

Summary
I den här artikeln beskriver vi elektrokemiska, electron paramagnetiska resonans och ultraviolett-synligt och infrarött spektroelektrokemisk metoder att analysera organiska föreningar för tillämpning i organisk elektronik.
Abstract
Cyklisk voltametri (CV) är en teknik som används i analysen av organiska föreningar. När denna teknik kombineras med electron paramagnetiska resonans (EPR) eller ultraviolett-synligt och infrarött (UV-Vis-NIR) spektroskopier, få vi användbar information såsom Elektronfrändskap, jonisationspotential, band-gap energier, typ av Laddningsbärare och nedbrytning information som kan användas för att syntetisera stabila organiska elektroniska enheter. I denna studie presenterar vi elektrokemisk och spektroelektrokemisk metoder för att analysera de processer som sker i aktiva skikten av en organisk enhet samt genererade laddningsbärarna.
Introduction
Hela världen, söker forskare ständigt efter nya organiska material som kan användas i organisk elektronik med önskvärda prestanda eller stabilitet, som sjunker på grund av utökad användning. När det gäller ekologiska produkter är det viktigt att förstå beteendet hos de laddningsbärare fullt veta reglerna körning enhetens beteende. Analys av effekten av molekylära struktur på generering av laddningsbärare och den dynamik och underhåll av balansen av injicerade laddningsbärare, både positiva (hål) och negativa (elektroner), är avgörande för att förbättra effektivitet och stabilitet av organiska enheter. Detta säkerställer den effektiva rekombination av dessa enskilda avgifter och följaktligen betydligt förbättrar fotoluminescens effektiviteten i den organiska Tända-utsändande av dioder (OLED)1,2. För organiska solceller (OPVs)3,4 , samt ekologiska fält effekt transistorer (OFETs)5,6är det nödvändigt att ha material med hög avgift transportören rörlighet. Utöver analysen av laddningsbärare, flera viktiga parametrar av organiska elektroaktiva material hjälper förutsäga där materialet kan användas: jonisationspotential (IP), electron affinitet (EA) energinivåer och band-klyftan mellan dem7 ,8,9,10.
I detta arbete presenterar vi en metod för effektiv mätning av cyklisk voltametri (CV) som kan användas i analysen av alla typer av elektroaktiva material. Denna teknik ger information om redox boenden, mekanismen för dopning/dedoping, stabilitet, konvertering och lagring av energi, etc. Det gör också för skattning av elektron affinitet och jonisering energi av föreningarna som test på ett mycket billigare och snabbare sätt jämfört med andra höga vakuum metoder. De ovan nämnda parametrarna korrelerar med energinivåerna i högsta ockuperade molekylorbital (HOMO) och lägst obesatt molekylorbital (LUMO).
Metoden presenteras i denna artikel kan användas för att analysera alla typer av konjugerade föreningar såsom de med delokaliserade π-elektronerna i sina strukturer. Konjugerad föreningar kan vara små molekyler med stora polymera kedjor. Små molekyler kan också vara monomerer; under den första reaktionen kan (fotokemiska, elektrokemiska eller kemisk) monomerer bilda polymerer. I OLED ansökan är energinivån värdena nödvändiga för att möjliggöra användning av rätt värd för sändaren i en termiskt aktiverade försenas fluorescens (TADF) gäst-värd system eller för att bestämma med vilka föreningar exciplex givare-acceptor lagret kan vara bildas och vilka ytterligare lager (elektron transport layer (ETL), hål transport layer (HTL), electron blockerande skikt (EBL) och hål blockerande skikt (HBL)) kommer att vara nödvändiga för att synthesize stabil effektivt debiteras balanserad OLED enheter11 , 12 , 13 , 14 , 15 , 16 , 17. ytterligare elektrokemiska mätningar tillåta utredning av eventuella reaktioner under nedbrytningen av det aktiva lagret och bildandet av låg mobil Ladda bärare (bipolarons)18,19 ,20,21,22.
Koppling elektrokemisk och spektroelektrokemisk metoder möjliggör enkel, exakt och tillförlitlig bestämning av graden av oxidation eller reduktion av konjugerade föreningar och deras nedbrytning potential, vilket är avgörande för stabiliteten23 , 24 , 25 , 26 , 27 , 28. Ultraviolett-synligt och infrarött (UV-Vis-NIR) spektroskopi tillsammans med elektrokemi kan karaktärisera grundläggande kromatisk egenskaperna för alla nya konjugerade föreningar, såsom byte av absorptionsbandet under dopning 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30.
I en studie som relaterade till mekanismen för dopning, är det viktigt att definiera vilken typ av laddningsbärare. I denna process, två klasser av laddade quasiparticles delta, en med okompenserad spin (polarons) och det andra är diamagnetic (bipolarons); paramagnetiska resonans (EPR) elektronspektroskopi ger ovärderlig hjälp, som direkt gör att man kan följa och spåra ändringar i populationer av paramagnetiska polarons29,30,31,32 . I små molekyler, är det svårt att bilda bipolarons, men dessa molekyler kan vara ganska konjugerat och har bipolaron-inducerande egenskaper. Det är viktigt att kontrollera om och där potentiella polarons och bipolarons bildas i strukturen. Bipolarons är minst en ordning lägre rörlighet än polarons; Därför, om bipolarons bildas i fungerande enheter, då det kan leda till en obalanserad förhållandet av laddningsbärarna, vilket skulle resultera i hög ström och överhettning av OLED enheten eller kanske centrerar av nedbrytning33.
Mätmetoden som föreslås i denna studie är billig och snabbare och möjliggör bestämning av de mest värdefulla operativa parametrarna för ett stort antal elektroaktiva material utan behov av speciella anordningar som baseras på nyligen syntetiseras material för att kontrollera dess prestanda. Genom att tillämpa elektrokemi och spectroelectrochemistry, är det möjligt att välja ett material som är riktigt lovande från hundratals nya material. Dessutom är det möjligt att erhålla detaljerad information om processerna av dopning och deras effekter på den kemiska strukturen av testet konjugerade system med elektrokemisk och spektroelektrokemisk metoder, som gör det möjligt att bygga mer effektiv organisk elektronik enheter.
Protocol
1. beredning av experimentet
- Förbereda 25 mL 0,1 M elektrolytlösning.
Obs: Beroende på föreningarna under utredning, använda olika elektrolyter som blandningen av ekologisk salt och vätska: mest gemensamma salter används i analysen av organiska molekyler är tetrabutylammonium hexafluorofosfat (Bu4NPF6 ) eller tetrabutylammonium tetrafluoroborat (Bu4NBF4). vanligaste lösningsmedlet används i analysen är diklormetan, acetonitril eller tetrahydrofuran.- Välj elektrolyt lösningsmedlet baserat på lösligheten av föreningarna som test. Det bör vara en lösning under undersökningen men vara i fast form när materialet används i skapandet av enheten, som en tunn film som deponeras på elektroden arbetsytor.
