

Summary
Immunolabeling metodi per analizzare popolazioni distinte dei microtubuli in zebrafish sviluppo del cervello sono descritti qui, che sono ampiamente applicabili ad altri tessuti. Il primo protocollo delinea un metodo ottimizzato per immunolabeling stabile e dinamica dei microtubuli. Il secondo protocollo fornisce un metodo per immagine e quantificare i microtubuli nascenti in particolare.
Abstract
Microtubuli (MTs) sono strutture dinamiche e fragile che stanno sfida a immagine in vivo, in particolare in embrioni vertebrati. Immunolabeling metodi sono descritte qui per analizzare popolazioni distinte di MTs nel tubo neurale in via di sviluppo dell'embrione di zebrafish. Mentre l'attenzione è il tessuto neurale, questa metodologia è ampiamente applicabile ad altri tessuti. Le procedure sono ottimizzate per presto ad metà-somitogenesis-stage embrioni (1 somite a 12 somiti), tuttavia possono essere adattati ad una gamma di altre fasi con relativamente piccoli aggiustamenti. Il primo protocollo fornisce un metodo per valutare la distribuzione spaziale delle MTs stabile e dinamico ed eseguire un'analisi quantitativa di queste popolazioni con il software di elaborazione delle immagini. Questo approccio si integra con gli strumenti esistenti per immagine microtubule dynamics e distribuzione in tempo reale, utilizza linee transgeniche o espressione transitoria di costrutti con tag. Infatti, tali strumenti sono molto utili, tuttavia non facilmente distinguere MTs dinamica e stabile. La capacità di immagine e analizzare queste popolazioni distinte microtubule ha implicazioni importanti per la comprensione dei meccanismi alla base di polarizzazione cellulare e morfogenesi. Il secondo protocollo delinea una tecnica per analizzare in particolare il nascente MTs. Questa operazione viene eseguita mediante l'acquisizione le proprietà di crescita de novo di MTs nel tempo, seguendo la depolimerizzazione dei microtubuli con il nocodazole di droga e un periodo di recupero dopo interruzione del farmaco. Questa tecnica non è ancora stata applicata allo studio di MTs in embrioni di zebrafish, ma è un saggio prezioso per indagare la funzione in vivo delle proteine implicate in microtubuli.
Introduction
Microtubuli (MTs) sono polimeri di α - e β-tubulina che assemblare in protofilamenti lineari, molte delle quali si combinano per formare un tubo cavo1,2. Le MT sono strutture polarizzate, con rapida crescita plus estremità ed a crescita lenta meno le estremità che sono ancorate al centrosoma o altri di organizzazione dei microtubuli (MTOC) centro3. De novo Formazione di MT è iniziata da nucleazione presso l'anello di γ-tubulina complessa (γ-TURC), che fornisce un modello per MT montaggio4. In ogni cellula, due popolazioni di MTs possono essere distinte che girano sopra ai tassi differenti. MTs dinamico esplorare il loro ambiente cellulare passando tra le diverse fasi di crescita e di ritiro in un processo noto come instabilità dinamica5. A differenza di dinamica MTs, MTs stabile sono non-crescita e hanno un'emivita più lunga rispetto a dinamiche MTs6.
Decenni di ricerca in biologia cellulare ha fornito una sofisticata gamma di strumenti per studiare la funzione e la struttura MT e ha provocato un grande corpo di conoscenza su questi elementi del citoscheletro. Per esempio, MTs gioca un ruolo centrale nella creazione e mantenimento della polarità cellulare, che è attribuibile non solo alla loro polarità intrinseca, ma anche per la distribuzione subcellulare differenziale della scuderia contro dinamico MTs7, 8. al contrario, molto meno è capito circa MT architettura e funzione in ambienti (3D) tridimensionale più complessi, come l'embrione dei vertebrati, in parte dovuto la sfida di imaging del citoscheletro MT ad alta risoluzione. Nonostante questa limitazione, la più recente generazione di transgenici esprimenti GFP linee quell'etichetta MTs o transitoria espressione di marcatori di MT fluorescente-Tag ha aumentato la nostra comprensione dei cambiamenti dinamici che MTs subiscono e loro cellulare e ruolo dello sviluppo nell'embrione di zebrafish. L'intera rete MT può essere imaged in linee transgeniche in cui tubulina è sia direttamente con etichetta9 o tubulina polimeri sono etichettati indirettamente utilizzando proteine associate MT Doublecortin-like-chinasi (Dclk) o Ensconsin (EMTB)10, 11. Altre linee (e costrutti) sono stati generati che consentire la valutazione della polarità intrinseca MT identificando specificamente MT più finisce o ancorato centrosoma meno estremità11,12,13, 14. la potenza di questi strumenti si trova nella capacità di studiare la dinamica di MT in diretta, lo sviluppo di organismi. Tali studi hanno rivelato, ad esempio, la distribuzione spaziale e dinamica di MTs in popolazioni di cellule specifiche, l'orientamento di mitotic mandrini in tessuti in fase di morfogenesi (un indicatore del piano di divisione delle cellule), la polarità del polimero MT per quanto riguarda processi quali allungamento delle cellule e la migrazione e tasso di crescita di MT determinata dalla cometa velocità9,13,15. La limitazione di questi strumenti è che essi non prontamente discriminano tra popolazioni MT stabile e dinamici.
Attingendo alla letteratura biologia cellulare ricca, immunolabeling metodi di immagine stabile e dinamico MTs nell'embrione di zebrafish sono descritti qui, che sono complementari all'uso di linee transgeniche. L'uso molto diffuso di tali metodi immunolabeling in zebrafish è stata un po' ostacolata dalla difficoltà nel preservare l'integrità MT durante la procedura di fissazione. 1 protocollo delinea un metodo ottimizzato per immunolabeling totale, dinamico, e stabile MTs in sezioni trasversali del hindbrain zebrafish in via di sviluppo. Inoltre, un metodo diretto utilizzando commercialmente disponibile software è descritto per quantificare queste popolazioni di MT. MTs stabile si distinguono da MTs dinamico basato su diverse modificazioni post-traduzionali di α-tubulina, quali acetilazione e detyrosination, che si accumulano su MTs stabile nel tempo16,17. Nell'embrione di zebrafish, acetilazione si verifica su MTs ciliare e axonal ma non su stabile interfase MTs18, limitando l'utilità di questo indicatore a un sottoinsieme di MTs stabilizzato. Al contrario, detyrosination sembra verificarsi su tutte le MTs stabile nel quartiere di embrione di zebrafish18. Questa modificazione post-traduzionale espone l'acido glutammico carbossi-terminale di α-tubulina (detyrosinated tubulina)18 e può essere rilevata utilizzando anti-Glu-tubulina19. Anche se detyrosination si verifica preferenzialmente su MTs stabile, la prova sperimentale indica che questa modificazione post-traduzionale è un risultato di, piuttosto che una causa di, MT stabilità16. La popolazione di reciproco MT, composta da MTs dinamico, si distingue usando un anticorpo, anti-Tyr-tubulina, che riconosce specificamente la forma di termine di α-tubulina19. A seguito di immunolabeling con questi marcatori e imaging confocale, l'analisi quantitativa di MTs (lunghezza, numero e abbondanza relativa) può essere eseguita in regioni definite del tubo neurale in via di sviluppo. Qui vi attende un metodo semplificato per l'esecuzione di questa analisi utilizzando software di elaborazione delle immagini 3D. Questo metodo può essere applicato per rispondere alle domande per quanto riguarda la morfogenesi e l'istituzione o la maturazione delle cellule polarità20. Infatti, l'elaborazione di matrici polarizzate di MTs stabile accompagna molti eventi inerenti allo sviluppo, tra cui fotorecettore morfogenesi21, epithelialization delle cellule nello sviluppo del tubo neurale18 e assone formazione8.
