

Summary
In questo articolo, descriviamo elettrochimica, risonanza paramagnetica elettronica e ultravioletto-visibile e vicino infrarosso Spettroelettrochimica metodi per analizzare composti organici per applicazioni in elettronica organica.
Abstract
Voltammetria ciclica (CV) è una tecnica utilizzata nell'analisi di composti organici. Quando questa tecnica è combinata con risonanza paramagnetica elettronica (EPR) o ultravioletto-visibile e vicino infrarosso spettroscopie (UV-Vis-NIR), otteniamo informazioni utili come affinità dell'elettrone, potenziale di ionizzazione, energie di spacco della fascia, il tipo di elementi portanti della carica e informazioni di degrado che possono essere utilizzati per sintetizzare dispositivi elettronici organici stabili. In questo studio, presentiamo elettrochimica e Spettroelettrochimica metodi per analizzare i processi che avvengono in strati attivi di un dispositivo organico così come elementi portanti della carica generata.
Introduction
In tutto il mondo, i ricercatori sono continuamente alla ricerca di nuovi materiali organici che possono essere utilizzati in elettronica organica con prestazioni desiderabile o stabilità, che scende a causa di un uso prolungato. Nel caso di dispositivi organici, è importante comprendere il comportamento dell'elemento portante della carica completamente conoscere le regole del comportamento del dispositivo di guida. Analisi dell'effetto di molecolare struttura sulla generazione dell'elemento portante della carica e la dinamica e il mantenimento dell'equilibrio degli elementi portanti di carica iniettata, sia positivi (fori) e negative (elettroni), è fondamentale migliorare l'efficienza e la stabilità i dispositivi organici. Questo assicura la ricombinazione efficace di queste spese individuali e di conseguenza migliora notevolmente l'efficienza di fotoluminescenza del organic light emitting diodi (OLED)1,2. Per il fotovoltaico organico (OPVs)3,4 , nonché biologico campo effetto transistor (OFETs)5,6, è necessario disporre di materiali con mobilità di elemento portante ad alta carica. Oltre all'analisi di elementi portanti della carica, diversi parametri importanti di materiali organici elettroattivi aiutano nel predire dove potrebbe essere utilizzato il materiale: potenziale di ionizzazione (IP), i livelli di energia di elettrone affinità (EA) e band-gap tra li7 ,8,9,10.
In questo lavoro, vi presentiamo un metodo per la misurazione efficiente di voltammetria ciclica (CV) che può essere utilizzato nell'analisi di tutti i tipi di materiali elettroattivi. Questa tecnica fornisce informazioni sulle proprietà redox, il meccanismo di doping/dedoping, la stabilità, la conversione e la conservazione di energia, ecc. Permette anche per la stima dell'energia di ionizzazione e affinità dell'elettrone dei composti della prova in un modo molto più economico e più veloce rispetto ad altri metodi di alti vuoto. I suddetti parametri correlano con i livelli di energia del più alto orbitale molecolare occupato (HOMO) e più basso non occupato orbitale molecolare (LUMO).
Il metodo presentato in questo articolo può essere utilizzato per analizzare tutti i tipi di composti coniugati come quelli con gli elettroni π delocalizzati nelle loro strutture. Composti coniugati possono essere piccole molecole con grandi catene polimeriche. Piccole molecole possono anche essere monomeri; durante la reazione iniziale monomeri (fotochimiche, elettrochimiche o chimiche) possono formare i polimeri. In applicazione di OLED, i valori del livello di energia sono necessari per consentire l'utilizzo di host corretto per l'emettitore in un'attivazione termica ritardata sistema guest host a fluorescenza (TADF) o per decidere con quali composti lo strato di donatore-accettore exciplex potrebbe essere formato e quali ulteriori strati (strato (ETL), foro trasporto strato (HTL), strato di blocco dell'elettrone (EBL) e strato ostruente foro (HBL) di trasporto dell'elettrone) sarà necessari sintetizzare stabile in modo efficiente a carico bilanciato dispositivi OLED11 , 12 , 13 , 14 , 15 , 16 , 17. ulteriori misure elettrochimiche consentono lo studio di reazioni secondarie possibili durante il processo di degradazione dello strato attivo e la formazione del mobile basso pagare vettori (bipolaroni)18,19 ,20,21,22.