- Ren elektroderna ska användas i experiment29.
- För elektrokemiska mätningar, använda en 1 mm diameter platina disk elektrod som arbetselektroden (WE), en platina spole eller tråd som hjälpelektroden (AE) och en silver/silverklorid (Ag/Granulatfyllda) elektrod som referenselektroden (RE).
- För EPR spektroelektrokemisk mätningar, använda platina tråd som i WE, en platina spole eller wire som AE och Ag/Granulatfyllda som RE.
- För UV-Vis-NIR spektroelektrokemisk mätningar, använda en kvarts indium tinoxiden (ITO) eller fluor-dopade tinoxiden (FTO) som vi, en platina tråd som AE, och en Ag/Granulatfyllda elektrod som RE.
- Polska platina disk elektroden genom att gnugga nackdelen med elektroden (platina elektrod arbetsområdet) på en polering pad täckt med 1 µm aluminiumoxid flytgödsel i 3 min.
- Skölj elektroden med avjoniserat vatten för att ta bort alla slam från elektroden och rengör i ett ultraljudsbad (320 W, 37 kHz) med avjoniserat vatten i 15 min.
- Skölj elektroden med en 1 mL spruta med isopropanol (3 x 1 mL) och sedan med aceton (3 x 1 mL).
- Använda en pappershandduk för att ta bort alla fasta återstoder och torka i luften i 3 min.
- Städa på ITO och FTO elektroder med avjoniserat vatten. Placera dem i ett ultraljudsbad (320 W, 37 kHz) i aceton för 15 min och sedan i isopropanol för nästa 15 min.
- Bränna den platina elektroder, ledningar och spolar med hjälp av en gaslåga med hög temperatur (> 1000 ° C) för 1 min. vara försiktig! Använda pincett för att hålla elektroderna. Vänta 5 min efter bränning och innan du använder elektroderna.
- Ren de elektrokemiska och spektroelektrokemisk cellerna (figur 1) med vatten och sedan med aceton. Använd hushållspapper för att avlägsna alla de fasta återstoder. Rengör alla andra element (ca. PTFE delar) med aceton och torka i luften innan användning.
2. CV-analys
- Slå på potentiostat och dator.
- Fyll en elektrokemisk cell med 1,5 mL elektrolytlösning som skall analyseras.
- Sätta alla tre elektroderna (WE, AE, och RE) i en cell (cellens cap är utrustad med PTFE elektrodhållare, så satte elektroder genom hålen i denna hållare) och Anslut den till en potentiostat. Hålla WE och RE som nära varandra som möjligt (figur 1). Följ informationen om anslutningen av varje tråd potentiostaten till dess respektive elektrod.
- Om det behövs (till exempel under minskning analys), sätta argon (eller kvävgas) röret (genom en ytterligare hål i elektrodhållare) och börja bubblande lösningen för minst 5 min. Efter detta flyttar argon (eller kvävgas) röret ovanför lösningens nivå och hålla gasflödet kör för mätning.
Obs: Cellen ser stängda men kan inte vara helt täta; därför en metod att ta bort den överflödiga mängden gas från cellen måste vara tillgängliga (t.ex., av en ytterligare små hål i elektrodhållare). Trycket beror på röret används; Ställ in trycket till hög som den är skyldig att ange långsam gasflödet. Om mycket flyktiga lösningsmedel används i experimentet (t.ex. diklormetan) eller experimentet tar lång tid (mer än 30 min), sedan använda Drechselflaskan för att mätta argon (eller kvävgas) gas med lösningsmedel används i mätningen. - Köra programvaran potentiostat, välja förfarandet som CV och använder följande inställningar: starta potential 0,00 V, övre vertex potential 2.0 V, lägre vertex potential 0,00 V (om utreda processen att minska, då en lägre vertex potential −2.5 V), stoppa potential 0,00 V, antal stopp passage av 6, och skanna hastighet av 0.05 V/s.
- I avsnittet Exportera ASCII data, definiera ett filnamn och välja en mapp att spara data i, och tryck på START -knappen (öka eller minska den nedre och övre vertex potentialen att passa fönstret elektrokemiska eller provet elektrokemisk aktivitet)
- Om någon topparna syns i plusområdet potential, sedan upprepa rengöringsproceduren. Om det finns en topp i intervallet negativa potentiella, sedan sätta argon (eller kvävgas) röret i lösningen och börja bubblande för en ytterligare 5 min.
- Lägg en droppe av 1 mM Ferrocen (i de lösningsmedel som används för att förbereda elektrolyten) till elektrolytlösningen med hjälp av en spruta.
- Ange start potential att 0,00 V, övre vertex potentiella till 0,85 V, den nedersta brytpunkten potentiella till 0,00 V, stop potential att 0,00 V antalet stopp passage till 10, och sökhastighet till 0.05 V/s. I avsnittet Exportera ASCII data, ändra filnamn och tryck på START -knappen.
- Efter mätningen, avsluta med att upprepa de rengöring förfaranden som nämns i steg 1.2 och 1.3.
- Förbered 4 mL av 1 mM test förening i beredda elektrolyten (steg 1.1).
- Fyll den elektrokemiska cellen med sammansatta provlösningen (1,5 mL); satte alla tre elektroder i en cell och Anslut till potentiostaten (steg 2,4). För att undersöka processen för minskning, avlägsna syre (steg 2.5).
- Ställ in start potentiella till 0,00 V, den övre brytpunkten potentiella till 0,50 V (eller 0,00 V vid utredning av minskning processen), den nedersta brytpunkten potentiella till 0,00 V (−0, 50 V vid utredning av minskning processen), stop potential att 0,00 V , antal stopp passage till 10, och sökhastighet till 0,05 V. I avsnittet Exportera ASCII data, ändra filnamn och tryck på START -knappen.
- Upprepa steg 2.13 genom att öka övre vertex potentialer (eller genom att minska lägre vertex potential) med 0,1 V fram till peak registrering (figur 2a). Om successiva genomsökningen skiftas i potential (figur 2b), sedan rengör RE och lämna den i elektrolytlösningen. Upprepa mätningen.
- Mäta både oxidation och reduktion processer på samma CV för bestämning av IP och EA: Ange start potential att 0,00 V, övre vertex potential att 1.00 V, lägre vertex potential att −2.70 V och stop potential till 0,00 V. Välj den övre och nedre vert ex potential på ett sätt som registrerar full reduktion och oxidation toppar och undviker ytterligare redox åtgärder (om sådana finns) (figur 2a).