Protocollo n. 2 descrive un adattamento in vivo di un'analisi di biologia delle cellule per analizzare MTs durante il loro montaggio fase (nucleazione/anchorage e crescita)22,23. Nascente MTs sono nucleate presso il centrosoma e successivamente ancorati alla subdistal appendici della madre Centriolo23. È descritto un metodo per analizzare la nascente ricrescita MT seguendo la depolimerizzazione. Questo protocollo fornisce dettagli sul trattamento nocodazole a depolymerize MTs, la procedura di washout di droga e il periodo di recupero post-trattamento. MT. re-crescita viene monitorata ad intervalli regolariwashout post s da immunolabeling con marcatori per MTs totale (anti β-tubulina) al fianco di marcatori per il centrosoma (anti γ-tubulina) e nucleo (4', 6-diamidino-2-phenylindole (DAPI)), secondo le procedure generali descritte nel protocollo n. 1. Il passo di depolimerizzazione MT del presente protocollo è essenziale in quanto permette la valutazione della crescita MT de novo piuttosto che estensione di preesistenza MTs. Questa tecnica è quindi distinta da altre procedure pubblicate per misurare i tassi di crescita di MT (in assenza di depolimerizzazione) utilizzando un marcatore con punta plus come fine proteina 3 fusa alla proteina fluorescente verde (EB3-GFP), come mostrato in Tran et al., 201211. Inoltre, questo test è particolarmente utile per l'analisi di embrioni difettosi in de novo MT assieme, come ad esempio i mutanti NEDD1 precedentemente segnalati in cui il reclutamento di γ-tubulina per il centrosoma è alterato, conseguente incompleta formazione del tubo neurale e difetti di un neurone24.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Dichiarazione etica: le procedure descritte di seguito Segui l'Università delle linee guida di cura degli animali di Maryland Baltimore County.
1. analisi di stabile e dinamico MTs utilizzando Immunolabeling (protocollo 1)
- dechorionation manuale degli embrioni prima della fissazione
- ottenere appena generato embrioni versando l'acqua in eccesso di sistema e quindi raccogliere embrioni rimanenti in una capsula di Petri di plastica (vedere Tabella materiali).
- Rimuovi eventuali detriti dagli embrioni di acqua e il trasferimento del sistema ad un nuovo piatto riempito con il mezzo di embrione (vedere Tabella materiali) per garantire che gli embrioni si sviluppano in un ambiente pulito.
- Consentire gli embrioni a sviluppare fino alla lavorazione in un incubatore termostatato a 28,5 ° C.
- Embrioni posto meno di 24h post-fertilizzazione (hpf) in un piatto di vetro prima di dechorionation.
Nota: Embrioni di Dechorionate prima della fissazione per massimizzare la penetrazione rapida dell'integrità MT fissativo e riserva. Utilizzo medio di embrione invece dell'acqua del sistema di fornire le ulteriori Ca 2 + richiesto durante dechorionation. - Rimuovere manualmente il chorions da embrioni mentre nella capsula di Petri, utilizzando una pinzetta sotto un microscopio per dissezione.
- Una piccola zona nel corion rotonda, trasparente che circonda l'embrione con un paio di pinze e delicatamente tirare forcipe a pezzi per creare una rottura della membrana di pizzicare.
- Ingrandire l'apertura facendo leva delicatamente il chorion rotto usando il forcipe. Fare attenzione a non toccare l'embrione con il forcipe, come potrebbe rompere.
- Fissazione degli embrioni in fasi
- trasferimento in scena, dechorionated embrioni per provette per centrifuga da 1,5 mL. Rimuovere quanto più medio di embrione possibili utilizzando un pipetta Pasteur di vetro.
Nota: Eseguire trattamenti di fissazione e droga sugli embrioni di giovani (metà-somitogenesis), prima della formazione dei centri neurali che mediano la sensazione di dolore, che non richiedono alcuna procedura di additonal per alleviare il dolore durante l'eutanasia. Fasi di sviluppo sono definite a Kimmel et al., 1995 25. Le fasi di somite 4-5 e 11-12 sono state utilizzate per ottenere immagini di figure 2 e 3. - Preparare 4% paraformaldeide (PFA) /MT Assemblea fissativo buffer (MAB) (vedere Tabella materiali) combinando 1 mL 8% PFA / 1 mL 2 X MAB e aggiungendo 2 µ l 100% Triton X-100 per 1 mL di volume totale.
Attenzione: Indossare guanti durante la manipolazione di soluzioni contenenti PFA e Triton X-100, che sono irritanti per la pelle. - Fix embrioni in 1 mL 4% PFA/MAB di fissativo per 5 min a 28,5 ° C. aspirato il fissativo con una pipetta, sostituirlo con 1 mL di fissativo fresco e incubare per 3 ore a temperatura ambiente (TA) su un agitatore meccanico.
Nota: Campioni devono essere risolto rapidamente alla loro temperatura biologica (28,5 ° C per zebrafish) per prevenire la depolimerizzazione di temperatura-dipendente MT.
- trasferimento in scena, dechorionated embrioni per provette per centrifuga da 1,5 mL. Rimuovere quanto più medio di embrione possibili utilizzando un pipetta Pasteur di vetro.
- Aspirare fissativo e aggiungere 1 mL 1 x soluzione fisiologica tamponata con NP40 buffer (TBS-NP40). Agitare delicatamente a RT su un rocker tre volte per 5 minuti ciascuno. Conservare gli embrioni a 4 ° C in 1 mL 1 fresco X TBS-NP40 per non più di 7 giorni.
Attenzione: Indossare guanti durante la manipolazione di soluzioni contenenti NP-40, irritante per la pelle.
Agarosio - embrioni di sezionamento per immunolabeling
- calore RT 4% basso punto di fusione (LMP) mezzo di inclusione in un contenitore chiuso fino a quando la soluzione diventa chiara utilizzando una piastra calda impostata a 50 ° C, posizionato nei pressi di un microscopio per dissezione . Tenere il contenitore chiuso tra campioni e riscaldata tutto il processo di incorporamento (passi 1.4.4-1.4.6).
- Trasferire gli embrioni da 1,5 mL provette per una capsula di Petri con una pipetta di vetro da centrifuga e riempirlo con 1 X TBS-NP40.
- Rimuovere le cellule di grande tuorlo da embrioni in fase somitogenesis (4-5 e 11-12 somiti) di Petri utilizzando una pinzetta sotto l'ingrandimento di un microscopio per dissezione 26. Tenere l'embrione per il germoglio di coda con un paio di pinze e staccarsi le cellule di tuorlo con l'altra coppia al fine di preservare il tessuto del hindbrain. Trasferire gli embrioni de-massicce ad un'area di Petri senza residui di tuorlo.
Nota: Incorporare embrioni nello stampo pieno di agarosio singolarmente per evitare il rapido indurimento di agarosio LMP. - Riempimento un 12 x 5 mm x 3 mm ben dello stampo sezionamento con 200 µ l fuso LMP agarosio utilizzando una micropipetta. Eseguire la procedura 1.4.5.-1.4.6. rapidamente (entro 20 s di riempimento dello stampo) per incorporare l'embrione prima l'agarosio LMP si raffredda a RT e solidifica.
