

Summary
Immunolabeling metoder for å analysere ulike bestander piskehale som henger i utviklingsland sebrafisk hjernen er beskrevet her, som er bredt aktuelt andre vev. Første protokollen skisserer en optimalisert metode for immunolabeling stabile og dynamisk piskehale som henger. Andre protokollen inneholder en metode for å image og kvantifisere begynnende piskehale som henger spesielt.
Abstract
Piskehale som henger (MTs) er dynamisk og skjøre strukturer som er vanskelig å image i vivo, spesielt i virveldyr embryoer. Immunolabeling metoder er beskrevet her for å analysere forskjellige populasjoner av MTs i den utvikle medfødte av sebrafisk fosteret. Mens fokus er på neural vev, er denne metodikken generelt gjelder andre vev. Prosedyrene er optimalisert for tidlig til midten av-somitogenesis-scene embryo (1 somite til 12 somites), men de kan tilpasses en rekke andre faser med relativt små justeringer. Første protokollen inneholder en metode for å vurdere den romlige fordelingen av stabil og dynamisk MTs og utføre en kvantitativ analyse av disse gruppene med bildebehandling programvare. Dette utfyller eksisterende verktøy til bildet microtubule dynamikk og distribusjon i sanntid, bruke transgene linjer eller forbigående uttrykk for merket konstruksjoner. Faktisk er er slike verktøy meget nyttig, men de ikke lett skiller mellom dynamisk og stabil MTs. Bilde og analysere populasjonene distinkte microtubule har viktige implikasjoner for forstå mekanismene bak celle polarisering og morphogenesis. Andre protokollen beskriver en teknikk for å analysere begynnende MTs spesielt. Dette gjøres ved å fange de novo vekst egenskapene for MTs over tid, etter microtubule depolymerization med narkotika-nocodazole og en restitusjonsperiode etter narkotika bleke. Denne teknikken har ennå ikke brukt til studiet av MTs i sebrafisk embryoer, men er en verdifull analysen for å undersøke funksjonen i vivo proteiner involvert i microtubule montering.
Introduction
Piskehale som henger (MTs) er polymerer av α - og β-tubulin som monterer til lineær protofilaments, hvorav flere sammen danner et hult rør1,2. MTs er polarisert strukturer med raskt voksende pluss ender og sakte voksende minus ender som er forankret i centrosome eller andre microtubule-organisere center (MTOC)3. De novo MT-formasjonen er initiert av nucleation på γ-tubulin ring kompleks (γ-TURC), som gir en mal for MT montering4. I en gitt celle, kan to bestander av MTs skilles det slå til ulike priser. Dynamisk MTs utforske sine mobile miljøet ved å bytte mellom faser av vekst og krymping i en prosess som kalles dynamisk ustabilitet5. I motsetning til dynamisk MTs, stabil MTs er ikke hurtigvoksende og har en lengre halveringstid enn dynamisk MTs6.
Tiår med forskning i cell biology har gitt et sofistikert utvalg av verktøy for å studere MT struktur og funksjon og resulterte i en stor mengde kunnskap om disse cellen cytoskjelett elementene. For eksempel MTs spille en sentral rolle i etableringen og vedlikeholdet av cellen polaritet, som skyldes ikke bare til deres iboende polaritet, men også til differensial subcellular distribusjon av stabil versus dynamisk MTs7, 8. derimot langt mindre er forstått om MT arkitektur og funksjon i mer komplekse tredimensjonale (3D) miljøer som virveldyr embryoet, delvis på grunn av utfordringen med imaging MT cytoskeleton med høy oppløsning. Til tross for denne begrensningen, den siste generasjonen av GFP-uttrykke transgene linjer denne etiketten MTs eller forbigående uttrykk for fluorescently-merket MT markører har økt vår forståelse av de dynamiske endringene som MTs gjennomgå og deres cellular og utviklingsmessige rolle i sebrafisk fosteret. Hele MT nettverket kan avbildes transgene linjene i hvilke tubulin er enten direkte merket9 eller tubulin polymerer indirekte merket med MT-assosiert proteiner Doublecortin-lignende-kinase (Dclk) eller Ensconsin (EMTB)10, 11. Andre linjer (og konstruksjoner) har blitt generert som aktiverer vurdering av MT iboende polaritet ved spesielt merking MT pluss ender eller centrosome-forankret minus ender11,12,13, 14. kraften i disse verktøyene ligger i evnen til å studere MT dynamikk i live, utvikle organismer. Slike undersøkelser har avdekket, for eksempel romlige og dynamisk distribusjon av MTs bestemt celle bestander, retningen på mitotisk spindles vev gjennomgår morphogenesis (en indikator på flyet celledeling), polariteten til MT polymer i tilknytning til prosesser som cellen forlengelse og migrasjon, og MT vekstrate bestemmes av kometen hastighet9,13,15. Begrensning av disse verktøyene er at de lett ikke diskriminerer mellom stabil og dynamisk MT bestander.
Tegning fra de rike celle biologi litteraturen, er immunolabeling metoder å image stabil og dynamisk MTs i sebrafisk embryo beskrevet her, som er komplementære til bruken av transgene linjer. Den utstrakte bruken av slike immunolabeling metoder i sebrafisk har vært noe hemmet av vanskeligheten i å bevare MT integritet under fiksering prosedyren. Protokollen 1 skisserer en optimalisert metode for immunolabeling totalt, dynamisk, og stabil MTs i tverrsnitt av det utvikle sebrafisk-hindbrain. Videre er en enkel metode bruke kommersielt tilgjengelig programvare beskrevet for å kvantifisere populasjonene MT. Stabil MTs er forskjellig fra dynamisk MTs basert på flere post-translasjonell modifikasjoner av α-tubulin, som acetylation og detyrosination, som samler seg på stabil MTs over tid16,17. I sebrafisk embryoet skjer acetylation på ciliary og axonal MTs men ikke stabil interphase MTs18, begrense nytten av denne indikatoren til et delsett med stabilisert MTs. Derimot vises detyrosination på alle stabil MTs sebrafisk embryoet18. Denne post-translasjonell modifikasjon eksponerer carboxy-terminal Glutaminsyre α-tubulin (detyrosinated tubulin)18 og kan oppdages ved hjelp av anti-Glu-tubulin19. Selv om detyrosination forekommer fortrinnsvis på stabil MTs, indikerer eksperimentelle bevis at denne post-translasjonell modifikasjon er et resultat av, snarere enn en årsak til, MT stabilitet16. Gjensidige MT befolkningen, består av dynamisk MTs, kjennetegnes ved hjelp av et antistoff, anti-Tyr-tubulin, som anerkjenner spesielt tyrosinated form av α-tubulin19. Etter immunolabeling med disse markører og AC confocal imaging, kvantitativ analyse av MTs (lengde, antall og relative overflod) kan utføres i definerte regioner i den utvikle medfødte. En strømlinjeformet metoden tilbys her for å utføre denne analysen med 3D bildebehandling programvare. Denne metoden kan brukes å svare på spørsmål om morphogenesis og etablering eller modning av cellen polaritet20. Faktisk følger utarbeidelsen av polarisert matriser av stabil MTs mange utviklingsmessige arrangementer, inkludert photoreceptor morphogenesis21, epithelialization celler i utviklingsland medfødte18 og axon formasjon8.