Obs: Uppskatta IP och EA genom att mäta uppkomsten potentiella. Det finns ett par olika sätt att beräkna parametrarna från CV men det mest användbara är att beräkna potentiella uppkomsten. Ibland är det inte möjligt att observera reversibel redox par, särskilt för både n och p dopning. Blandning av två olika tekniker är också inte önskvärt eftersom det kan ge fel resultat och slutsatser.- För att mäta uppkomsten potentiella, markera tangentlinje till CV peak (figur 3). Beräkna de önskade värdena från debut potential att minska eller oxidationsprocess med hjälp av ekvationer (ekvationer 1 och 2)10,18,19,36,37 , 38.
 (1)
(1)
där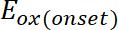 är uppkomsten av oxidation potentiella kalibreras med Ferrocen*
är uppkomsten av oxidation potentiella kalibreras med Ferrocen*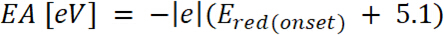 (2)
(2)
där är uppkomsten av den potentialen för minskning som kalibreras med Ferrocen*
är uppkomsten av den potentialen för minskning som kalibreras med Ferrocen*
* Potential som kalibreras med Ferrocen innebär att värdet av potential från mätningen minskas genom oxidation potential Ferrocen.
- För att mäta uppkomsten potentiella, markera tangentlinje till CV peak (figur 3). Beräkna de önskade värdena från debut potential att minska eller oxidationsprocess med hjälp av ekvationer (ekvationer 1 och 2)10,18,19,36,37 , 38.
- Dra ut elektroderna och utgjuta provlösningen i behållaren för korrekt avfall.
- Upprepa rengöringsproceduren (steg 1.2 och 1.3).
- Upprepa steg 2,9 – 2.12 med 0,25, 0,10 och 0,20 V/s Skanna priser.
- Tillsätt en droppe av 1 mM Ferrocen (i de lösningsmedel som används för att förbereda elektrolytlösningen) till provlösningen med hjälp av en spruta. Ange start potential att 0,00 V, övre vertex potentiella till 0,60 V, lägre vertex potential att 0,00, stop potential att 0,00 V, antalet stopp passage till 6 och sökhastighet till 0,05 V. Tryck på START -knappen. När mätningen är klar, ändra övre vertex potentiella till ett värde som registrerar Ferrocen paret med den första toppen av oxidation av testet förening.
- Dra ut elektroderna och utgjuta provlösningen i behållaren för korrekt avfall.
- Om electropolymerization inträffar under elektrokemisk oxidation, tvätta vi noggrant med elektrolytlösningen sprutan (3 ´ 0,5 mL). Ren RE, AE och elektrokemisk cell med hjälp av standard procedur (steg 1.2-1.3).
- För att undersöka elektrokemiska produkter deponeras på vi, Fortsätt till steg 2,23. För att undersöka en lösning, gå vidare till steg 1.2.1 och starta igen från steg 2,3.
- Fyll den elektrokemiska cellen med en elektrolytlösning. Sedan satte alla tre elektroderna i en cell och ansluta dem till en potentiostat (steg 2,4). Att utreda minskning processen, deoxidate lösningen (steg 2.5).
- Upprepa steg 2.10, 2.12 och sedan steg 2.16.
 (1)
(1)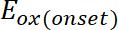 är uppkomsten av oxidation potentiella kalibreras med Ferrocen*
är uppkomsten av oxidation potentiella kalibreras med Ferrocen*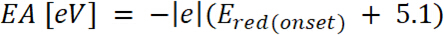 (2)
(2) är uppkomsten av den potentialen för minskning som kalibreras med Ferrocen*
är uppkomsten av den potentialen för minskning som kalibreras med Ferrocen*3. UV-Vis-NIR spektroelektrokemisk analys
- Mätning förberedelse
- Slå på spektrometer och potentiostat.
- Fyll spektroelektrokemisk cellen med 0,5 mL elektrolytlösning (steg 1.1).
- Lägg PTFE tätning på ena sidan av cellen; sedan sätta ITO elektroden till cellen i detta sätt att ha en ledande sida av elektroden vidrör PTFE tätning. Lägg resten av PTFE delar som visas i figur 1. Sätta de RE och AE genom PTFE elektrodhållare och täck ovansidan av ITO elektroden med kopparfolie som visas i figur 1 för bättre ledningsförmåga.
- Sätta den monterade cellen i innehavaren av spektrometern och Anslut alla elektroderna till en potentiostat. Följ informationen om anslutningen av varje tråd potentiostaten till dess respektive elektrod.
- Kör programvaran potentiostat och spektrometer.
- Välj fil i programvaran spektrometer, | Nya | Absorbansen mätning.
- Välj en av de lista detektorerna.
- Kontrollera att knappen på detektorerna är i ”öppet” läge. Klicka sedan på den glödande lampan; Stäng detektorn (flytta knappen till stängt läge) och klicka sedan på den mörka lampan.
- Välj andra detektorn och upprepa steg 3.1.7.
- Återanslut elektroder från potentiostaten och dra ut elektroderna och andra delar av cellen spektroelektrokemisk.
- Upprepa rengöring procedurer (steg 1.2 och 1.3).
- Fylla den spektroelektrokemisk cellen med en utspädd (1´10−5 M) lösning av testet förening i elektrolytlösningen om testar den liten molekylen. Fyll spektroelektrokemisk cellen med elektrolyten om testar polymera (Oligomera) lagret deponeras på ITO elektroden.
- Montera spektroelektrokemisk cellen (steg 3.1.3).
- Klicka på Spara.
- Välj en av de lista detektorerna.
- Definiera Spara plats och namn filen (inkludera den valda detektorn).
- Välj andra detektorn och upprepa steg 3.1.15.
- Potentiostatic analys18.
- Tillämpa en neutral potential (dvs. 0,00 V). Detta spektrum kommer att starta spektra. Öka med 0,1 V och vänta ca. 10 s tills processen stabiliserar och registrerar Absorptionsspektra (steg 3.1.13–3.1.16). Fortsätta mätningen till den första förändringen i UV-Vis-NIR spektra; vänta för 10 s och sedan spara spektra (steg 3.1.13–3.1.16).
- Öka potentialer av 0.05 V; vänta för 10 s och spara (steg 3.1.13–3.1.16).
- Upprepa 3.2.2 tills att uppnå potentialen i första eller andra oxidation potential (dessa potentialer är kända från CV mätning); sedan vända potential och gå tillbaka till apparatens nollinställning.
- När mätningarna genomförs, mäta CV av provlösningen. Lägg till 1 mM Ferrocen (10 μL) och mät CV igen. Värdet av Ferrocen kommer att uppskatta potentialen i processen och förena data.
- Denna analys ger information angående den optiska band-gap, maximal våglängd av undoped och dopade stater och isosbestic punkter av elektrokemiska processen. Beräkna de optiska band-luckorna från debut värden för π-π* band på UV-Vis spektra och använda ekvation 318,19,23,24.