- Utilizzare una pinzetta per trasferire un embrione de-massicce dalla tailbud dalla capsula di Petri per la muffa dell'agarosi-riempita verso la sua fine conico sotto un microscopio per dissezione.
- Il forcipe fine uso per orientare l'embrione nello stampo, tale che il vibratome taglia dentro il piano desiderato. Creare sezioni trasversali orientando l'embrione, tale che il tessuto del hindbrain corre parallelo alla lunghezza dello stampo con la sua superficie dorsale rivolta verso il bordo e la sua superficie anteriore rivolto verso l'estremità affusolata regione. Ripetere i passaggi 1.4.4-1.4.6 per i rimanenti embrioni.
- Consenti l'incorporamento dell'agarosi per solidificare per 5 min a RT.
- Generare 40 µm sezioni dell'asse più alto di agarosio incorporato embrioni (misure 1.4.1-1.4.7) usando un vibratomo con il taglio piatto riempito con 1 x TBS-NP40. Trasferimento sezioni di interesse a una piastra a 24 pozzetti in 500 µ l 1 x TBS-NP40 utilizzando una pinzetta. Inserire le sezioni di un solo embrione per pozzetto.
Nota: Fare riferimento per fare riferimento a 18 per maggiori dettagli. Assicurare che le sezioni restano idratate a tutte le volte in almeno 250 µ l di tampone e roccia a bassa velocità (10-25 rpm) per i passaggi rimanenti per impedire la separazione dall'incorporamento di agarosio. Detergenti presentano in blocco e soluzioni di lavaggio dovrebbero ridurre la tensione superficiale del liquido di coltura e consentire sommersione delle sezioni. Controllare che le sezioni restano nei pozzetti durante e dopo tutte le manipolazioni. Usare cautela per evitare che accidentalmente scartando sezioni durante lavaggi.
- Rimuovere il buffer e aggiungere 500 µ l di soluzione bloccante. Roccia per almeno 1 h a RT.
Nota: Utilizzare una soluzione di blocco che contiene siero di 5% da specie ospiti di ogni anticorpo secondario da utilizzare (vedere Tabella materiali). - Incubare in anticorpi primari di 300 µ l diluiti in tampone bloccante per 36-72 h a 4 ° C su un agitatore meccanico. Lavare due volte a 600 µ l 1 x TBS-NP40 su un rocker per 30 minuti ciascuno, a TA.
Nota: Sezioni doppio-etichetta incubando in anticorpi primari contro totale MTs (anti β-tubulina, o anti α-tubulina) e stabile MTs (anti-Glu-tubulina) o dinamici MTs (tubulina anti-Tyr). Selezionare gli anticorpi primari che sono stati sollevati in specie differenti ospite quando doppia etichettatura per totale e traduzionalmente modificati popolazioni di α-tubulina. Fare riferimento alla Tabella materiali per diluizioni di anticorpo. - Incubare a 300 µ l di anticorpi secondari coniugati fluoroforo diluito in tampone bloccante su un rocker per 16-24 h, a 4 ° C al buio. Lavare due volte a 600 µ l 1 x TBS-NP40 su un rocker per 30 minuti ciascuno, a TA.
Nota: Avvolgere il piatto multi-pozzetto contenente anticorpo secondario in un foglio da questo punto in poi e dopo ogni manipolazione per evitare l'estinzione. Selezionare anticorpi secondari che reagiscono con l'immunoglobulina di host dell'anticorpo primario. Scegliere fluorofori anticorpo secondario che hanno separati, non sovrapposte spettri di emissione. Fare riferimento alla Tabella materiali per diluizioni di anticorpo. - Incubare gli embrioni in 500 µ l di soluzione DAPI su un rocker per 30 min, a RT. Wash tre volte in TBS-NP40 dondolo a temperatura ambiente per 5 minuti ciascuno.
Nota: Etichettatura nucleare fornisce contesto cellulare per la quantificazione di MT eseguita nel passaggio 1.12. - Porre una goccia di mezzo di montaggio con agente anti-dissolvenza al centro di una diapositiva privo di polvere. Utilizzare una pinzetta per trasferire sezioni la gocciolina medio di montaggio. Porre un coprivetrino privo di polvere sopra il campione. Archiviare le diapositive in un luogo asciutto, buio e fresco, avvolto in carta stagnola, finché non viene eseguita la formazione immagine.
Nota: Girando le sezioni sul retro della diapositiva utilizzando un pennarello indelebile punta fine prima di formazione immagine vi aiuterà a identificare le sezioni quando si utilizza il microscopio. - Imaging confocale
- sezioni di supporto su un laser invertito microscopio confocale a scansione apponendo il vetrino sul palco con il coprioggetto rivolto verso l'obiettivo. Determinare l'ottica appropriata (obiettivo, il laser e le impostazioni di canale come guadagno e offset) su un vetrino di controllo e tenerli coerente tra campioni 27. Evitare oversaturating i pixel per evitare perdite di dati.
- Cattura immagini confocal Z-stack utilizzando impostazioni di canale per l'anticorpo secondario selezionato fluorofori e salvare il file di immagine 27. Acquisire Z-stack per ogni sezione.
Nota: Replicare i parametri utilizzati per acquisire le immagini nelle figure 2 e 3 utilizzando le seguenti impostazioni di acquisizione: modalità = XYZ; ingrandimento dell'obiettivo = 63 X obiettivo d'immersione in olio; apertura numerica dell'obiettivo = 1.4; Z-passo = 0,1 µm; Profondità Z = 16,23 µm. utilizzare le seguenti impostazioni di canale: eccitazione DAPI con 20% UV-gamma laser, intervallo di filtro di emissione = 430-480 nm, fotomoltiplicatore (PMT) guadagno = 525 V e PMT offset =-1.72%; 448 nm fluoroforo (vedere Tabella materiali) eccitazione con 20% laser a 488 nm, intervallo di filtro di emissione = 493-573 nm, guadagno PMT = 689 V e PMT offset = -0,2%; eccitazione di fluoroforo 594 nm con 32% 594 nm del laser, intervallo di filtro di emissione = 608-706 nm, guadagno PMT = 768 V e PMT offset = -6,8%. - Salvare file di dati raw con nomi di file univoci, descrittivo e creare una copia per la modifica nel software di analisi di immagine.
- Compilation di Z-stack per la visualizzazione di massime proiezioni
- aprire la copia del file di dati utilizzando software di analisi immagine 3-d di pubblico dominio (ad es., ImageJ). Verificare che ogni canale viene visualizzato come una sequenza di immagini singole (Z-stack).
- Dividere canali dell'immagine utilizzando la seguente sequenza di menu: “ canali di immagini/colore/Split ”.
- Creare un'immagine unita sovrapponendo i canali di interesse utilizzando la seguente sequenza di menu: " canali di immagini/colore/Merge. " selezionare il 594 nm, 488 nm e canali DAPI per essere falso colore rosso, verde e blu, rispettivamente. Verifica " composito crea " e selezionare " OK " 28.