Protokollen 2 beskriver i vivo tilpasning av en celle biologi analysen analysere MTs under deres montering fase (nucleation/anchorage og vekst)22,23. Begynnende MTs er nucleated på centrosome og senere forankret til subdistal vedheng mor centriole23. En metode for å analysere begynnende MT gjengroing følge depolymerization er beskrevet. Denne protokollen gir detaljer om nocodazole behandling til depolymerize MTs, narkotika bleke prosedyren og etter behandling restitusjonsperiode. MT ettervekst overvåkes med jevne mellomroms innlegg bleke av immunolabeling med merker for totalt MTs (anti β-tubulin) sammen med markører for centrosome (anti γ-tubulin) og kjernen (4', 6-diamidino-2-phenylindole (DAPI)), i henhold til de generelle fremgangsmåtene i protokollen 1. MT depolymerization trinnet av denne protokollen er viktig ettersom vurdering av de novo MT vekst ikke utvidelsen av eksisterende MTs. Denne teknikken er derfor forskjellig fra andre publiserte prosedyrer å måle MT vekstrater (i fravær av depolymerization) ved hjelp av en pluss tips markør som end bindende protein 3 smeltet grønne fluorescerende protein (EB3-GFP), som vist i Tran et al., 201211. Videre denne analysen er spesielt nyttig for å analysere embryo defekt i de novo MT montering, som tidligere rapporterte NEDD1 mutanter der rekruttering av γ-tubulin til centrosome er svekket, noe som resulterer i ufullstendige medfødte dannelse og neuronal defekter24.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
etikk uttalelse: fremgangsmåten beskrevet nedenfor følger University of Maryland Baltimore County dyr omsorg retningslinjer.
1. analyse av stabil og dynamisk MTs bruker Immunolabeling (protokollen 1)
- manuell dechorionation av embryo før fiksering
- Hent ferske framsatt embryo helle av overflødig system vann og deretter samle gjenværende embryo i en plast Petriskål (se Tabell av materialer).
- Fjern alle partikler fra system vann og overføre embryo til en ny rett fylt med embryoet medium (se Tabell for materiale) slik at embryoene utvikle i et rent miljø.
- Tillater embryoene å utvikle til det ønskede scenen i et temperaturkontrollert inkubator på 28.5 ° C.
- Sted embryo yngre enn 24 timer etter befruktning (hpf) i et glass rett før dechorionation.
Merk: Dechorionate embryo før fiksering å maksimere rask gjennomtrengning av bindemiddel og bevare MT integritet. Bruk embryoet medium i stedet for system vann å gi det ekstra Ca 2 + kreves under dechorionation. - Fjerne manuelt i chorions fra embryo i Petriskål, med fine tang under dissecting mikroskop.
- Klemme et lite område i den runde, gjennomsiktig plasmocitara som omgir embryoet med et par pinsett og trekke Tang fra hverandre til å opprette et brudd i membranen.
- Forstørre åpningen av delikat nysgjerrige på sprukket plasmocitara ved hjelp av pinsett. Være forsiktig med å ta fosteret med tang som det kunne ruptur.
- Fiksering av iscenesatt embryo
- overføring iscenesatt, dechorionated embryo til 1,5 mL sentrifuge rør. Fjerne så mye embryoet medium som mulig med et glass Pasteur pipette.
Merk: Utføre fiksering og narkotika på unge (midt-somitogenesis) embryo, før dannelsen av nevrale sentrene formidling smerteopplevelsen, som krever ingen ytterligere prosedyre for å lindre smerte under euthanasia. Utviklingsstadier som definert i Kimmel et al., 1995 25. De 4-5 og 11-12 somite trinnene ble brukt til å hente bilder for tall 2 og 3. - Forberede 4% paraformaldehyde (PFA) /MT montering buffer (MAB) bindemiddel (se Tabell for materiale) ved å kombinere 1 mL 8% PFA per 1 mL 2 X MAB og legge 2 µL 100% Triton X-100 per 1 mL av totalvolumet.
FORSIKTIG: Bruk hansker under behandling løsninger som inneholder PFA og Triton X-100, som er hud irritanter. - Fastsette embryo i 1 mL 4% PFA/MAB bindemiddel i 5 min på 28.5 ° C. leveringstanken bindemiddel med en pipette, erstatte den med 1 mL frisk bindemiddel og ruge for 3t ved romtemperatur (RT) på en rocker.
Merk: Eksempler være faste raskt på biologiske temperaturen (28.5 ° C for sebrafisk) å hindre temperaturen-avhengige MT depolymerization.
- overføring iscenesatt, dechorionated embryo til 1,5 mL sentrifuge rør. Fjerne så mye embryoet medium som mulig med et glass Pasteur pipette.
- Sug opp bindemiddel og legge 1 mL 1 x Tris-bufret saline med NP40 (SS-NP40) buffer. Forsiktig røre på RT på en rocker tre ganger i 5 minutter. Lagre embryoer på 4 ° C i 1 mL frisk 1 X TBS-NP40 for mer enn 7 dager.
FORSIKTIG: Bruk hansker når du håndterer løsninger som inneholder NP-40, en hudirriterende. - Sectioning embryo for immunolabeling
- varme RT 4% lavt Smeltepunkt (LMP) agarose innebygging medium i en lukket beholder til løsningen blir klart med en varm plate satt til 50 ° C plassert nær dissecting mikroskop . Beholderene stengt mellom prøvene og oppvarmet hele innebygging prosessen (trinn 1.4.4-1.4.6).
- Overføring embryoer fra det 1,5 mL sentrifuge rør til en Petriskål ved hjelp av en glass pipette og fyll den med 1 X SS-NP40.
- Fjerne store eggeplomme celler fra somitogenesis scenen embryo (4-5 og 11-12 somites) i Petriskål med fine tang under forstørrelsen av en dissecting mikroskop 26. Hold embryoet hale bud med et par pinsett og løsner eggeplomme cellene med den andre par for å bevare hindbrain vev. Overføre de de-yolked embryoene til et område i Petriskål gratis eggeplomme rusk.
Merk: Bygge embryo i agarose-fylt mold å forhindre tidlig herding av LMP agarose. - Fylle en 12 x 5 mm x 3 mm godt av snitting mold med 200 µL smeltet LMP agarose ved hjelp av brønnene. Utføre trinn 1.4.5.-1.4.6. raskt (innen 20 s fylle mold) å legge embryo før LMP agarose kjøler til RT og stivner.