 (3)
(3)
där h är Plancks konstant, λdebut är sammansatta absorption debut, och c är ljushastigheten i vakuum. - Tid-löst Potentistatic analys19,32,33
- Förbereda mätningen liknar den metod som beskrivs i 3.1.1–3.1.16 och sätta argon (eller kvävgas) röret (genom en ytterligare hål i elektrodhållare) och bubblande lösning minst 5 min före mätningen. Efter detta flyttar argon röret ovanför lösning och hålla gasflödet kör för mätning.
- Köra programvaran potentiostat, välja det kronoamperometri förfarandet med följande inställningar: potential starta potential till 0,00 V, övre potential till 1,0 V, lägre potential att −1.00 V, varaktighet som 5 s, tidsintervall som 0,010 s, antal repetitioner till 10, och förseningar tid till 10 s. I avsnittet Exportera ASCII data, ange ett filnamn och välja en mapp att spara data.
- Välj fil i programvaran spektrometer, | Nya | Höghastighetståg förvärv. Ett nytt fönster kommer att visas; Ställ in följande parametrar: Integration tid — 10 ms, skannar till medelvärdet — 1, Boxcar Wirth — 0, antal av File — 10000. Klicka inte på knappen gå .
- Tryck på START -knappen i potentiostat-programvaran. När nedräkningen sjunker till 1 s, tryck på knappen gå i programvaran spektrometer.
- Observera att det inte är möjligt att se absorption resultat tills processen är klar.
- Analysera data för att tillhandahålla parametrar. Använda den information som sparas i filen sådan transmittans, ström, spänning och strömtätheten att tillhandahålla fungerande parametrar:
- Beräkna optiska densitet (ΔOD), absorbans testet förening under dopning (dedoping) med ekvation 4:
 (4)
(4)
där är Tb transmittans dopade form, ”blekmedel” staten [%] och Tc transmittans i undoped form, en ”färgade” stat [%]. - Beräkna Coloration effektivitet (CE, η), mängden elektrokroma färg bildas av den använda mängden kostnadsfritt och som anslutning mellan injiceras/matas ut avgiften som en funktion av elektroden område (Qd) och ändringen i optisk täthet (ΔOD) värde vid en viss våglängd (λmax) med ekvation 5. Dess värde beror på våglängden valts för studien, så värdet mäts vid λmax av optiska absorptionsbandet färgade statens.
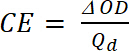 (5)
(5)
där ΔOD är den optiska densiteten [-] och Qd är laddningstätheten [C/cm2]. - Beräkna kontrast baserat (CR), uppmätta intensiteten i färgen förändras under elektrokemiska dopning och brukar karakteriseras av λmax av färgade form med ekvation 6:
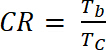 (6)
(6)
där är Tb transmittans dopade form, ”blekmedel” staten [%] och Tc transmittans i undoped form, ”färgade” staten [%].
- Beräkna optiska densitet (ΔOD), absorbans testet förening under dopning (dedoping) med ekvation 4:
 (3)
(3) (4)
(4)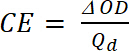 (5)
(5)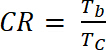 (6)
(6)4. EPR spektroelektrokemisk analys
- Mätning förberedelse
- Slå på spektrometern och potentiostaten.
- Fylla den spektroelektrokemisk cellen med elektrolyt (steg 1.1).
- Set 1 mW och tune resonator kan uppnå en skarp, centrerad signal i Q-dip (Använd alternativet automatiskt justerade).
- Ställ markören Mn (mangan standard) till 600 och Stäng fönstret Q-dip. Om det finns ingen Mn markör, sedan hoppa över detta steg och gå till steg 4,8.
- Ange ”sweep bredd ±” till 2.5´ 10 mT, ”mod bredd ±” till 1.0´0.1 mT, ”amplitud” till 5´100, ”tidskonstant” till 0,03 s, och ”center fältet” värde ca. 338 mT och starta mätningen.
- Om inte registrerar sex spektral-fodrar av Mn markör, sedan ändra värdet ”center fält”.
- När du registrerar dig sex spektral-fodrar av Mn markör (figur 4), ”center fält” värdet mellan tredje och fjärde raden och minska värdet för ”sopa bredd ±” till ett värde som täcker endast dessa två rader. Starta mätningen igen.
- Gå till alternativet Q-Dip, Mn markör 0, och Stäng fönstret Q-dip.
- Mät ren elektrolyten som bakgrund och kontrollera om det finns några signaler. Om signalerna är synliga, kan då signalen vara från kontaminering, låg oxidation potentiella föreningar, eller från glaset.
- Ren spektroelektrokemisk cellen (steg 1.3).
- Utredning av små molekyler (lösningar).
- Fylla den spektroelektrokemisk cellen med en utspädd (1´10−5 M) lösning av testet förening i elektrolyten; satte alla tre elektroder genom elektrodhållare till cellen på detta sätt så att WE och RE är inuti spiralen AE (figur 1).
- Sätta WE nära botten av cellen och RE på ovansidan av aktivt (metall inte omfattas av PTFE) en del av vi (figur 1). Placera cellen utrustad med en elektrod inne i provet håligheten i spektrometern och anslut elektroderna med en potentiostat (Följ informationen om anslutningen av varje tråd potentiostaten till dess respektive elektrod).
- Tillämpa den potential motsvarande uppkomsten av den första redox-toppen (värde känd från CV mätning). Om en signal visas är då utrustningen korrekt inställd.
- Gå till alternativet Q-Dip, ställa Mn markören till 600, Stäng fönstret Q-dip och starta mätningen. Registreringen av signal med signalen från Mn markör tillåter beräkning av g-faktor på den mottagna signalen.
- Gå till alternativet Q-Dip, ställa Mn markören till 0, Stäng fönstret Q-dip och vänta ca. 5 min.
- Ange värdet ”center fält” i mitten av signalen, minska värdet för ”sopa bredd ±” till ett värde som täcker signalen, men inte klippa bort början och slutet av signal (när ”sweep bredd ±” ändras, kontrollera hela signalen) och minska värdet för ”m OD bredd ± ”att få väl löst spectra.
- Om signalen är små, sedan öka ”amplitud” värde och ”tid” (förvärv tid) av mätningen.
- För att minska signal-brusförhållande, inställt förvärvet 4, 9 eller 16 beroende på hur bullriga signalen är. Om signalen är mycket bullriga, Välj 16.
- Fylla den spektroelektrokemisk cellen med en utspädd (1´10−5 M) lösning av testet förening i elektrolyten; satte alla tre elektroder genom elektrodhållare till cellen på detta sätt så att WE och RE är inuti spiralen AE (figur 1).
- Utredningar av Polymera lagret deponeras på ytan av WE.
- Fyll spektroelektrokemisk cellen med en elektrolytlösning (steg 1.1).
- Satte elektroder in i cellen. Var noga med att inte förstöra det polymera lagret på WE. Anslut elektroden med potentiostaten (steg 4.2.1).
- Tillämpa neutrala potential (dvs. 0,00 V) för att föreningen ska vara i en neutral stat; Detta spektrum kommer att starta EPR spektra.