Nota: Il canale DAPI per meglio convogliare dettaglio specifico per MTs in una proiezione massima come in Figura 2 e 3, a solo selezionando falsi colori per gli altri due canali di omettere. - Esaminare lo stack di Z Unito e prendere nota di inizio e fine posizioni di Z-piani migliori interiori per tutti i canali visibili. Allontanare i Z-piani esterni che in genere hanno segnale non ottimale a causa di superfici irregolari della sezione. Fare riferimento per fare riferimento a 29 per maggiori info.
- Visualizzare lo Z-stack Unite come un'unica immagine 2D eseguendo una proiezione di massima intensità dello Z-stack utilizzando la seguente sequenza di menu analisi immagine 3-d: " immagini/pila/Z-progetto. " inserire posizioni dei migliori interiore l'iniziale e finale Z-aerei dal passaggio 1.11.3 come il " inizio fetta " e " Stop fetta, " rispettivamente. Selezionare " intensità Max " come il tipo di proiezione e fare clic su " OK ". Fare riferimento per fare riferimento a 28 per maggiori dettagli.
- MT analisi etichettatura
- aprire il software di analisi di immagine 3D commerciale. Selezionare " Crea libreria " e fornire un nome descrittivo per la libreria di immagini. Fare clic su " crea. " trascinare file immagine raw generati dal microscopio confocale nella libreria. File di dimensioni maggiori richiedono più tempo per trasferire.
- Selezionare un file da analizzare. Scegliere " messa a fuoco estesa " dalla " vista " menu per visualizzare l'immagine canale-fuse nella finestra principale.
- Regolare la soglia trascinando lo strumento slider per ogni canale verso sinistra o verso destra fino a quando il segnale di fondo è ridotto e il vero segnale è robusto. Osservare che ogni canale mostra un vero segnale per la molecola etichettato (ad es., canale DAPI che mostrano nuclei oblunghi o mitotici ma non auto-fluorescenza dal citoplasma o agarosio).
- Seleziona il " Freehand regione di interesse (ROI) " tool e delineare la regione di interesse per essere analizzati. Selezionare il " azioni " scheda seguita da " Ritaglia immagine " per ritagliare l'immagine. Salvare il file di immagine ritagliata con un nuovo nome. Fare clic sul " misure " scheda per creare il protocollo per il filtraggio di oggetti specifici rilevanti per l'analisi 3D.
- Drag " trovare oggetti " alla finestra del protocollo. Rinominare il primo protocollo " DAPI. " selezionare il canale DAPI nel menu a discesa. Trascinare le seguenti impostazioni per il protocollo DAPI e metterli sotto " trovare oggetti " nell'ordine seguente (tabella 1): " riempire buchi in ObjectsŔ " Separata toccare ObjectsŔ " Escludere gli oggetti da SizeŔ " Escludere non toccare ROIs ".
Nota: L'obiettivo delle impostazioni nella tabella 1 sono innanzitutto impostare una soglia che rigetti segnali cui distribuzione e dimensione non sono coerenti con le dimensioni degli oggetti analizzati. Ad esempio, eliminare un segnale non abbastanza grande da essere un nucleo quando si contano i nuclei. - Eseguire la sequenza (passo 1.12.9) per assemblare il restante filtri per β-tubulina e altri marcatori utilizzando le impostazioni nella tabella 1.
- Selezionare " misura " nella parte inferiore di ogni protocollo. Scegliere " intensità e Volume misura " e " lunghezza scheletrica " per tutte le tubuline etichettatura, ma soltanto il primo per il segnale DAPI.
- Disegnare un ROI intorno alla regione da misurare. Osservare le misure sotto il " Riassunto " scheda dopo il software elabora la regione. Copiare i dati e salvarli in un foglio di calcolo realizzabile, come illustrato nella tabella 2. Creare una copia di backup del foglio di calcolo per analisi successive.
- Selezionare misure di interesse (ad esempio, la lunghezza del fascio di MT, numero di fasci di MT/nucleo, come rivelato con diversi marcatori) nel foglio di calcolo e analizzare per determinare i valori medi per ogni gruppo.
Nota: La media MT Lunghezza pacco = somma del ' significa lunghezza scheletrica per β-tubulina ' per ciascun embrione diviso per il numero totale di embrioni. Fare riferimento alla riga 20 della tabella 2. Formattare il foglio di calcolo, in modo che le variabili e gruppi sperimentali sono facilmente graphed.
2. De Novo MT Assembly Assay (protocollo n. 2)
- test multi-flusso-attraverso apparato ( Figura 1) 2 giorni prima dell'esperimento e costrutto.
Nota: L'apparecchio consente di interruzione simultanea di più gruppi sperimentali dopo il trattamento di nocodazole utilizzando forniture da Tabella materiali. Sigillante siliconico richiede almeno 24 ore di tempo di asciugatura prima di esso non pone alcun rischio di tossicità per gli embrioni.- Dividere un tubo di centrifuga da 50 mL a metà nel senso della lunghezza, utilizzando un jig o sega.
- Semicerchi cut 7, con un raggio di 3 cm, fuori 70 µm in nylon mesh e ritagliarli per adattare strettamente in una metà della spaccatura Centrifugare la provetta. Incollare i semicerchi nella provetta da centrifuga parallela per le marcature di gradazione 10 mL utilizzando sigillante silicone acquario-cassetta di sicurezza. Consentire al dispositivo di asciugare per 2 giorni e risciacquare immergendo in un bicchiere d'acqua per 2-3 h.
- Linea estremità superiore (filettati) del tubo taglio centrifuga con argilla di modellistica, tale che l'altezza del liquido conservato nel dispositivo di scorrimento ha una profondità di ¼ di pollice ( Figura 1, viste 2 e 3).
- Preparare l'apparato di sbiaditura rimuovendo lo stantuffo da una siringa da 50 mL e inserendo 12 pollici del tubo bene nella punta. Spingere il tubo quanto andrà e guarnizione attorno al giunto mediante modellazione argilla.
- Pre-bagnato la mesh usando embrione mezzo per permettere al liquido di scorrere attraverso l'intero flusso-attraverso il dispositivo. Angolo del dispositivo sul ghiaccio così quel liquido piscine in tutti i compartimenti, ma ancora si svuota la parte anteriore dove si trova il cerchio di argilla. Utilizzando un supporto da anello, sospendere l'apparato di sbiaditura sopra il dispositivo di scorrimento su ghiaccio ( Figura 1).
- Chill 200 mL di mezzo di embrione sul ghiaccio e versare abbastanza nell'apparato di interruzione per garantire che tutte le bolle d'aria vengono cancellate e che la portata è di circa 7 mL/min. regolare la portata modificando l'altezza della siringa.
- Enzimaticamente gli embrioni dechorionate
- fare una soluzione di lavoro di non-specifiche proteasi diluendo 1 mL di 10 mg/mL non specifiche proteasi magazzino in 20 mL di terreno di embrione.
- Eseguire dechorionation chimici sugli embrioni 1 h prima del timepoint quando si prevede di raggiungere lo stadio di sviluppo desiderato. Chorions digest rimuovendo medio di embrione da 100mm di Petri contenente in scena gli embrioni e l'aggiunta di 20 mL di soluzione di lavoro di proteasi aspecifiche.
- Embrioni di incubare a 37 ° C per 5 min.
Nota: Non superare i 5 min o utilizzare una maggiore concentrazione di soluzione non specifiche proteasi, come questo si tradurrà in embrioni che cade a pezzi una volta trattato con nocodazole. - Rapidamente Pipettare fuori della proteasi aspecifiche e riempire piatti con circa 25 mL di liquido di embrione. Ripetere un' volta.