- Bruke fine tang overføre en de-yolked embryoet av tailbud fra Petriskål til agarose-fylt mold mot koniske enden under dissecting mikroskop.
- Bruk fint tang å orientere embryoet i mold slik at vibratome kutt i ønsket flyet. Opprett tverrgående deler av orientere embryoet slik at hindbrain vev går parallelt med lengden av mold dorsal overflaten mot kanten og fremre overflaten overfor konisk regionen slutten. Gjenta trinn 1.4.4-1.4.6 for de gjenværende embryoene.
- Tillat agarose innebygging for å stivne i 5 min på RT.
- Genererer 40 µm deler av høyeste aksen av agarose innebygd embryo (trinn 1.4.1-1.4.7) bruker en vibratome med snitting parabolen fylt med 1 x SS-NP40. Overføre deler av interesse til en 24-vel plate i 500 µL 1 x TBS-NP40 ved hjelp av fine pinsett. Plasserer delene på bare én fosteret per brønn.
Merk: Se referanse til 18 for mer informasjon. Sikre at deler forblir hydrert på alle tider i minst 250 µL av bufferen og rock ved lav hastighet (10-25 rpm) for trinnene for å forhindre at agarose bygger inn. Vaskemidler bør i blokkering og vask løsninger redusere overflatespenning av flytende medium tillate neddykking delene. Kontroller at inndelinger beholdes i brønnene under og etter alle manipulasjoner. Vær forsiktig for å forhindre tilfeldigvis utstøting deler under vasker.
- Fjerner bufferen og legge 500 µL av blokkering løsning. Rock minst 1t på RT.
Merk: Bruk en blokkering løsning som inneholder 5% sera fra verten arter av hvert sekundære antistoff brukes (se Tabell av materialer). - Incubate i 300 µL primære antistoffer fortynnet i blokkering buffer for 36-72 h på 4 ° C på en rocker. Vask to ganger i 600 µL 1 x TBS-NP40 på en rocker i 30 minutter hver, på rett
Merk: Dobbel-label deler av incubating inne primære antistoffer mot totalt MTs (anti β-tubulin eller anti α-tubulin) og stabil MTs (anti-Glu-tubulin) eller dynamiske MTs (anti-Tyr tubulin). Velg primære antistoffer som har vært reist i andre arter når dobbel merking for totalt og post-translationally endret α-tubulin bestander. Se Tabellen for materiale for antistoff fortynninger. - Incubate i 300 µL av fluorophore-konjugerte sekundære antistoffer fortynnet i blokkering buffer på en rocker for 16-24 h, på 4 ° C i mørket. Vask to ganger i 600 µL 1 x TBS-NP40 på en rocker i 30 minutter hver, på rett
Merk: Pakk flere godt parabolen som inneholder sekundære antistoff folie herfra måles arb videre og etter hver manipulasjon å hindre slukker. Velg sekundær antistoffer som reagerer med verten immunglobulin den primære antistoff. Velg sekundær antistoff fluorophores som har egen, ikke-overlappende utslipp spectra. Se Tabellen for materiale for antistoff fortynninger. - Ruge embryo i 500 µL DAPI løsning på en rocker i 30 min, på RT. Wash tre ganger i SS-NP40 rocking på RT i 5 min.
Merk: Kjernefysiske merking skaper mobilnettet kontekst for MT kvantifiseringen utført i trinn 1.12. - Plasser en dråpe montering medium med anti-fade agent på midten av et støvfritt lysbilde. Bruke fine tang overføre deler til montering middels slippverktøyet. Plass en støvfritt dekkglassvæske på prøven. Lagre lysbilder i et tørt, mørkt og svalt sted, innpakket i folie, til bildebehandling utføres.
Merk: Sirkle delene bak på lysbildet ved hjelp av en tynn permanent markør før bildebehandling vil bidra til å identifisere delene ved mikroskopet. - AC confocal Imaging
- Mount deler på en invertert laserskanning AC confocal mikroskop ved påføring lysbildet til scenen med dekkglassvæske mot målet. Fastslå riktige optikk (mål, laser og Kanalinnstillinger som få og forskyvning) en kontroll lysbildet og hold dem konsekvente mellom prøver 27. Unngå oversaturating piksler for å forhindre datatap.
- Ta Z-stabler AC confocal bilder med Kanalinnstillinger for den valgte sekundære antistoff fluorophores og lagre bilde filer 27. Erverve Z-stabler for hver inndeling.
Merk: Gjenskape parameterne brukes til å hente bilder i tall 2 og 3 ved hjelp av følgende oppkjøpet innstillinger: modus = XYZ; objektive forstørrelse = 63 X olje nedsenking linsen; objektive numeriske blenderåpning = 1.4; Z-trinn = 0,1 µm; Z-dybde = 16.23 µm. Bruk følgende Kanalinnstillinger: DAPI eksitasjon med 20% UV-range laser, utslipp filter området = 430-480 nm, photomultiplier (avdrag) gevinst = 525 V og avdrag forskyvning =-1.72%. 448 nm fluorophore (se Tabell for materiale) eksitasjon med 20% 488 nm laser, utslipp filter området = 493-573 nm, avdrag gevinst = 689 V og avdrag offset =-0.2%. 594 nm fluorophore eksitasjon med 32% 594 nm laser, utslipp filter området = 608-706 nm, avdrag gevinst = 768 V og avdrag offset =-6.8%. - Lagre rådataene filene med unike, beskrivende filnavn og lage en kopi for redigering i analyseprogramvare.
- Samling av Z-stabler for å vise maksimal anslag
- Åpne data fil bruke offentlige 3D image analyseprogramvare (f.eks ImageJ). Kontroller at hver kanal vises som en enkelt bildesekvens (Z-stabler).
- Dele bildekanaler ved hjelp av følgende menyen rekkefølge: “ bilder/farge/del kanaler ”.
- Opprette et sammenslått bilde ved overliggende kanaler rundt ved hjelp av følgende menyen rekkefølge: " bilder/farge/Flett kanaler. " Velg 594 nm, 488 nm og DAPI kanaler å være falske rød, grønn og blå, henholdsvis. Sjekk " opprette kompositt " og velg " OK " 28.
Merk: Utelate DAPI kanal og bedre formidle detaljert gjelder MTs i en maksimal projeksjon som i figur 2 og 3, ved bare å velge falske farger for de andre to kanalene. - Undersøk flettede Z-stakken og gjøre oppmerksom på begynnelsen og slutten posisjoner i indre beste Z-flyene for alle synlige kanaler. Avvise ytre Z-flyene som vanligvis har suboptimal tapsfrie ujevne overflater i delen. Se referanse til 29 informasjon.