- Öka möjligheterna med 0.10 V. vänta ca 10 s tills processen stabiliserar och registrera EPR spektra.
- Upprepa steg 4.3.4 när EPR signalen visas.
- Öka risken av 0,05 V. vänta ca 10 s tills processen stabiliserar och registrera EPR spektra.
- Upprepa steg 4.3.6 tills först eller andra oxidation potential uppnås och så vidare (värden av dessa potentialer är kända från CV mätning); sedan vända potential och gå tillbaka till Start potentiella.
- Tillämpa den potential som EPR signal dök upp i föregående cykel (4.3.5), gå till alternativet Q-Dip, ställa Mn markören till 600, Stäng fönstret Q-dip och starta mätningen. Registrera signalen med tredje och fjärde spectral raden av Mn markör (4.1.7).
- När mätningen är klar, analysera data.
- Några av analysen som ett antal spins kan utföras endast för föreningar deponeras (olösliga polymerer) på WE. För att beräkna antalet dragningar, dubbel-integrera signalen EPR, följt av en jämförelse av erhållna värdet med kalibrerad Mn intern standard. Beräkna antalet av upprepade enheter bildar polymeren på elektroden med hjälp av ekvation 724,32,33:
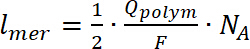 (7)
(7)
där lmer – nummer av upprepade enheter i polymeren, Qpolym är polymerisation laddningen, F är Faradays konstant och NA är antalet Avogadro. - För att beräkna dopning avgift, använda CV av deponerade polymer registreras i en spektroelektrokemisk cell innan EPR mätning, med hjälp av ekvation 8:
 (8)
(8)
där Qd är dopning laddningen, är nuvarande , E är potentiella, E0 är apparatens nollinställning, v är sökhastighet och e är den elementära laddningen. - Använda beräknade dopning kostnad för att beräkna dopning nivå, med hjälp av ekvation 9:
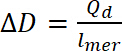 (9)
(9)
där ∆D- dopning nivå, Qd är dopning kostnad och lmer – antal upprepade enheter i polymeren - För att beräkna antalet bipolarons, anta ett inledande dopning nivå lika med noll. Använd antal polarons per enhet från koncentrationen av snurrar, så antalet bipolarons bör beräknas med hjälp av ekvation 10. I beräkningarna förutsätter att det ursprungliga beloppet av bipolarons är noll och att faradaic nuvarande är mycket högre än för icke-faradaic ström.
 (10)
(10)
där zjag är kostnad flygbolagets elementär laddning, niär antalet laddningsbärare polymer styck, (p) är polarons och (b) är bipolarons. - Extrahera den g-faktor värde från EPR spektra. Bestämma med Mn som intern standard (steg 4.2.3 och 4.3.8), att veta att den fjärde mangan raden i det har en g-faktor på 2.03324. Beräkna linjebredden EPR signal som avståndet i mT mellan minimum och maximum av EPR signalen. Detta värde är viktigt eftersom det kan berätta var (på vilken atom) radikalerna bildas.
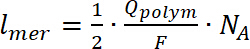 (7)
(7) (8)
(8)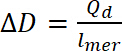 (9)
(9) (10)
(10)Representative Results
Den vanligaste tillämpningen av CV analysen är uppskattningen av IP och EA. Även om det finns ett par sätt att få CV data, rekommenderas det starkt att beräkna dem baserat på uppkomsten av redox toppar (figur 3). Detta tillvägagångssätt gör att unifying beräkningsproceduren. Inte alla av de testa materialen genomgår reversibel oxidation/reduktion processer (figur 3). i sådana situationer är det inte möjligt att beräkna baserat i genomsnitt potentialer (genomsnitt från potentialer motsvarar högst katodiskt-minskning och anodisk-oxidation toppar). Dock är det nästan alltid möjligt att köra tangens till en topp som visas i figur 3. Med korsningen med bakgrundslinje och ekvationer 1och 2, beräknas värdena IP och EA som 5.35 eV och −2.90 eV, respektive. Det finns också flera olika skalor som används för att utvärdera IP och EA på CV mätningar. De vanligaste skalorna för organiska material är −4.8 och −5.1 eV som motsvarar 0,00 V kontra Normal väte elektrod (NHE). Alla skalor är dock bara en approximation; Kom ihåg detta när man jämför olika resultat. Avgörande är att ange vilka parametrar som ansågs för beräkningar. I det här fallet den värde −5.1 eV har valts som det bör motsvara den formella potentialen hos Ferrocen redox par; i Fermi skalan är det 0,40 V kontra mättad kalomel elektrod (SCE) i acetonitril, som är överens med den tidigare mätningen.
Det finns många artiklar publicerade angående analysen av CV. häri, vi visar när processen inte kommer att vara som det förväntas. Analysen är baserad på tiopen derivat: NtVTh (struktur visas i figur 4), som genomgår nedbrytning vid oxidation (figur 5).
NtVTh har två oxidation potential: först på 0,70 V och andra på 0,84 V (figur 5). Under den första cykeln beaktas minskning toppen inte, som anger en oåterkallelig process. Elektrokemiska egenskaper av NtVTh visar att polymerisation inträffar inte och efter den första oxideringspotential, vissa electro-inaktiva lager av reaktionsprodukter insättningar på elektrod ytan, därmed hindra den polymerisation processen. Vad är synliga är reaktionen med radikala cationen om vinyl bond, där molekylen förlorar dess konjugering och bildar dimerer på elektroden.
Det är svårt att extrahera information om laddningsbärarna från CV, är det möjligt att skilja mellan polarons och bipolarons stöds av en UV-Vis-NIR-spektrometer. Den neutrala poly (OjagPrThEE) präglades av två brett absorption π | π * övergångar band med toppar maxima på λmax1 = 363 nm och λmax2 = 488 nm, relaterade till aromatiska form av undoped polytiofen derivatan. Under den oxidativa dopning genereras nya polaronic och bipolaronic band. UV-Vis spektrumet erhållits under poly (OjagPrThEE) oxidation avslöjade minskande neutrala polymer absorptionsbandet (300-550 nm) (figur 6) tillsammans med bildandet av ett nytt absorption band (550-950 nm) av de radikala kationerna av bithiophene och p- phenylenevinylene med maxima på 692 nm. Oxidationsprocessen isosbestic var belägen vid 604 nm. Bipolaronic bandet verkade mellan 950 nm och 1700 nm med högst belägna vid 1438 nm.