- Utilizzando una pipetta di vetro da 1 mL, trasferire gli embrioni più giovane di 24 hpf al vetro piatti per proteggerli da eventuali danni.
- Completa dechorionation rimuovendo manualmente utilizzando una coppia di una pinzetta, chorions, come descritto al punto 1.1.5.
- Posto vetro piastre Petri contenenti embrioni di dechorionated in un'incubatrice di 28,5 ° C per un minimo di 30 min fino a raggiungere lo stadio di sviluppo desiderato.
- Depolymerize il MTs esistente
- preparare una soluzione di lavoro di 5 µ g/mL nocodazole unendo 50 µ l 1 mg/mL stock nocodazole con 10 mL di terreno di ghiaccio freddo embrione.
Attenzione: Utilizzare guanti durante la manipolazione nocodazole, irritante per la pelle. - Scambiare il mezzo di embrione del gruppo di trattamento nocodazole con 10 mL di soluzione di lavoro freddo nocodazole. Posto di Petri sul ghiaccio per un tempo adeguato per lo stadio di sviluppo (ad esempio, 1 h per embrioni somite 4-5). Mettere da parte gli embrioni di controllo non trattato in una capsula di Petri sul ghiaccio per essere fissato al fianco di campioni di colori attenuati nel passaggio 2.3.4.1.
- Trasferimento embrioni usando una pipetta di vetro lucido 1 mL di fuoco per l'apparato di flusso continuo, con compartimenti separati per ogni gruppo sperimentale. Avviare la sbiaditura di nocodazole per mezzo di embrione freddo ghiaccio versando nella parte superiore della siringa 50ml.
Nota: Utilizzare almeno 30 embrioni per ogni gruppo sperimentale. Gruppi sperimentali potrebbero essere costituita da embrioni di controllo o una varietà di morpholino o embrioni iniettati di RNA. La sbiaditura richiederà un totale di circa 150 mL di mezzo di embrione da aggiungere ogni 8-10 minuti di interruzione il nocodazole pur continuando a inibire la crescita di MT con ghiaccio. Mantenere gli embrioni su ghiaccio è essenziale per il successo di questo test perché MTs sono instabili a freddo e freddo ritarda lo sviluppo di embrioni in anticipo. - Consentire MTs a ricrescere dopo 20 min di washout a RT con il trasferimento di embrioni di vetro Petri contenente caldo medio di embrione (28,5 ° C) utilizzando una pipetta di vetro lucido 1ml di fuoco. Non appena gli embrioni vengono trasferiti, avviare un timer.
- Difficoltà gli embrioni di controllo e di interruzione a 1 min, 5 min e 10 min pipettando circa 10 embrioni in un 1,5 mL Centrifuga tubo riempito con correzione di 1 mL 4% PFA/MAB (28,5 ° C) e seguendo le indicazioni in passo 1.2.3.
- preparare una soluzione di lavoro di 5 µ g/mL nocodazole unendo 50 µ l 1 mg/mL stock nocodazole con 10 mL di terreno di ghiaccio freddo embrione.
- Preparare campioni per immunolabeling come descritto nelle sezioni 1.3-1.5.
- Marcatura galleggiante sezioni ed embrioni di immagine come descritto nelle sezioni 1.6-1.10 con le seguenti modifiche alle specifiche di anticorpo primario: uso 1: 500 coniglio anti-γ-tubulina e 1: 200 del mouse anti-β-tubulina.
- Processo e analizzare immagini utilizzando software di analisi di immagine 3D, come descritto al punto 1.12.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Analisi di MTs stabile e dinamico utilizzando immunolabeling
Nel protocollo n. 1, la distribuzione delle sub-popolazioni MT durante prima (chiglia neurale) e fine (neurali asta) fasi di sviluppo del tubo neurale sono rivelate, utilizzando Glu-tubulina e Tyr-tubulina come marcatori per MTs stabile e dinamico, rispettivamente. MTs dinamico predominano in del hindbrain nella fase di chiglia neurale (4-5 somiti) (Figura 2A-D). Come la chiglia si sviluppa nell'asta neurale (11-12 somiti), una fase di avanzata epithelialization, qualitativamente meno MTs sono immunoreactive con l'anticorpo anti-Tyr-tubulina (Figura 2E-H), soprattutto nello stelo ventrale. Al contrario, Glu-tubulina è sparse e punctate in tutta la chiglia neurale (Figura 3A-D), ma si arricchisce del tondino neurale ventrale lungo i tratti di MT (Figura 3E-H). Punte di freccia indicano specifici MT fasci o strutture dove etichettatura è aumentato.
Anche se gli anticorpi anti-Glu-tubulina e di anti-Tyr-tubulina sono state prodotte nella stessa specie ospite (evitando un doppio esperimento d'etichettatura), questi risultati indicano che marcatori MT stabili e dinamici si sovrappongano raramente il hindbrain di zebrafish. In primo luogo, l'asta neurale ventrale è più stabile (Figura 3F) di dinamica (Figura 2F) MTs. La tendenza è invertita del tondino neurale dorsale, coerenti con un modello di zebrafish neurulation in cui il tessuto dorsale rimane dinamico fino a quando il tubo neurale è formato20. In secondo luogo, mentre fusi mitotici sono completamente etichettate con l'anticorpo di Tyr-tubulina nella chiglia neurale (Figura 2D, punte di freccia), solo la base del mandrino, coincida con il centrosoma, è etichettate con l'indicatore di stabilità (Glu-tubulina Figura 3 D, punte di freccia). Immunofluorescenza di β-tubulina, comune a entrambe le analisi, informa lo sperimentatore della distribuzione di tutte le MTs e fornisce una base per allontanare il contrassegno non specifico.
Misurazione di oggetti utilizzando software di analisi di immagine 3D si traduce in una grande quantità di dati che possono essere organizzati in una comoda tabella (tabella 2). Per rendere la lunghezza, conteggio e misure di zona, stiamo usando solo un sottoinsieme dei dati che sono disponibili per analizzare. Uno dei componenti dei dati che non analizziamo più ulteriormente è il numero di oggetti identificati. Questo numero viene utilizzato come controllo qualità interno, come il numero non dovrebbe variare ampiamente tra come sezioni e il rapporto dei nuclei per MTs deve rimanere simili in una condizione di singolo trattamento. Un outlier è un indicatore che l'analisi deve essere rieseguita con filtri regolati o che l'immagine è troppo mal etichettato per analizzare. Così, tutte le immagini di valore erratico dovrebbero essere analizzati nuovamente con impostazioni modificate. La sezione di valore erratico dovrebbe essere esaminata per segni di scarsa etichettatura o danni fisici che potrebbero tradursi in oggetto insolito conteggi. Una volta terminata l'analisi e qualità controllata, informazioni utili possono essere recuperati da dati grezzi come durata media della MTs totale e stabile MTs o il rapporto di stabile MTs MTS totale (tabella 3). Oltre a queste misure, molte altre metriche possono essere ottenuti utilizzando il software di analisi di immagine 3D che può essere utilizzato per trarre inferenze su MTs o loro relazione per altre strutture cellulari (nucleo, centrosoma, ecc.).
