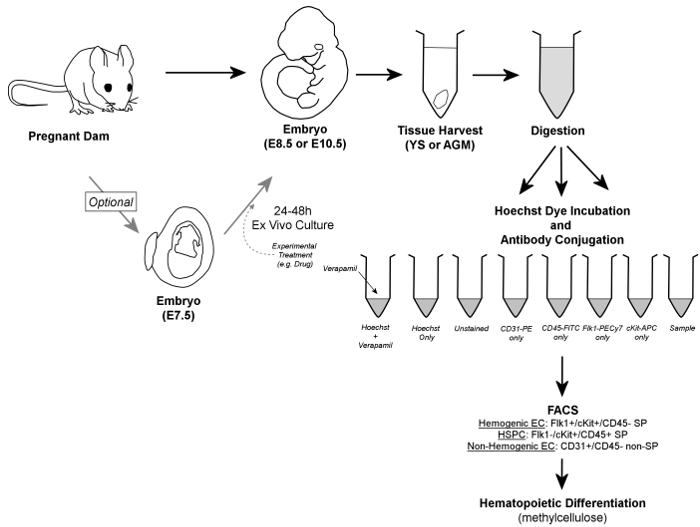
Figure 1. Overall Workflow. In brief, embryos are removed from pregnant dams, and YS or AGM tissues are harvested. Embryos may optionally be cultured for 24 – 48 hr ex vivo prior to tissue harvest. Harvested YS or AGM tissues are digested to a single cell suspension, aliquoted into control and sample tubes, and incubated in the presence of Hoechst dye and/or fluorescently-conjugated antibodies. Verapamil, a calcium channel inhibitor, is also used to generate a negative control essential for verification of accurate gating of the SP fraction. Hemogenic endothelial cells are identified by FACS as Flk1+/cKit+/CD45- SP cells, while HSPC are contained within the Flk1-/cKit+/CD45+ SP fraction of cells; both cell types are sorted onto methylcellulose for confirmation of hemogenic potential. Additionally, non-hemogenic endothelial cells can be discriminated (and retrieved, if so desired) as CD31+/CD45- non-SP cells. Please click here to view a larger version of this figure.

Figure 2. Dissection of YS and AGM Tissues. A) The YS is dissected away from E8.5 embryos, and placed whole into sterile HBSS+ for subsequent digestion. B) The trunk of E10.5 embryos is isolated by making horizontal cuts below both the forelimb and hindlimb buds. The AGM is then separated from the limb buds and ventral tissue using forceps. C) Bright-field images show dissection of both YS and AGM from an E10.5 embryo (scale = 1 mm). Please click here to view a larger version of this figure.
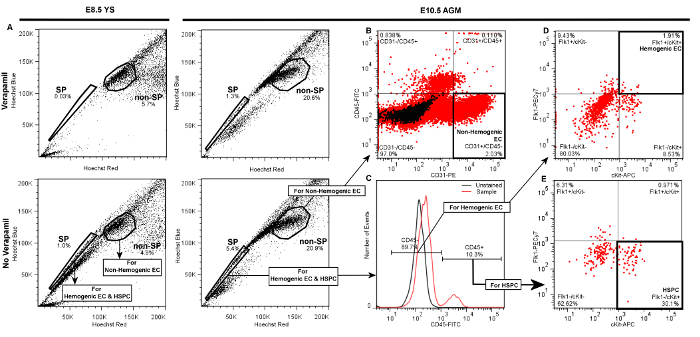
Figure 3. Representative Plots Demonstrating Gate Hierarchy for Discrimination of Hemogenic Endothelial Cells (Flk1+/cKit+/CD45- SP cells), HSPC (Flk1-/cKit+/CD45+ SP cells), and Non-hemogenic Endothelial Cells (CD31+/CD45- non-SP) by FACS from E8.5 YS or E10.5 AGM. Following live cell and doublet discrimination of cells from either YS (left panels) or AGM (right panels) by forward and side scatter (not shown), A) the side population (SP) gate is drawn and verified by a significant decrease of the SP fraction in the Verapamil-treated negative control. The non-SP population is identified as the dense cluster of cells adjacent to the SP. In each of the presented plots 20,000 events are displayed. B) Non-hemogenic endothelial cells are identified as CD31+/CD45- cells within the non-SP fraction, and represent 2 – 5% of non-SP cells whether obtained from AGM (shown) or YS (not shown). To discriminate Hemogenic EC or HSPC, C) daughter gates are drawn from the SP fraction to identify CD45+ and CD45- cells. D) For identification of hemogenic endothelial cells from either AGM (shown) or YS (not shown), additional daughter gates are drawn from the CD45- fraction to distinguish cKit+ (vs. cKit-) and Flk1+ (vs. Flk1-) cells. Hemogenic EC from either YS or AGM are typically ~1 – 3% of CD45- events. E) HSPC are identified from the CD45+ fraction as cKit+ and Flk1-, and typically represent 20 – 30% of CD45+ cells whether sorted from AGM (shown) or YS (not shown). Please click here to view a larger version of this figure.

Figure 4. Visualization of Hematopoietic Differentiation following Culture of Hemogenic Endothelial Cells and HSPC on Methylcellulose. A) Colonies form from sorted individual cells within 7 days of plating onto methylcellulose. Multi-lineage hematopoietic potential can be confirmed by observation of multiple hematopoietic colony types, identified by assessment of distinct colony morphologies11. B) Phase microscopic imaging of sorted hemogenic EC and HSPC from AGM (or YS, not shown) show multi-lineage hematopoietic colony formation after 7 days of culture in methylcellulose culture medium. Non-hemogenic EC from AGM (or YS, not shown) do not demonstrate growth under these conditions. At higher magnification, adherent cells with classical "cobblestone" endothelial cell morphology (white arrow) can be seen giving rise to clusters of hematopoietic cells in cultures of hemogenic EC; no such endothelial cells are observed in cultures of sorted HSPC (scale = 100 μm). Please click here to view a larger version of this figure.