EPR spektroskopi är den teknik som upptäcker material med en oparad elektron, Detta inkluderar organiska radikaler39. Det finns flera parametrar som kunde utvinnas från EPR spectra, men en av de mest intressanta är att uppskatta där radikalerna är lokaliserade. Elektroner, protoner, liknande besitter spin. Genom att placera en elektron i ett yttre magnetfält, detta spinn kan delas upp på två sätt: parallella och antiparallel till det magnetiska fältet, ger två energinivåer. Detta fenomen kallas de Zeeman verkställer40,41. Vid organiska radikaler interagerar oparade elektronen inte bara med externa magnetfält, men också med magnetiska kärnor (kärnor som har en noll spinn; I≠0). Ett antal degenererade energinivåer är lika 2I + 1, där jag är den snurrandequantumen numrerar av kärnan som interagerar oparade elektronen42. Samspelet mellan oparade elektronen med ett större antal magnetiska kärnor leder till ytterligare uppdelning av energin jämnar och hyperfine strukturera av EPR spectra registrering43 (figur 7).
För molekyler där oparade elektronen interagerar med ett ännu större antal kärnor, enskilda spectral linjen kunde överlappar varandra, vilket resulterar i registrering av en enda, bred signal44,45,46 (figur 8). Detta är typiskt för konjugerade polymerer, där den genererade radikala ion under en redox process är delokaliserade47,48.
Kombinationen av EPR-spektroskopi med elektrokemiska metoder tillåter karakterisering av Laddningsbärare (radikala Jon) genereras under processen redox samt bestämning av mekanismen av dessa processer49,50 . Om väl löst (toppar är åtskilda, inte i form av en bred topp) spektra registreras, som i fallet med elektrokemisk reduktion av s -tetrazine derivat (figur 7), sedan analysen av hyperfine strukturera av spectra leder till slutsatser om lokaliseringen av oparad elektron. Ett sätt att analysera denna typ av spectra är att genomföra simulering med speciell programvara och för att passa simulerade spectra med den experimentella en51. Detta är särskilt användbart när hyperfine strukturera är komplex på grund av samspelet mellan oparade elektronen med stort antal protoner. Vid s- tetrazine derivatan visas i figur 7, visar simulering av EPR spectra (röda linjen) samspelet mellan oparade elektronen med fyra kväveatomer av s- tetrazine.

Figur 1: elektrokemisk och spektroelektrokemisk celler som används för mätningar. Figuren visar systemet installationen av elektrokemisk/spektroelektrokemisk celler som använder cyklisk voltametri, ultraviolett-synligt och infrarött (UV-Vis-NIR) och elektron paramagnetiska resonans (EPR) spektroelektrokemisk mätningar. Klicka här för att se en större version av denna siffra.

Figur 2: cyklisk voltametri (CV) av korrekt uppmätta förening COPO1 (a) och CV med en instabil referera potentiella dibenzothiophene-S, S-koldioxid med Ferrocen (b)52. Figuren visar två cykliska voltamogrammen. (a) presenterar korrekt registrerad CV och (b) visar en voltamogrammen registrerade använder en referenselektrod med ingen stabil potentiella. Klicka här för att se en större version av denna siffra.

Figur 3: cyklisk voltametri (CV) av sammansatta COPO1 i ett brett utbud av potentialer. Uppskattning av debut potentialen för EA och IP-beräkningar av COPO1 föreningar52. EA = −2.90 eV och IP = 5.35 eV. Klicka här för att se en större version av denna siffra.

Figur 4. EPR sex spectral linje mangan standard. Paramagnetiska signalen mangan som används för kalibrering av shift signal. Klicka här för att se en större version av denna siffra.

Figur 5: cyklisk voltametri (CV) av sammansatta NtVTh. Cyklisk voltamogrammen 15 mm NtVTh i 0.1 M Bu4NBF4/CH3KN och i förhållande till Ferrocen standard presenterar nedbrytningsprocessen som är inblandade på arbetselektroden. Sökhastighet 0.05 V/s: (en) togs i intervallet −1, 4 V till 0,8 V och (b) i den intervall −1, 4 V till 0,9 V. vänligen klicka här för att visa en större version av denna siffra.

Figur 6: Ultraviolett-synligt och infrarött (UV-Vis-NIR) spectroelectrochemistry av poly (OjagPrThEE) derivat. UV-Vis-NIR spektra presentera utvecklingen av absorptionsbandet genom generering av laddningsbärare på polymer struktur. Klicka här för att se en större version av denna siffra.

Figur 7: EPR spektroelektrokemisk analys av tetrazine derivat. (a) struktur av s- tetrazine derivat. (b) EPR spectra registrerade under elektrokemisk reduktion av s- tetrazine derivat (svart linje-experimentell och röd linje simulerade spektrum). Klicka här för att se en större version av denna siffra.

Figur 8: EPR spektroelektrokemisk polaron signal av konjugerad polymer. Electron paramagnetiska resonans (EPR) spektra registrerade under det första steget av oxidation av konjugerad polymer (EPR spectra polaron arter). Klicka här för att se en större version av denna siffra.

Figur 9: cyklisk voltametri (CV) av Ferrocen och decamethylferrocene. Jämförelse av två elektrokemiska standarder som ren och blandningen visar förskjutningen av potential. Klicka här för att se en större version av denna siffra.
Discussion
Elektrokemisk och spektroelektrokemisk tekniker har inga begränsningar. man kan analysera både SSD och flytande lösningar i ett brett spektrum av temperatur och andra tillstånd med dessa tekniker. Viktigaste i alla dessa fall är att föreningar/material analyseras enligt tillämpad potential, replikera verkliga förutsättningar för arbetar organisk elektronik enheter. Den enda skillnaden är att i elektrokemi, observeras bildandet av laddningsbärare.
De metoder som presenteras här visar nyttan av analysen av laddade bärare genereras i organiska föreningar som korrelerar med deras tillämplighet i organisk elektronik. Dessutom de elektrokemiska och spektroelektrokemisk teknikerna är billigare och mindre krävande än för typiska metoder som används i kostnad flygbolaget analys, men det finns några kritiska steg och ändringar av det protokoll som behövs beroende på den erhållna resultat.
Under elektrokemisk karaktärisering, börja alltid med en viss koncentration. Om en uppsättning av föreningarna som jämförs, sedan måste alla material ha samma molar koncentration. Bäst är att börja med 1 mM koncentration och 50 mV/s Skanna hastighet som anges i protokollet i denna studie, men det är bra att veta att koncentrationen i provet på observerade elektrokemiska beteendet. Alltid försöka mäta minst tre skanningar. De två första skanningar är oftast annorlunda eftersom de börja villkor (jämvikt) är olika. Andra och tredje skanningar bör vara samma. Om andra och tredje skanningar är samma, sedan finns det förmodligen ingen sida som observerats i detta system (figur 2a). I en oxidationsprocess visas en ny topp på en lägre potential visar att det ledande materialet deponerades på WE18,19,24,25,29,30 , 31 , 32. om höjden av den lägsta toppen ökar i på varandra följande skanningar, då förmodligen konjugerad polymer var insatta18,19,24,25,29 , 30 , 31 , 32. om alla strömmarna minskning av på varandra följande skanningar, då oledande produkten av nedbrytning deponerades på elektroden. Om en mycket liten topp observeras före huvudsakliga oxidation eller reduktion peak (särskilt för polymerer), är detta förmodligen avgift-svällning processen19,23,31,34. Om en mycket skarp dedoping topp av oxidation eller reduktion observeras, sedan orsakas detta förmodligen av nedbrytning av kristallina strukturer på en elektrod som bildas genom electrocrystallization processen vid oxidation35.