De novo Analisi di montaggio MT
Il trattamento nocodazole depolymerizes MTs conseguente etichettatura diffusa (Figura 4A, 4 D e 4G). Come far ricrescere le MTs, si estendono dal centrosoma (Figura 4B, 4E e 4 H), tuttavia, questo potrebbe non essere evidente in un unico piano a causa della loro traiettorie non planare (Figura 4C, 4F e 4I). Tuttavia, alcuni software di analisi di immagine sono in grado di misurare lunghezze in 3-d, permettendo una valutazione della crescita MT dopo l'interruzione di nocodazole (tabella 4). Un'importante osservazione che possa essere ottenuta dal dataset in tabella 4 è che la lunghezza media del MTs sembra aumentare nel tempo dopo l'interruzione di nocodazole in tutte le regioni del tubo neurale analizzati. Come accennato in precedenza, altri tipi di metriche ottenute dal software di analisi di immagine 3D è in grado di fornire contesto cellulare per interpretare i dati di MT (ad esempio, il rapporto di MTs per nucleo).
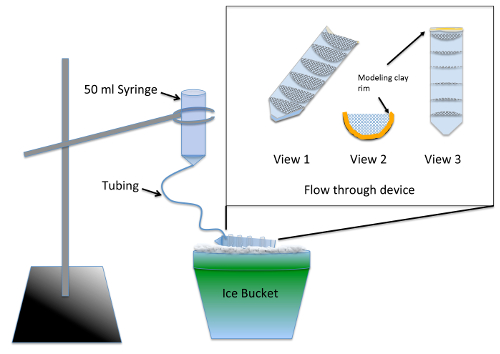
Figura 1 : Illustrazione di apparecchi di interruzione per de novo Analisi di montaggio MT. L'inserto è un primo piano del flusso-attraverso il dispositivo fatto da una maglia incollata in una provetta da centrifuga da 50 mL tagliata longitudinalmente. La maglia compartmentalizes il dispositivo di scorrimento tale che più gruppi sperimentali possono essere trattate contemporaneamente. Durante l'uso, embrione medio viene aggiunto alla siringa e scorre lentamente attraverso il tubo per riempire il dispositivo di scorrimento, fornendo un risciacquo costante a tutti i gruppi sperimentali. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 2: utilizzo di immunolabeling all'immagine dinamica MTS. Dechorionated embrioni sono stati fissati alle fasi appropriate (4-5 in A-D e 12-13 somiti in E-H), sezionati trasversalmente attraverso il hindbrain e immunolabeled con anticorpi diretti contro β-tubulina (verde in A ed E) per contrassegnare tutte le MTs e termine α-tubulina (rosso in B e F) per rivelare dinamiche popolazioni di MT. MTs altamente dinamico può essere visto nelle immagini unite (C, G) e loro ingrandimenti superiori (D, H) come aree dove etichetta gialla è visibile (punte di freccia in D, H). Scala bar = 25 µm (A-C ed E-G) e 10 µm (D e H). Clicca qui per visualizzare una versione più grande di questa figura.

FIGURA 3: utilizzo di immunolabeling all'immagine stabile MTS. Dechorionated embrioni sono stati fissati, sezionato tramite il hindbrain e immunolabeled a stadi appropriati (4-5 somiti in A -D e 12-13 somiti in E-H). Stabile MTs sono etichettati con anticorpi contro la forma detyrosinated di α-tubulina (Glu-tubulina) (rosso in B e F) mentre totale MTs sono stati visualizzati con un anticorpo generale β-tubulina (verde in A ed E). Segnali rossi e gialli in immagini unite (C, G) e loro ingrandimenti superiori (D, H) rappresentano le aree di alta stabilità di MT (punte di freccia in D, H). Scala bar = 25 µm (A-C ed E-G) e 10 µm (D e H). Clicca qui per visualizzare una versione più grande di questa figura.

Figura 4: utilizzo di immunolabeling all'immagine nascente MTS. Dechorionated embrioni erano fissi alle 4-5 somiti e sezionati trasversalmente attraverso il hindbrain. Sezioni immunolabeled con β-tubulina (D, E e F) per contrassegnare MTs in crescita e γ-tubulina (A, Be C) per contrassegnare il punto di nucleazione/centrosoma. Una regione dorsale del tubo neurale è confezionata in (A, D; B, E e C-F) e mostrato a maggiore ingrandimento (G, H, I, rispettivamente) a rivelare i nuclei (DAPI, blu), centrioli (γ-tubulina, rosso) e totale MTs (β-tubulina, verde). Bianco punte di freccia: colocalizzazione di MTs e centrioli; giallo di punte di freccia: il Centriolo secondo di una cella è visibile. Scala bar = 25 µm (A-F) e 10 µm (G-I). Clicca qui per visualizzare una versione più grande di questa figura.

Tabella 1: Le impostazioni predefinite per filtrare gli oggetti nel software di analisi di immagine 3D.

Tabella 2: rappresentante i set di dati grezzi ottenuti mediante analisi di immagine 3D software per analizzare stabile MTs. Ogni colonna rappresenta misure da un'unica sezione. Min: la misura più piccola; Max: più grande misura; SD: deviazione standard; SE: errore standard.

Tabella 3: esempi di set di dati che possono essere ottenuti da 3-d immagine software di analisi per quantificare stabile MTs. Selezionare le misure della lunghezza media del totale (β-tubulina) e stabile (Glu-tubulina) MTs calcolato prendendo la media della lunghezza media scheletrica per l'etichetta rilevante da tutti i campioni (Vedi tabella 2) e il rapporto di scuderia per totale (MTs Glu-tubulin striature a striature di β-tubulina) calcolato prendendo il conteggio medio β-tubulina, diviso per il numero medio di Glu-tubulina.

Tabella 4: Esempi di set di dati che possono essere ottenuti dal software di analisi di immagine 3D per analizzare de novo Assemblea di MT. Risultati rappresentativi dell'Assemblea de novo MT esperimento, confronto tra DataSet ottenuto per tre punti di tempo di recupero (1, 5 e 10 min) dopo nocodazole washout. Per ogni punto di tempo, misure ottenute per conteggio nucleare, centrioli (γ-tubulina puncta), numero di totale MTs (striature di β-tubulina), sono indicati per selezionato regioni del software analizzati (sezione trasversale del tubo neurale in via di sviluppo).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Attualmente ci sono molti metodi per imaging dinamiche MT nello sviluppo iniziale di zebrafish, che vanno da formazione immagine diretta di molecole con tag a immunolabeling di fisso tessuto11,12,13,14. Anche se MTs in una singola cella può esistere in stati stabili o dinamici, epithelialization è un processo in cui MTs sono progressivamente stabilizzate nel corso del tempo. Usando gli indicatori per MTs stabile e dinamico offre un modo per visualizzare questo fenomeno. Il metodo presentato qui sfrutta la potenza del software di imaging 3D per quantificare la transizione dalla dinamica di popolazioni stabili di MT in una sezione trasversale del tessuto embrionale zebrafish. Nel protocollo n. 2, il metodo viene utilizzato per etichettare una popolazione distinta di MTs nascente e seguire il loro straordinario di nucleazione e crescita.