- Visualisere flettede Z-stakken som en enkelt 2D-bilde ved å utføre en maksimal intensitet projeksjon av Z-stakken bruke følgende 3-D bilde analyse menyen sekvens: " bilder/stabel/Z-prosjekt. " angi start- og posisjoner av de indre beste Z-fly fra trinn 1.11.3 den " Start bit " og " stopp skive, " henholdsvis. Velg " Max intensitet " projeksjon type og klikk " OK ". Se referanse til 28 for mer detaljer.
- Analysere MT merking
- Åpne kommersielle 3-D bilde analyseprogramvare. Velg " opprette bibliotek " og angi et beskrivende navn for bildebiblioteket. Klikk " opprette. " dra raw-bildefiler generert fra AC confocal mikroskopet inn i biblioteket. Større filer krever mer tid å overføre.
- Velge filen du vil analysere. Velg " utvidet fokus " fra den " visningen " menyen for å vise kanal flettede bildet i hovedvinduet.
- Juster terskelverdi ved å dra glidebryteren for for hver kanal til venstre eller høyre til bakgrunnen signalet er redusert og sanne signalet er robust. Observere at hver kanal viser et sant signal for molekylet merket (f.eks, DAPI kanalen viser avlang eller mitotisk kjerner men ikke auto-fluorescens fra cytoplasma eller agarose).
- Velg den " Frihånd område av interesse (ROI) " verktøyet og skissere regionen rundt som skal analyseres. Velg den " handlinger " kategorien etterfulgt av " avling utvalg " å beskjære bildet. Lagre filen beskåret bilde med et nytt navn. Klikk i " målinger " kategorien opprette protokollen for filtrering bestemte objekter relevant for 3D Analyser.
- Dra " finne objekter " til vinduet protokollen. Endre første protokollen " DAPI. " Velg DAPI kanalen på rullegardinmenyen. Dra følgende innstillinger for DAPI-protokollen, og plasser dem under " finne objekter " i følgende rekkefølge (tabell 1): " fylle hullene i ObjectsŔ " Separat berøre ObjectsŔ " Utelate objekter etter SizeŔ " Utelukker ikke berører ROIs ".
Merk: Målet med innstillingene i tabell 1 er å først angi en terskel som forkaster signaler som distribusjon og størrelse stemmer ikke overens med størrelsen på objekter blir analysert. For eksempel fjerne et signal ikke stor nok til å være en kjerne ved telling atomkjerner. - Utføre sekvensen (trinn 1.12.9) å montere de resterende filtre for β-tubulin og andre indikatorer ved hjelp av innstillingene i tabell 1.
- Velg " mål " på bunnen av hver protokoll. Velg " intensitet og volummåling " og " skjelett lengde " for alle tubulin merking, men bare tidligere DAPI signalkabelen.
- Tegn en avkastning rundt regionen skal måles. Se målene under den " Sammendrag " kategorien etter programvaren prosessene regionen. Kopiere dataene og lagre dem på en gjennomførbar regneark, som i tabell 2. Opprette en sikkerhetskopi av regnearket for senere analyse.
- Velg mål av interesse (for eksempel lengden på MT bunt, antall MT pakker/kjernen, som med forskjellige markører) i regnearket og analysere for å finne gjennomsnitt for hver gruppe.
Merk: Den gjennomsnittlige MT bunt lengden = summen av det ' bety skjelettlidelser lengde for β-tubulin ' for hver fosteret delt på totalt antall embryoer. Se rad 20 i tabell 2. Formaterer regnearket slik at variabler og eksperimentelle grupper er lett grafisk.
2. De Novo MT montering analysen (Protocol 2)
- konstruksjon og test flere godt gjennomflytsenhet apparatet ( figur 1) 2 dager før eksperimentet.
Merk: Apparatet aktiveres samtidig bleke for flere eksperimentelle grupper etter nocodazole behandling med forsyninger fra Tabellen for materiale. Silikon lakk krever minst 24 h tørketid før det utgjør ingen toksisitet risiko til embryoene.- Dele en 50 mL sentrifuge rør i halvparten lengderetningen, bruker en gigg eller båndsag.
- Kutte 7 semi sirkler, med en radius på 3 cm av 70 µm nylon maske og klippe dem til å passe tett til en sentrifuge halvparten av Splits rør. Fest semi-sirkler i sentrifuge røret parallelle 10 mL gradering markeringene bruker akvarium-safe silisium sealer. Tillat enheten å tørke i 2 dager og skyll av soaking i et beger vann for 2-3 h.
- Linje den øverste (gjengede) enden av klippe sentrifuge røret med modell-leire slik at høyden på væsken beholdt til flyten gjennom enheten har en dybde på ¼ tomme ( figur 1, visninger 2 og 3).
- Forberede bleke apparatet ved å fjerne stempelet fra en 50-mL sprøyte og sette inn 12 inches av fine rør i spissen. Skyv slangen så langt som mulig, og forsegle rundt felles bruker modellering leire.
- Våt pre mesh ved hjelp av embryoet medium slik at væsken å kjøre gjennom hele gjennomflytsenhet enheten. Vinkel enheten på isen slik at flytende bassengene i alle rom, men fortsatt tømmer ut foran der leire felgen ligger. Bruke en ring stativ, suspendere bleke apparatet over gjennomflytsenhet enheten på is ( figur 1).
- Chill 200 mL av embryoet medium på is, og hell nok i den bleke apparatet å sikre at alle luftbobler fjernes og at infusjonshastigheten er ca 7 mL/min. justere infusjonshastigheten ved å endre høyden på sprøyten.
- Enzymatisk dechorionate embryo
- lage en fungerende løsning av ikke-spesifikk protease fortynne 1 mL 10 mg/mL uspesifisert protease lager i 20 mL embryoet medium.
- Utføre kjemisk dechorionation på embryo 1t før timepoint når de er ventet å nå de ønskede utviklingsstadiet. Ufullstendig chorions ved å fjerne fosteret medium fra 100 mm Petri retter som inneholder iscenesatt embryoer og legge til 20 mL uspesifisert protease fungerende løsning.
- Incubate embryo ved 37 ° C i 5 min.
Merk: Ikke overstige 5 min eller bruk en høyere konsentrasjon av ikke-spesifikk protease løsning, da dette vil resultere i embryoer faller fra hverandre når behandlet med nocodazole. - Raskt Pipetter ut uspesifisert protease og fylle retter med ca 25 mL embryoet medium. Gjentas én gang.
- Bruker en 1-mL glass pipette, overføring embryo yngre enn 24 hpf til glass retter for å beskytte dem fra skader.
- Fullført dechorionation ved å manuelt fjerne chorions med en fin tang, som beskrevet i trinn 1.1.5.
- Plasserer glass Petri retter som inneholder dechorionated befruktede egg i en inkubator 28,5 ° C i minst 30 min før de når ønsket utviklingsstadiet.