Kontrollera alltid uppförandet av sammansatta före, under och efter redox toppar testet. Det innebär att minst tre CV skanningar bör registreras: med övre (i fallet oxidation) eller lägre vertex potentialen lägre eller högre, respektive potential av peak maximala, med övre eller nedre vertex potential ange till exakt på maximalt toppvärde och med vertex potentialer högre (oxidation) och nedre (minskning) än potentialen hos peak maximalt. Observerade processen kan variera och ibland två processer kan observeras under en peak teoretiskt. Jämför alltid den insamlade cykliska voltamogrammen av elektrolyten (steg 2,6), Ferrocen (steg 2,9), föreningen (steg 2.13) och Ferrocen med förening (steg 2.19); i området i närheten finns det flera frågor att ta hänsyn till.
Jämför alltid CV signalerar av elektrolyten och testet förening, om några signaler från elektrolyten är synlig på den cykliska voltamogrammen av uppmätta föreningen, då elektrolyten måste ändras eftersom dess elektrokemiska fönster är för låg, eller elektrolyten är förorenad. Om signalen (redox par) av Ferrocen (steg 2,9) och Ferrocen med förening (steg 2.19) är i samma position, sedan utförs allt korrekt. Om topparna skiftas mellan varandra, sedan kontrollera RE och upprepa mätningen. Om signalen (oxidation, reduktion eller redox par potentiella) av testet förening med extra Ferrocen (steg 2.19) är på en högre potential än de rena förening (steg 2.13), sedan överväga värden (oxidation, reduktion eller redox par potentiella) från den cykliska voltamogrammen av ren föreningen. Shift orsakas av den högsta mängden Ferrocen i lösningen. När två oxidering processer observeras, kan den första processen (oxidation eller reduktion) som alltid är på vi påverka den aktiva ytan; Detta kan orsaka en ökning i oxidation potentialen hos den andra processen (figur 9).
Disclosures
Författarna har något att avslöja.
Acknowledgments
Författarna tacksamt erkänna finansiellt stöd till ”Excilight” projekt ”givare-Acceptor Light Emitting Exciplexes som material för lätt-till-skräddare ultraeffektiv OLED Lightning” (H2020-behöriga myndigheters-ITN-2015/674990) finansieras av Marie Skłodowska-Curie Åtgärder inom ramprogrammet för forskning och innovationer ”Horisont 2020”.
Materials
| Name | Company | Catalog Number | Comments |
| Potentiostat | Metrohm | Autolab PGSTAT100 | |
| EPR | JEOL | JES-FA200 | |
| UV-Vis detector | Oceanoptics | QE6500 | |
| NIR detector | Oceanoptics | NIRQuest | |
| Dichloromethane (DCM) | Sigma-Aldrich | 106048 | |
| Tetrabutylammonium tetrafluoroborate (Bu4NBF4) | Sigma-Aldrich | 86896 | |
| 2-propanol, 99.9% | Sigma-Aldrich | 675431 | |
| Acetone, 99.9% | Sigma-Aldrich | 439126 | |
| Ultrasonic Bath | Elma | S30H | |
| Tetrahydrofuran >99.9% | Sigma-Aldrich | 401757 | |
| ferrocene >98% | Sigma-Aldrich | F408 | |
| decamethylferrocene >97% | Sigma-Aldrich | 378542 |
References
- O'Brien, D. F., Baldo, M. A., Thompson, M. E., Forrest, S. R. Improved energy transfer in electrophosphorescent devices. Applied Physics Letters. 74, 442-444 (1999).
- Chin, B. D., et al. Effects of interlayers on phosphorescent blue organic light-emitting diodes. Applied Physics Letters. 86, 133505-133507 (2005).
- Cardon, C. M., Li, W., Kaifer, A. E., Stockdale, D., Bazan, G. C. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bang, A. -M., et al. A novel random terpolymer for high-efficiency bulk-heterojunction polymer solar cells. RSC Advances. 7, 1975-1980 (2017).
- Tybrandt, K., Kollipara, S. B., Berggren, M. Organic electrochemical transistors for signal amplification in fast scan cyclic voltammetry. Sensors and Actuators B: Chemical. 195, 651-656 (2014).
- Schaur, S., Stadler, P., Meana-Esteban, B., Neugebauer, H., Sariciftci, N. S. Electrochemical doping for lowering contact barriers in organic field effect transistors. Organic Electronics. 13, 1296-1301 (2012).
- Mullen, K., Scherf, U. Organic Light Emitting Devices: Synthesis, Properties and Applications. , Wiley-VCH. Weinheim. (2006).
- Monk, P. M. S., Mortimer, R. J., Rosseinsky, D. R. Electrochromism and Electrochromic Devices. , Cambridge University Press. Cambridge. (2007).
- Skotheim, T. A., Elsenbaumer, R. L., Reynolds, J. R. Handbook of Conducting Polymers. , Marcel Dekker. New York. (1998).
- Data, P., et al. Kesterite Inorganic-Organic Heterojunction for Solution Processable Solar Cells. Electrochimica Acta. 201, 78-85 (2016).
- Data, P., Pander, P., Okazaki, M., Takeda, Y., Minakata, S., Monkman, A. P. Dibenzo[a,j]phenazine-Cored Donor-Acceptor-Donor Compounds as Green-to-Red/NIR Thermally Activated Delayed Fluorescence Organic Light Emitters. Angewandte Chemie International Edition. 55 (19), 5739-5744 (2016).
- Jankus, V., et al. Highly Efficient TADF OLEDs: How the Emitter-Host Interaction Controls Both the Excited State Species and Electrical Properties of the Devices to Achieve Near 100% Triplet Harvesting and High Efficiency. Advanced Functional Materials. 24 (39), 6178-6186 (2014).
- Data, P., et al. Exciplex Enhancement as a Tool to Increase OLED Device Efficiency. Journal of Physical Chemistry C. 120 (4), 2070-2078 (2016).
- Dias, F. B., et al. The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Advanced Science. , (2016).
- Okazaki, M., et al. Thermally Activated Delayed Fluorescent Phenothiazine-Dibenzo[a,j]phenazine-Phenothiazine Triads Exhibiting Tricolor-Changing Mechanochromic Luminescence. Chemical Science. 8, 2677-2686 (2017).
- Data, P., et al. Efficient p-phenylene based OLEDs with mixed interfacial exciplex emission. Electrochimica Acta. 182, 524-528 (2015).