Le MT sono notoriamente difficili da immagine nel loro stato nativo a causa della loro tendenza a depolymerize. Così, il componente chiave di questo metodo è il fissaggio rapido di MTs in tutta l'intero embrione. Ciò si ottiene a partire la fissazione alle temperature fisiologiche e utilizzando un buffer che stabilizza il MTs e aumenta la permeabilità dell'embrione. Il tempo di fissazione è anche importante come fissazione accorciata non riesce ad arresto MTs mentre over-fissazione può mascherare gli epitopi, interferire con il grippaggio dell'anticorpo. Il tempo di fissazione suggerito di 3-4 h funziona con gli embrioni che sono a metà gastrulazione fino alla post-fecondazione 24 h. Embrioni verso la fine più giovane della scala temporale è opportuno fissare per più vicino a 3 h mentre gli embrioni più anziani possono avere bisogno l'intero 4 h. Anche con adeguata fissazione, la MTs verrà depolymerize con il tempo così sezionamento e immunolabeling deve avvenire entro una settimana di fissaggio.
Una volta che il tessuto sia fissato correttamente, possono verificarsi problemi con immunolabeling. Il problema più frequente è stata scarsa penetrazione attraverso il centro del tessuto, in particolare, se troppe sezioni vengono incubati nel pozzo stesso. L'aumento della concentrazione del tempo dell'anticorpo e incubazione primaria per anticorpi primari e secondari, combinata con la crescente i detergenti per migliorare la permeabilizzazione degli embrioni migliorerà la maggior parte dei problemi immunolabeling. Se l'anticorpo ha esito negativo a causa di questioni di fissazione o immunolabeling problemi di etichettatura, è possibile determinare la causa esaminando il modello di contrassegno dell'anticorpo. Povera fissazione comporta intenso etichettatura nella membrana e diffusa etichettatura nel citoplasma, mentre over-fissazione si tradurrà in debole etichettatura che mantiene architettura MT. Scarsa penetrazione dell'anticorpo, tuttavia, verrà visualizzato come aree nel centro del tessuto senza etichettatura.
La capacità di analizzare MT immagini in modo significativo è dipendente su formazione immagine di alta qualità. Per acquisire MT di lunghezza in 3-d, la dimensione minima di Z-passo possibile per l'obiettivo e l'apertura numerica deve essere utilizzata. Immagini mostrate qui sono state catturate con un 63 X obiettivo emersione olio con apertura numerica 1,4 producendo i seguenti: pixel = 240 nm, Z-passo = 0,1 µm, dimensione dello stack Z = 16.252 µm. Poiché la larghezza di una singola MT è 25 nm, circa 10 volte sotto il limite di risoluzione di un microscopio ottico, questa metrico non può essere accuratamente misurato utilizzando questa tecnica. Invece, possono essere misurate sola lunghezze MT uguale o maggiore della dimensione minima in pixel in tutte le tre dimensioni sono raggiungibili. Linea e/o frame in media può migliorare la definizione del segnale di MT. Analisi di MT dovrebbero essere riservato per le sezioni di alta qualità. Mentre il tessuto con la fissazione del povera non può essere ripreso e analizzato, overfixation lieve può essere controbilanciato da attentamente aumentando l'intensità del laser e guadagno per rilevare il segnale debole pur mantenendo una buona gamma dinamica. Penetrazione dell'anticorpo poveri, mentre non ottimale, può essere corretto limitando l'acquisizione di immagini di regioni ben identificate, risultante in una sezione più sottile (5-10 µm) di imaging. Alta priorità bassa da etichettatura può essere compensata regolando le impostazioni del filtro. Tuttavia, se tutte queste regolazioni sono fatte, è necessario verificare che la soglia di filtri in modo accettabile su ogni aereo dello Z-stack.
Software di analisi di immagine 3D permette lo sperimentatore di quantificare MT lunghezza, area, angolo, abbondanza e altre metriche nello spazio 3-d delle sezioni fisse del tessuto. Il metodo qui descritto fornisce indicazioni per l'ottenimento di tali dati utilizzando il software disponibile in commercio. Tuttavia, i moduli filtranti possono essere adattati al pubblico dominio software migliorato con plugin pertinenti e/o macro, rendere disponibile a tutti l'analisi. Prima dell'analisi, immagini raw devono essere con soglia per evitare di includere segnali aspecifici e sfondo nella quantificazione. Una volta che viene completata l'analisi e i dati vengono trasferiti in un foglio di calcolo realizzabile, molte illazioni possono essere fatto da set di dati. Uno dei calcoli fatti qui era striature di Glu-tubulina per β-tubulina striature o il rapporto di stabile MTs MTS totale dove 1 rappresenta che il citoscheletro MT intero è stato stabilizzato in un ROI. Se lo sperimentatore ha voluto integrare i dati quantitativi, generazione di formato di un lucido tagged image file (TIFF) immagine con barre della scala è senza sforzo con il software di analisi di immagini 3D.
Questo test permette l'analisi funzionale delle proteine implicate nell'assembly MT, in vivo. Se immunolabeling viene eseguita su alternando sezioni di serie, questo protocollo potrebbe essere usato per studiare la dinamica e stabile MTs nell'embrione stesso. In futuro, le modifiche come detergenti aumentati o alterati angoli incorporamento permetterà l'uso di questi metodi per embrioni più anziani e una gamma più ampia di domande anatomiche.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori non hanno nulla a rivelare.
Acknowledgments
Il microscopio confocale è stato acquistato con fondi dal US National Science Foundation (NSF), grant #DBI-0722569. La ricerca è stata sostenuta da l'US National istituti di salute/National Institute of General Medical Sciences (NIH/NIGMS) grant #GM085290 e U.S. dipartimento della difesa (DOD) grant n #W81XWH-16-1-0466 assegnato a R.M. Brewster. E. Vital è stato sostenuto da una sovvenzione per UMBC da Howard Hughes Medical Institute attraverso il pre-college e il programma di educazione scientifica dello studente non laureato, concedere #52008090. S.P. Brown è stato sostenuto da un US Dipartimento di educazione GAANN Fellowship, una borsa di studio Meyerhoff finanziato dalla sovvenzione di NIH/NIGMS #GM055036 e un assistentato di ricerca finanziato da l'US DOD grant n #W81XWH-16-1-0466.