- Depolymerize eksisterende MTs
- forberede en fungerende løsning av 5 µg/mL nocodazole ved å kombinere 50 µL 1 mg/mL lager nocodazole med 10 mL isen kalde embryoet medium.
Advarsel: Bruk hansker når du håndterer nocodazole, en hudirriterende. - Bytte embryoet mediet av gruppen nocodazole behandling med 10 mL kaldt nocodazole fungerende løsning. Plasser Petri retter på is etter en passende tid for det utviklingen scenen (for eksempel 1 h for 4-5 somite embryo). Satt av ubehandlet kontroll embryo i en Petriskål på is fikses sammen med bleke prøver i trinn 2.3.4.1.
- Overføring embryo bruker en brann polert 1-mL glass pipette til gjennomflytsenhet apparatet, med separate seksjoner for hver forsøksgruppen. Starte nocodazole bleke av helle isen kalde embryoet medium øverst på 50 mL sprøyte.
Merk: Bruke minst 30 embryo per forsøksgruppen. Eksperimentell grupper kan bestå av kontroll embryo eller en rekke morpholino eller RNA-injisert embryoer. Bleke krever totalt ca 150 mL av embryoet medium legges hver 8-10 min. bleke nocodazole mens du fortsetter å hemme MT vekst med is. Holde embryoene på isen er avgjørende for suksessen til denne analysen fordi MTs er ustabil på kalde temperaturer og kalde forsinkelser utviklingen i tidlig embryo. - Lar MTs å regrow etter 20 min på utvasking på RT ved å overføre embryo glass Petri retter som inneholder varme (28.5 ° C) embryoet mediet bruker en brann polert 1 mL glass pipette. Så snart embryoene overføres, start en tidtaker.
- Reparere de kontroll og bleke embryoene på 1 min, 5 min og 10 min etter pipettering ca 10 embryo i en 1,5 mL sentrifuge rør fylt med 1 mL 4% PFA/MAB fix (28.5 ° C) og følge instruksjonene i trinn 1.2.3.
- forberede en fungerende løsning av 5 µg/mL nocodazole ved å kombinere 50 µL 1 mg/mL lager nocodazole med 10 mL isen kalde embryoet medium.
- Forberede prøver for immunolabeling som beskrevet i delene 1.3-1.5.
- Immunolabel flytende seksjoner og bilde embryo som beskrevet i delene 1.6-1.10 med følgende endringer primære antistoff spesifikasjoner: Bruk 1:500 kanin anti-γ-tubulin og 1:200 mus anti-β-tubulin.
- Prosessen og analysere bilder med 3-D bilde analyseprogramvare, som beskrevet i trinn 1.12.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Analyse av stabil og dynamisk MTs bruker immunolabeling
I protokollen 1, er distribusjon av MT undergrupper under tidlig (nevrale keel) og slutten (nevrale stav) stadier av medfødte utvikling avslørt, bruke Glu-tubulin og Tyr-tubulin som markører for stabil og dynamisk MTs, henholdsvis. Dynamisk MTs dominerer i hindbrain på neural kjøl scenen (4-5 somites) (figur 2A-D). Kjølen utvikler seg til nevrale stangen (11-12 somites), et stadium av forbedret epithelialization, er kvalitativt færre MTs immunoreactive med Tyr-tubulin antistoffer (figur 2Eh), spesielt i ventrale stangen. I kontrast, Glu-tubulin er spredt og vises punctate gjennom nevrale kjølen (Figur 3Ad), men er beriket i ventrale nevrale stangen langs MT områder (Figur 3Eh). Pilspisser peker til bestemte MT pakker eller strukturer der merking er økt.
Selv om både anti-Glu-tubulin og anti-Tyr-tubulin antistoffer ble produsert i samme verten Art (hindre en dobbel merking eksperimentet), indikerer disse resultatene at stabil og dynamisk MT markører sjelden overlapper i sebrafisk-hindbrain. Først har ventrale nevrale stangen mer stabil (Figur 3F) enn dynamisk (figur 2F) MTs. Trenden er motsatt i dorsal nevrale stangen, samsvarer med en modell av sebrafisk neurulation der dorsal vevet forblir dynamiske til medfødte er dannet20. For det andre, mens mitotisk spindler er fullt merket med Tyr-tubulin antistoffer i nevrale kjølen (figur 2D, pilspisser), bare bunnen av spindelen, samtidig med centrosome, er merket med stabilitet markøren Glu-tubulin ( Figur 3 D, pilspisser). Β-tubulin immunofluorescence, felles for begge analyser, informerer eksperimentator av fordelingen av alle MTs og gir et grunnlag for å fjerne ikke-spesifikk merking.
Måle objekter med 3-D bilde analyseprogramvare gir en stor mengde data som kan bli organisert i en praktisk tabell (tabell 2). For å gjøre lengde, antall og området mål, bruker vi bare et delsett av dataene som er tilgjengelige til å analysere. En av komponentene i dataene som vi ikke ytterligere analysere er antall objekter som er identifisert. Dette nummeret brukes som en intern kvalitetskontroll, så hvor ikke skal varierer mye mellom som inndelinger og forholdet mellom kjerner til MTs bør holde lignende i en enkelt behandling tilstand. En avvikende er en indikator som enten analyse må kjøres med justerte filtre eller bildet er så dårlig merket for å analysere. Dermed bør alle avvikende bilder være reanalyzed med justerte innstillinger. Delen avvikende bør undersøkes for underskriver av dårlig merking eller fysisk skade som kan resultere i uvanlig objekt teller. Når analysen er fullført og kvalitetssikret, nyttig informasjon kan gjenopprettes fra rådataene som gjennomsnittlig lengde på totalt MTs og stabilt MTs eller andelen stabil MTs til totalt MTs (tabell 3). I tillegg til disse målingene, kan mange andre beregninger fås med 3-D bilde analyseprogramvare som kan brukes til å trekke slutninger om MTs eller relasjonen til andre cellulære strukturer (kjernen, centrosome, osv.).
De novo MT montering analysen
Nocodazole behandling depolymerizes MTs resulterer i diffus merking (Figur 4A, 4 D, og 4 G). Som MTs regrow, de strekker seg fra centrosome (Figur 4B, 4E, og 4 H), men dette kan ikke være opplagt i en enkelt fly på grunn av deres ikke-plane baner (Figur 4C, 4F, og 4I). Likevel noen analyseprogramvare er i stand til å måle lengder i 3D, slik at en vurdering av MT vekst etter nocodazole bleke (Tabell 4). En viktig observasjon som kan fås fra dataset i Tabell 4 er at mener lengden på MTs synes å øke over tid etter nocodazole bleke i alle regioner i den medfødte analysert. Som nevnt ovenfor, kan andre typer beregninger fra 3D image analyseprogramvare gir mobilnettet sammenheng å tolke MT data (for eksempel forholdet mellom MTs per kjerne).