- Goushi, K., Yoshida, K., Sato, K., Adachi, C. Organic light-emitting diodes employing efficient reverse intersystem crossing for triplet-to-singlet state conversion. Nature Photonics. 6, 253-258 (2012).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of heteroaryl group on electrochemical and spectroscopic properties of conjugated polymers. Electrochimica Acta. 83, 271-282 (2012).
- Data, P., Motyka, M., Lapkowski, M., Suwinski, J., Monkman, A. Spectroelectrochemical Analysis of Charge Carries as a Way of Improving Poly(p-phenylene) Based Electrochromic Windows. Journal of Physical Chemistry C. 119, 20188-20200 (2015).
- Enengl, C., et al. Explaining the Cyclic Voltammetry of a Poly(1,4-phenylene-ethynylene)-alt-poly(1,4-phenylene-vinylene) Copolymer upon Oxidation by using Spectroscopic Techniques. ChemPhysChem. 18, 93-100 (2017).
- Gudeika, D., et al. Hydrazone containing electron-accepting and electron-donaiting moieties. Dyes Pigments. 91, 13-19 (2011).
- Swist, A., Cabaj, J., Soloducho, J., Data, P., Lapkowski, M. Novel acridone-based branched blocks as highly fluorescent materials. Synthetic Metals. 180, 1-8 (2013).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of alkyl chain on electrochemical and spectroscopic properties of polyselenophenes. Electrochimica Acta. 87, 438-449 (2013).
- Laba, K., et al. Electrochemically induced synthesis of poly(2,6-carbazole). Macromolecular Rapid Communications. 36, 1749-1755 (2015).
- Pluczyk, S., et al. Unusual Electrochemical Properties of the Electropolymerized Thin Layer Based on a s-Tetrazine-Triphenylamine Monomer. Journal of Physical Chemistry C. 120, 4382-4391 (2016).
- Blacha-Grzechnik, A., Turczyn, R., Burek, M., Zak, J. In situ Raman spectroscopic studies on potential-induced structural changes in polyaniline thin films synthesized via surface-initiated electropolymerization on covalently modified gold surface. Vibrational Spectroscopy. 71, 30-36 (2014).
- Laba, K., et al. Diquinoline derivatives as materials for potential optoelectronic applications. Journal of Physical Chemistry C. 119, 13129-13137 (2015).
- Enengl, S., et al. Spectroscopic characterization of charge carriers of the organic semiconductor quinacridone compared with pentacene during redox reactions. Journal of Materials Chemistry C. 4, 10265-10278 (2016).
- Data, P., et al. Electrochemically Induced Synthesis of Triphenylamine-based Polyhydrazones. Electrochimica Acta. 230, 10-21 (2017).
- Brzeczek, A., et al. Synthesis and properties of 1,3,5-tricarbazolylbenzenes with star-shaped architecture. Dyes Pigments. 113, 640-648 (2015).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Electrochemistry and spectroelectrochemistry of a novel selenophene-based monomer. Electrochimica Acta. 59, 567-572 (2012).
- Data, P., et al. Electrochemical and spectroelectrochemical comparison of alternated monomers and their copolymers based on carbazole and thiophene derivatives. Electrochimica Acta. 122, 118-129 (2014).
- Data, P., et al. Evidence for Solid State Electrochemical Degradation Within a Small Molecule OLED. Electrochimica Acta. 184, 86-93 (2015).
- Data, P., et al. Unusual properties of electropolymerized 2,7- and 3,6- carbazole derivatives. Electrochimica Acta. 1282, 430-438 (2014).
- Etherington, M. K., et al. Regio- and conformational isomerization critical to design of efficient thermally-activated delayed fluorescence emitters. Nature Communications. 8, 14987 (2017).
- Trasatti, S. The absolute electrode potential: an explanatory note. Pure and Applied Chemistry. 58, 955-966 (1986).
- Cardona, C. M., et al. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bredas, J. -L. Mind the gap! Mater Horiz. 1, 17-19 (2014).
- Gerson, F., Huber, W. Electron Spin Resonance Spectroscopy of Organic Radicals. , (2003).
- Brustolon, M., Giamello, E. Electron Paramagnetic Resonance. A Practitioner's Toolkit. , John Wiley & Sons Inc. New Jersey. (2009).
- Weil, J., Bolton, J. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. , John Wiley & Sons, Inc. (2007).
- Rieger, P. H. Electron Spin Resonance. Analysis and Interpretation. , The Royal Society of Chemistry. (2007).
- Roznyatovskiy, V. V., Gardner, D. M., Eaton, S. W., Wasielewski, M. R. Radical Anions of Trifluoromethylated Perylene and Naphthalene Imide and Diimide Electron Acceptors. Organic Letters. 16, 16-19 (2013).
- Pluczyk, S., Kuznik, W., Lapkowski, M., Reghu, R. R., Grazulevicius, J. V. The Effect of the Linking Topology on the Electrochemical and Spectroelectrochemical Properties of Carbazolyl Substituted Perylene Bisimides. Electrochimica Acta. 135, 487-494 (2014).
- Ledwon, P., et al. A Novel Donor-acceptor Carbazole and Benzothiadiazole Material for Deep Red and Infrared Emitting Applications. Journal of Materials Chemistry C. 4, 2219-2227 (2016).
- Heinze, J., Frontana-Uribe, B. A., Ludwigs, S. Electrochemistry of Conducting Polymers-Persistent Models and New Concepts. Chemical Reviews. 110, 4724-4771 (2010).
- Kurowska, A., Kostyuchenko, A. S., Zassowski, P., Skorka, L., Yurpalov, V. L., Fisyuk, A. S., Pron, A., Domagala, W. Symmetrically Disubstituted Bithiophene Derivatives of Properties. Journal of Physical Chemistry C. 118, 25176-25189 (2014).
- Zotti, G., Schiavon, G., Zecchin, S., Morin, J. F., Leclerc, M. Electrochemical, Conductive, and Magnetic Properties of 2,7-Carbazole-Based Conjugated Polymers. Macromolecules. 35, 2122-2128 (2002).
- Kaim, W., Fiedler, J. Spectroelectrochemistry: The Best of Two Worlds. Chemical Society Reviews. 38, 3373-3382 (2009).
- Duling, D. R. Simulation of Multiple Isotropic Spin-Trap EPR Spectra. Journal of Magnetic Resonance, Series B. 104, 105-110 (1994).
- Benson, C. R., et al. Multiplying the Electron Storage Capacity of a Bis-Tetrazine Pincer Ligand. Dalton Transactions. 43, 6513-6524 (2014).
- Nobuyasu, R. S., et al. Rational Design of TADF Polymers Using a Donor-Acceptor Monomer with Enhanced TADF Effi ciency Induced by the Energy Alignment of Charge Transfer and Local Triplet Excited States. Advanced Optical Materials. 4, 597-607 (2016).