Materials
| Name | Company | Catalog Number | Comments |
| Agarose | Used to treat petridishes. Prepare 1% agarose by heating a solution of 1 gram agarose per 100 ml 1X embryo medium in a microwave until polymerized. |
||
| Kpipes | Sigma | P7643 | |
| NaCl | Sigma | S7653 | |
| Tris-HCl | Sigma | T3253-500G | |
| KCl | Sigma | P9333-500G | |
| CaCl2·2H2O | Sigma | C5080 | |
| NP-40 | American Bioanalyticals | AB01424 | |
| EGTA | Sigma | E3889-25G | |
| MgCl2 | Sigma | M2670-500G | |
| Bovine serum albumin (BSA) | Fisher | BP1605 | |
| Triton-x | American Bioanalyticals | AB02025 | |
| Anti-Fade mounting medium | Invitrogen | P10144 | |
| Mouse anti-β-tubulin | Developmental studies Hybridoma Bank | E7 | 1/200 |
| Rabbit anti-γ-tubulin | Genetex | GTX113286 | 1/500 |
| Rabbit anti-α-tubulin | Genetex | GTX108784 | 1/1000* |
| Rabbit anti-detyrosinated-tubulin | Millipore | AB3201 | 1/200-1/1000* Titrate antibody with first use of new lot. |
| Rabbit anti-tyrosinated-tubulin | Millipore | ABT171 | 1/500 |
| Mouse anti-centrin | Millipore | 04-1624 | 1/1000 |
| Goat 488 anti-rabbit | Thermofisher | A11008 | 1/500 |
| Goat 594 anti-rabbit | Thermofisher | A11012 | 1/500 |
| Goat 594 anti-mouse | Thermofisher | A11005 | 1/500 |
| Goat 488 anti-mouse | Thermofisher | A11001 | 1/500 |
| Vibratome | Vibratome | 1500 | |
| Forceps | World Precision Instruments | 555227F | |
| 100 mm petri dish | Cell treat | 229693 | |
| 35 mm petri dish | Cell treat | 229638 | |
| 50 ml falcon tube | Fisher | 14-432-22 | |
| Woven nylon mesh 70 um | Amazon.com | B0043D1SZG | |
| Micropipette | Gilson | F123602 | |
| Glass pipette | Fisher | NC-999363-9 | |
| Aquarium sealant | Amazon.com, by MarineLand | Silicone Sealer 1 oz (Tube) | |
| Ring stand | Fisher | 14-675BO | |
| Microbore PTFE Tubing, 0.022"ID | Cole-Parmer | WU-06417-21 | |
| Modeling clay | Amazon.com | Sargent Art 22-4000 | Any wax or oil based non-toxic modeling clay will suffice |
| Clamp | Fisher | 02-215-466 | |
| 60ml syringe | Fisher | 14-820-11 | |
| Embryo medium (E3) | 34.8 g NaCl 1.6 g KCl 5.8 g CaCl2·2H2O 9.78 g MgCl2·6H2O To prepare a 60X stock, dissolve the ingredients in H2O, to a final volume of 2 L. Adjust the pH to 7.2 with NaOH. Autoclave. To prepare 1X medium, dilute 16.5 mL of the 60X stock to 1 L. |
||
| Blocking Solution | 50 ml TBS-NP-40 2.5 ml normal goat serum 1 g BSA 625 µl Triton-X |
||
| TBS-NP-40 (pH 7.6) | 155 mM NaCl 10 mM Tris HCl 0.1% NP-40 |
||
| 2x MAB (pH6.4) | 160 mM KPIPES 10 mM EGTA 2 mM MgCl2 |
||
| Commercial 3-D Image processing Software | PerkinElmer | Volocity (V 6.2) | |
| Dry block heater | VWR | 12621-108 | Used as a hot plate to melt agarose in Protocol 1. |
| Dissecting Microscope | Leica | MZ12 | |
| Confocal Microscope | Leica | SP5 | |
| Flat embedding mold | emsdiasum.com | BEEM 70904-01 | |
| Public domain image processing software | NIH | ImageJ (V 1.5) | |
| * Success varies by lot number | |||
References
- Akhmanova, A., Steinmetz, M. O. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 9 (4), 309-322 (2008).
- Conde, C., Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 10 (5), 319-332 (2009).
- Kaverina, I., Straube, A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 22 (9), 968-974 (2011).
- Kollman, J. M., Merdes, A., Mourey, L., Agard, D. A. Microtubule nucleation by γ-tubulin complexes. Nat Rev Mol Cell Biol. 12 (11), 709-721 (2011).
- Howard, J., Hyman, A. A. Growth, fluctuation and switching at microtubule plus ends. Nat Rev Mol Cell Biol. 10 (8), 569-574 (2009).
- Schulze, E., Kirschner, M. Dynamic and stable populations of microtubules in cells. J Cell Biol. 104 (2), 277-288 (1987).
- Gundersen, G. G., Kalnoski, M. H., Bulinski, J. C. Distinct populations of microtubules: Tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell. 38 (3), 779-789 (1984).
- Li, R., Gundersen, G. G. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat Rev Mol Cell Biol. 9 (11), 860-873 (2008).
- Asakawa, K., Kawakami, K. A transgenic zebrafish for monitoring in vivo microtubule structures. Dev Dyn Off Publ Am Assoc Anat. 239 (10), 2695-2699 (2010).
- Wühr, M., Tan, E. S., Parker, S. K., Detrich, H. W., Mitchison, T. J. A model for cleavage plane determination in early amphibian and fish embryos. Curr Biol CB. 20 (22), 2040-2045 (2010).
- Tran, L. D., Hino, H., et al. Dynamic microtubules at the vegetal cortex predict the embryonic axis in zebrafish. Development. 139 (19), 3644-3652 (2012).
- Butler, R., Wood, J. D., Landers, J. A., Cunliffe, V. T. Genetic and chemical modulation of spastin-dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis Model Mech. 3 (11-12), 743-751 (2010).
- Yoo, S. K., Lam, P. -Y., Eichelberg, M. R., Zasadil, L., Bement, W. M., Huttenlocher, A. The role of microtubules in neutrophil polarity and migration in live zebrafish. J Cell Sci. 125 (23), 5702-5710 (2012).
- Andersen, E. F., Halloran, M. C. Centrosome movements in vivo correlate with specific neurite formation downstream of LIM homeodomain transcription factor activity. Development. 139 (19), 3590-3599 (2012).
- Lee, S. -J. Dynamic regulation of the microtubule and actin cytoskeleton in zebrafish epiboly. Biochem Biophys Res Commun. 452 (1), 1-7 (2014).
- Bulinski, J. C., Gundersen, G. G. Stabilization and post-translational modification of microtubules during cellular morphogenesis. BioEssays. 13 (6), 285-293 (1991).
- Magiera, M. M., Janke, C. Chapter 16 - Investigating Tubulin Posttranslational Modifications with Specific Antibodies. Methods Cell Biol. 115, 247-267 (2013).
- Hong, E., Jayachandran, P., Brewster, R. The polarity protein Pard3 is required for centrosome positioning during neurulation. Dev Biol. 341 (2), 335-345 (2010).
- Westermann, S., Weber, K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 4 (12), 938-948 (2003).
- Jayachandran, P., Olmo, V. N., et al. Microtubule-associated protein 1b is required for shaping the neural tube. Neural Develop. 11, 1 (2016).
- Nam, S. -C. Role of Tau, a microtubule associated protein, in Drosophila photoreceptor morphogenesis. Genes N Y N 2000. 54 (11), 553-561 (2016).
- Abal, M., Piel, M., Bouckson-Castaing, V., Mogensen, M., Sibarita, J. -B., Bornens, M. Microtubule release from the centrosome in migrating cells. J Cell Biol. 159 (5), 731-737 (2002).
- Delgehyr, N., Sillibourne, J., Bornens, M. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci. 118 (8), 1565-1575 (2005).
- Manning, J. A., Lewis, M., Koblar, S. A., Kumar, S. An essential function for the centrosomal protein NEDD1 in zebrafish development. Cell Death Differ. 17 (8), 1302-1314 (2010).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Dev Dyn Off Publ Am Assoc Anat. 203 (3), 253-310 (1995).
- Beck, A. P., Watt, R. M., Bonner, J. Dissection and Lateral Mounting of Zebrafish Embryos: Analysis of Spinal Cord Development. JoVE J Vis Exp. (84), e50703 (2014).
- FÖldes-Papp, Z., Demel, U., Tilz, G. P. Laser scanning confocal fluorescence microscopy: an overview. Int Immunopharmacol. 3 (13-14), 1715-1729 (2003).
- Ferreira, T., Rasband, W. S. ImageJ User Guide - IJ 1.46. , Available from: https://imagej.nih.gov/ij/docs/guide/ (2010).
- Z-functions - ImageJ. , Available from: https://imagej.net/Z-functions (2017).