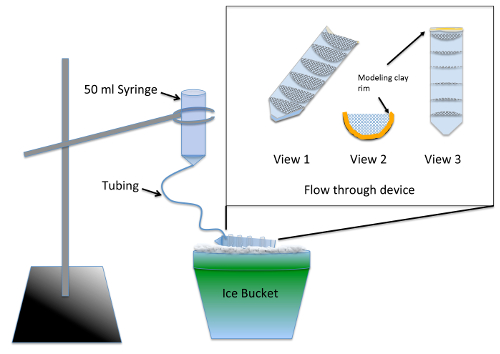
Figur 1 : Illustrasjon av bleke apparater for de novo MT montering analysen. Rammemargen er et nærbilde av gjennomflytsenhet enheten laget maske festet til en 50-mL sentrifuge tube skjær på langs. Mesh compartmentalizes gjennomflytsenhet enheten slik at flere eksperimentelle grupper kan bearbeides samtidig. Under bruk, fosteret medium er lagt til sprøyten og renner sakte gjennom slangen å fylle gjennomflytsenhet enheten, gir en konstant skylling for alle eksperimentelle. Klikk her for å se en større versjon av dette tallet.

Figur 2: Bruk av immunolabeling å image dynamisk MTs. Dechorionated embryo var fast på hensiktsmessige stadier (4-5 i A-D og 12-13 somites i Eh), tvers delt gjennom hindbrain og immunolabeled med antistoffer mot β-tubulin (grønn i A og E) merke alle MTs og tyrosinated α-tubulin (rød i B og F) for å vise dynamiske MT populasjoner. Svært dynamisk MTs kan sees i flettede bilder (C, G) og deres høyere forstørrelse (D, H) som områder der gul etikett er synlig (pilspisser i D, H). Skalere barer = 25 µm (vekselstrøm og E-G) og 10 µm (D og H). Klikk her for å se en større versjon av dette tallet.

Figure 3: bruk av immunolabeling å image stabil MTS Dechorionated embryo ble løst, delt gjennom hindbrain og immunolabeled på hensiktsmessige stadier (4-5 somites i A -D og 12-13 somites i Eh). Stabil MTs er merket med antistoffer mot detyrosinated form av α-tubulin (Glu-tubulin) (rød i B og F) mens totale MTs var visualisert med et generelt β-tubulin antistoff (grønn i A og E). Røde og gule signaler i flettede bilder (C, G) og deres høyere forstørrelse (D, H) representerer områder med høy MT stabilitet (pilspisser i D, H). Skalere barer = 25 µm (vekselstrøm og E-G) og 10 µm (D og H). Klikk her for å se en større versjon av dette tallet.

Figur 4: Bruk av immunolabeling å image begynnende MTs. Dechorionated embryo var fast på 4-5 somites og tvers delt gjennom hindbrain. Delene var immunolabeled med β-tubulin (D, E og F) merke voksende MTs og γ-tubulin (A, Bog C) merke nucleation punkt/centrosome. En dorsal region av medfødte er innestengt i (A, D; B, E og C-F) og vist på høyere forstørrelse (G, H, jeghenholdsvis) å avsløre kjerner (DAPI, blå), centrioles (γ-tubulin, rød) og totalt MTs (β-tubulin, grønn). Hvit pilspisser: colocalization av MTs og centrioles; gul pilspisser: den andre centriole i en celle vises. Skalere barer = 25 µm (A-F) og 10 µm (G-jeg). Klikk her for å se en større versjon av dette tallet.

Tabell 1: Standardinnstillinger for filtrering objekter i 3D image analyseprogramvare.

Tabell 2: representant rå datasett Hentet ved hjelp av 3D bildeanalyser å analysere stabil MTs. Hver kolonne representerer målinger for ett enkelt avsnitt. Min: minste måling; Max: største mål; SD: standardavvik; SE: standardfeil.

Tabell 3: eksempler på datasett som kan fås fra 3D image analyseprogramvare for å kvantifisere stabil MTs. Velg målinger av hvor mener totalt (β-tubulin) og stabil (Glu-tubulin) MTs beregnes ved å ta gjennomsnittet av mener skjelettlidelser lengden på relevante etiketten fra alle prøver (se tabell 2) og andelen stabil å summere MTs ( Glu-tubulin striper per β-tubulin striper) beregnet ved gjennomsnittlig β-tubulin antallet delt på gjennomsnittlig Glu-tubulin greven.

Tabell 4: Eksempler på datasett som kan fås fra 3D image analyseprogramvare å analysere de novo MT montering. Representant resultater fra samlingen de novo MT eksperimentere, sammenligne datasett innhentet for tre utvinning tid poeng (1, 5 og 10 min) etter nocodazole bleke. For hvert punkt vises målinger innhentet for kjernefysisk teller, centrioles (γ-tubulin puncta), antall totale MTs (β-tubulin striper), for valgt regioner i den fotografert analysert (tverrsnitt av den utvikle medfødte).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Det finnes mange metoder for bildebehandling MT dynamikk i tidlig sebrafisk utvikling, mellom live bildebehandling merket molekyler immunolabeling av fast vev11,12,13,14. Selv om MTs i en enkelt celle kan eksistere i dynamisk eller stabil stater, er epithelialization en prosess der MTs er gradvis stabilisert over tid. Bruke markører for stabil og dynamisk MTs tilbyr en måte å visualisere dette fenomenet. Metoden som presenteres her bruker kraften til 3D programvare å kvantifisere overgangen fra dynamisk til stabile MT bestander i et tverrsnitt av embryonale sebrafisk vev. I protokollen 2brukes metoden å merke forskjellige innbyggere begynnende MTs og følge overtiden nucleation og vekst.
MTs er notorisk vanskelig å bilde i sin opprinnelige tilstand på grunn av deres tilbøyelighet til å depolymerize. Dermed er nøkkelen komponenten av denne metoden rask fiksering av MTs gjennom hele fosteret. Dette oppnås starter fiksering på fysiologiske temperaturer og bruker en buffer som både stabiliserer MTs og øker permeabilitet av fosteret. Fiksering tidspunktet er også viktig som redusert fiksering mislykkes å arrestere MTs mens over fiksering kan maskere epitopes, forstyrrer antistoff bindende. Den foreslåtte fiksering tiden av 3-4 h fungerer med embryo som er i midten av gastrulation opptil 24 timer etter befruktning. Embryo mot yngre slutten av tidsskalaen skal være fast for nærmere 3t mens eldre embryo må hele 4 h. Selv med riktig fiksering, vil MTs depolymerize med tiden så snitting og immunolabeling må skje innen en uke med å fikse.
Når vev er riktig fast, kan det oppstå problemer med immunolabeling. Det vanligste problemet oppstod har vært dårlig gjennomtrenging gjennom midten av vev, spesielt hvis for mange inndelinger er ruges i samme brønnen. Øker konsentrasjonen av primære antistoff og inkubasjon tiden for primær og sekundær antistoffer, kombinert med økende vaskemidler for å forbedre permeabilization av embryoene vil forbedre de fleste immunolabeling problemene. Hvis antistoff merkingen mislykkes på grunn av fiksering spørsmål eller immunolabeling problemer, er det mulig å finne årsaken ved å undersøke antistoffer merking mønster. Dårlig fiksering vil føre til intens merking i membranen og diffuse merking i cytoplasma, mens over fiksering vil medføre svakt merking som beholder MT arkitektur. Dårlig gjennomtrengning av antistoffer, vises imidlertid som områder i midten av vevet uten merking.
Muligheten til å analysere MT bilder på en meningsfull måte er avhengig av høy kvalitet tenkelig. For å fange MT lengde i 3D, bør Z-trinn minimumsstørrelsen mulig for målet og numeriske blenderåpning brukes. Bilder vises her ble tatt med en 63 X emersion oljeobjektiv med 1,4 numeriske blenderåpning produsere følgende: pixel = 240 nm, Z-trinn = 0,1 µm, Z-stack størrelse = 16.252 µm. Fordi en enkelt MT er 25 nm, omtrent 10 ganger under grensen på oppløsning for en lett mikroskop, dette metrisk ikke kan være nøyaktig målt ved hjelp av denne teknikken. I stedet kan bare MT lengder lik eller større enn den minste pikselstørrelsen oppnåelig i alle tre dimensjoner måles. Og/eller ramme snitt kan forbedre MT signal definisjonen. MT analyse skal reserveres for høy kvalitet deler. Mens vev med dårlig fiksering ikke fotografert og analysert, mild overfixation kan være balansert med nøye øke laser og få for å gjenkjenne svak samtidig opprettholde en god dynamisk rekkevidde. Dårlig antistoff penetrasjon, kan mens ikke optimalt, korrigeres ved å begrense bildeopptak til godt merket områder, noe som resulterer i imaging en tynnere delen (5-10 µm). Høy bakgrunn fra merking kan kompenseres for ved å justere filterinnstillingene. Men hvis noen av disse justeringene er gjort, er det nødvendig å kontrollere at filtre terskelen akseptabelt på hvert plan av Z-stakken.
3D image analyseprogramvare lar eksperimentator å kvantifisere MT lengde, område, vinkel, overflod og andre beregninger i 3-D løpet av fast vev. Metoden beskrevet her gir retninger for å få slike data ved hjelp av kommersielt tilgjengelig programvare. Filtrering modulene kan imidlertid være tilpasset offentlige programvareforbedret med relevante plugins og/eller makroer, gjør analyse tilgjengelig for alle. Før analyse, må raw-bilder være thresholded å unngå inkludert bakgrunnen og ikke-spesifikk signaler i kvantifiseringen. Når analysen er fullført, og dataene overføres til en gjennomførbar regneark, kan mange slutninger være laget av datasettet. En av beregningene gjort her var Glu-tubulin striper per β-tubulin striper eller andelen stabil MTs til totalt MTs der 1 representerer at hele MT cytoskeleton ble stabilisert seg i en avkastning. Hvis eksperimentator ønsker å utfylle kvantitative data, generere en polert tagged image file format (TIFF) er bildet med skala barer enkel med 3-D bilde analyseprogramvare.
Denne analysen kan funksjonell analyse av proteiner involvert i MT montering, i vivo. Hvis immunolabeling er utført på vekslende føljetong deler, kan denne protokollen brukes til å studere dynamisk og stabil MTs i samme embryo. I fremtiden, vil endringer som økt vaskemidler eller endrede innebygging vinkler tillate bruk av disse metodene for eldre embryoer og et bredere spekter av anatomiske spørsmål.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Forfatterne ikke avsløre.
Acknowledgments
AC confocal mikroskop ble kjøpt med midler fra det amerikanske National Science Foundation (NSF), grant #DBI-0722569. Forskningen ble støttet av amerikanske nasjonale institutter for helse/National Institute of General Medical Sciences (NIH/NIGMS) gi #GM085290 og US Department of Defense (DOD) gi #W81XWH-16-1-0466 til RM Brewster. E. Vital ble støttet av et stipend til UMBC fra Howard Hughes Medical Institute gjennom pre college og Undergraduate Science Education Program, gi #52008090. S.P. Brown ble støttet av en US Department av utdanning GAANN fellesskap, en Meyerhoff Graduate Fellowship finansiert av NIH/NIGMS grant #GM055036, og en forskning Assistantship finansiert av den amerikanske DOD grant #W81XWH-16-1-0466.
Materials
| Name | Company | Catalog Number | Comments |
| Agarose | Used to treat petridishes. Prepare 1% agarose by heating a solution of 1 gram agarose per 100 ml 1X embryo medium in a microwave until polymerized. |
||
| Kpipes | Sigma | P7643 | |
| NaCl | Sigma | S7653 | |
| Tris-HCl | Sigma | T3253-500G | |
| KCl | Sigma | P9333-500G | |
| CaCl2·2H2O | Sigma | C5080 | |
| NP-40 | American Bioanalyticals | AB01424 | |
| EGTA | Sigma | E3889-25G | |
| MgCl2 | Sigma | M2670-500G | |
| Bovine serum albumin (BSA) | Fisher | BP1605 | |
| Triton-x | American Bioanalyticals | AB02025 | |
| Anti-Fade mounting medium | Invitrogen | P10144 | |
| Mouse anti-β-tubulin | Developmental studies Hybridoma Bank | E7 | 1/200 |
| Rabbit anti-γ-tubulin | Genetex | GTX113286 | 1/500 |
| Rabbit anti-α-tubulin | Genetex | GTX108784 | 1/1000* |
| Rabbit anti-detyrosinated-tubulin | Millipore | AB3201 | 1/200-1/1000* Titrate antibody with first use of new lot. |
| Rabbit anti-tyrosinated-tubulin | Millipore | ABT171 | 1/500 |
| Mouse anti-centrin | Millipore | 04-1624 | 1/1000 |
| Goat 488 anti-rabbit | Thermofisher | A11008 | 1/500 |
| Goat 594 anti-rabbit | Thermofisher | A11012 | 1/500 |
| Goat 594 anti-mouse | Thermofisher | A11005 | 1/500 |
| Goat 488 anti-mouse | Thermofisher | A11001 | 1/500 |
| Vibratome | Vibratome | 1500 | |
| Forceps | World Precision Instruments | 555227F | |
| 100 mm petri dish | Cell treat | 229693 | |
| 35 mm petri dish | Cell treat | 229638 | |
| 50 ml falcon tube | Fisher | 14-432-22 | |
| Woven nylon mesh 70 um | Amazon.com | B0043D1SZG | |
| Micropipette | Gilson | F123602 | |
| Glass pipette | Fisher | NC-999363-9 | |
| Aquarium sealant | Amazon.com, by MarineLand | Silicone Sealer 1 oz (Tube) | |
| Ring stand | Fisher | 14-675BO | |
| Microbore PTFE Tubing, 0.022"ID | Cole-Parmer | WU-06417-21 | |
| Modeling clay | Amazon.com | Sargent Art 22-4000 | Any wax or oil based non-toxic modeling clay will suffice |
| Clamp | Fisher | 02-215-466 | |
| 60ml syringe | Fisher | 14-820-11 | |
| Embryo medium (E3) | 34.8 g NaCl 1.6 g KCl 5.8 g CaCl2·2H2O 9.78 g MgCl2·6H2O To prepare a 60X stock, dissolve the ingredients in H2O, to a final volume of 2 L. Adjust the pH to 7.2 with NaOH. Autoclave. To prepare 1X medium, dilute 16.5 mL of the 60X stock to 1 L. |
||
| Blocking Solution | 50 ml TBS-NP-40 2.5 ml normal goat serum 1 g BSA 625 µl Triton-X |
||
| TBS-NP-40 (pH 7.6) | 155 mM NaCl 10 mM Tris HCl 0.1% NP-40 |
||
| 2x MAB (pH6.4) | 160 mM KPIPES 10 mM EGTA 2 mM MgCl2 |
||
| Commercial 3-D Image processing Software | PerkinElmer | Volocity (V 6.2) | |
| Dry block heater | VWR | 12621-108 | Used as a hot plate to melt agarose in Protocol 1. |
| Dissecting Microscope | Leica | MZ12 | |
| Confocal Microscope | Leica | SP5 | |
| Flat embedding mold | emsdiasum.com | BEEM 70904-01 | |
| Public domain image processing software | NIH | ImageJ (V 1.5) | |
| * Success varies by lot number | |||
References
- Akhmanova, A., Steinmetz, M. O. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 9 (4), 309-322 (2008).
- Conde, C., Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 10 (5), 319-332 (2009).
- Kaverina, I., Straube, A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 22 (9), 968-974 (2011).
- Kollman, J. M., Merdes, A., Mourey, L., Agard, D. A. Microtubule nucleation by γ-tubulin complexes. Nat Rev Mol Cell Biol. 12 (11), 709-721 (2011).
- Howard, J., Hyman, A. A. Growth, fluctuation and switching at microtubule plus ends. Nat Rev Mol Cell Biol. 10 (8), 569-574 (2009).
- Schulze, E., Kirschner, M. Dynamic and stable populations of microtubules in cells. J Cell Biol. 104 (2), 277-288 (1987).
- Gundersen, G. G., Kalnoski, M. H., Bulinski, J. C. Distinct populations of microtubules: Tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell. 38 (3), 779-789 (1984).
- Li, R., Gundersen, G. G. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat Rev Mol Cell Biol. 9 (11), 860-873 (2008).
- Asakawa, K., Kawakami, K. A transgenic zebrafish for monitoring in vivo microtubule structures. Dev Dyn Off Publ Am Assoc Anat. 239 (10), 2695-2699 (2010).
- Wühr, M., Tan, E. S., Parker, S. K., Detrich, H. W., Mitchison, T. J. A model for cleavage plane determination in early amphibian and fish embryos. Curr Biol CB. 20 (22), 2040-2045 (2010).
- Tran, L. D., Hino, H., et al. Dynamic microtubules at the vegetal cortex predict the embryonic axis in zebrafish. Development. 139 (19), 3644-3652 (2012).
- Butler, R., Wood, J. D., Landers, J. A., Cunliffe, V. T. Genetic and chemical modulation of spastin-dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis Model Mech. 3 (11-12), 743-751 (2010).
- Yoo, S. K., Lam, P. -Y., Eichelberg, M. R., Zasadil, L., Bement, W. M., Huttenlocher, A. The role of microtubules in neutrophil polarity and migration in live zebrafish. J Cell Sci. 125 (23), 5702-5710 (2012).
- Andersen, E. F., Halloran, M. C. Centrosome movements in vivo correlate with specific neurite formation downstream of LIM homeodomain transcription factor activity. Development. 139 (19), 3590-3599 (2012).
- Lee, S. -J. Dynamic regulation of the microtubule and actin cytoskeleton in zebrafish epiboly. Biochem Biophys Res Commun. 452 (1), 1-7 (2014).
- Bulinski, J. C., Gundersen, G. G. Stabilization and post-translational modification of microtubules during cellular morphogenesis. BioEssays. 13 (6), 285-293 (1991).
- Magiera, M. M., Janke, C. Chapter 16 - Investigating Tubulin Posttranslational Modifications with Specific Antibodies. Methods Cell Biol. 115, 247-267 (2013).
- Hong, E., Jayachandran, P., Brewster, R. The polarity protein Pard3 is required for centrosome positioning during neurulation. Dev Biol. 341 (2), 335-345 (2010).
- Westermann, S., Weber, K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 4 (12), 938-948 (2003).
- Jayachandran, P., Olmo, V. N., et al. Microtubule-associated protein 1b is required for shaping the neural tube. Neural Develop. 11, 1 (2016).
- Nam, S. -C. Role of Tau, a microtubule associated protein, in Drosophila photoreceptor morphogenesis. Genes N Y N 2000. 54 (11), 553-561 (2016).
- Abal, M., Piel, M., Bouckson-Castaing, V., Mogensen, M., Sibarita, J. -B., Bornens, M. Microtubule release from the centrosome in migrating cells. J Cell Biol. 159 (5), 731-737 (2002).
- Delgehyr, N., Sillibourne, J., Bornens, M. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci. 118 (8), 1565-1575 (2005).
- Manning, J. A., Lewis, M., Koblar, S. A., Kumar, S. An essential function for the centrosomal protein NEDD1 in zebrafish development. Cell Death Differ. 17 (8), 1302-1314 (2010).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Dev Dyn Off Publ Am Assoc Anat. 203 (3), 253-310 (1995).
- Beck, A. P., Watt, R. M., Bonner, J. Dissection and Lateral Mounting of Zebrafish Embryos: Analysis of Spinal Cord Development. JoVE J Vis Exp. (84), e50703 (2014).
- FÖldes-Papp, Z., Demel, U., Tilz, G. P. Laser scanning confocal fluorescence microscopy: an overview. Int Immunopharmacol. 3 (13-14), 1715-1729 (2003).
- Ferreira, T., Rasband, W. S. ImageJ User Guide - IJ 1.46. , Available from: https://imagej.nih.gov/ij/docs/guide/ (2010).
- Z-functions - ImageJ. , Available from: https://imagej.net/Z-functions (2017).