Accoppiamento elettrochimica e Spettroelettrochimica metodi consente facile, precisa e affidabile determinazione del grado di ossidazione o riduzione di composti coniugati e la loro degradazione potenziale, che è cruciale per la stabilità23 , 24 , 25 , 26 , 27 , 28. ultravioletto-visibile e vicino infrarosso spettroscopia (UV-Vis-NIR) accoppiata con elettrochimica può caratterizzare le proprietà cromatiche fondamentali di tutti i nuovi composti coniugati, come il cambio della banda di assorbimento durante il doping 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30.
In uno studio relazionato al meccanismo di anti-doping, è importante definire il tipo di elementi portanti della carica. In questo processo, due classi di quasiparticelle caricate prendono parte, uno con centrifuga non compensata (polaroni) e il secondo è diamagnetico (bipolaroni); spettroscopia di risonanza paramagnetica elettronica (EPR) fornisce assistenza inestimabile, che direttamente permette di osservare e rilevare le modifiche in popolazioni di polaroni paramagnetico29,30,31,32 . In piccole molecole, è difficile forma bipolaroni, ma queste molecole possono essere coniugate abbastanza e hanno proprietà che inducono bipolarone; è importante verificare se e a quali potenziali polaroni e bipolaroni sono formati nella struttura. Bipolaroni sono almeno un ordine in mobilità inferiore a quella di polaroni; Pertanto, se i bipolaroni sono formati in dispositivi di lavoro, quindi potrebbe portare a un rapporto squilibrato di elementi portanti della carica, che porterebbe a corrente elevata e il surriscaldamento del dispositivo OLED o può ben essere i centri di degradazione33.
Il metodo di misurazione proposto in questo studio è più veloce ed economico e permette per la determinazione dei parametri operativi più importanti per un gran numero di materiali elettroattivi senza la necessità di speciali dispositivi che si basano sulla nuova sintesi materiali per controllare le prestazioni. Applicando elettrochimica e spectroelectrochemistry, è possibile selezionare un materiale che è davvero promettente da centinaia di nuovi materiali. Inoltre, è possibile ottenere informazioni dettagliate per quanto riguarda i processi del doping e loro effetti sulla struttura chimica della prova coniugato con sistemi che utilizzano elettrochimica e Spettroelettrochimica metodi, che permette di costruire più dispositivi di elettronica organica efficiente.
Protocol
1. preparazione dell'esperimento
- Preparare 25 mL della soluzione dell'elettrolito 0.1 M.
Nota: A seconda dei composti in esame, utilizzare diversi elettroliti come la miscela di sale organico e solvente: il più comune sali utilizzati nell'analisi di molecole organiche sono Esafluorofosfato di tetrabutilammonio (Bu4NPF6 ) o tetrabutilammonio tetrafluoroborate (Bu4NBF4); il solvente più comune utilizzato nell'analisi sono diclorometano, acetonitrile o tetraidrofurano.- Scegliere il solvente elettrolita basato sulla solubilità dei composti della prova. Dovrebbe essere una soluzione durante inchiesta ma essere in uno stato solido quando il materiale è utilizzato nella fabbricazione del dispositivo, come un film sottile depositato sulla superficie dell'elettrodo di lavoro.
- Pulire gli elettrodi da impiegare nel esperimento29.
- Per misure elettrochimiche, utilizzare un elettrodo di platino disco diametro 1mm come l'elettrodo di lavoro (noi), una spiralina di platino o filo come l'elettrodo ausiliario (AE) e da un elettrodo di argento/cloruro di argento (Ag/AgCl) come l'elettrodo di riferimento (RE).
- Per le misure di Spettroelettrochimica EPR, utilizzare filo di platino come WE, una spiralina di platino o filo come AE e Ag/AgCl come il RE.
- Per le misure di Spettroelettrochimica UV-Vis-NIR, utilizzare un ossido di stagno di quarzo Indio (ITO) o ossido di stagno drogato con fluoro (FTO) come il WE, un filo di platino come l'AE e un elettrodo Ag/AgCl come il RE.
- Polacco l'elettrodo di platino disco sfregando l'inconveniente dell'elettrodo (l'area di lavoro di elettrodo di platino) su un pad lucidatrice coperto con 1 µm liquami di allumina per 3 min.
- Sciacquare l'elettrodo con acqua deionizzata per rimuovere tutti i residui dall'elettrodo e pulire in bagno a ultrasuoni (320 W, 37 kHz) con acqua demineralizzata per 15 min.
- Sciacquare l'elettrodo utilizzando una siringa da 1 mL con isopropanolo (3 x 1 mL) e poi con acetone (3 x 1 mL).
- Utilizzare un tovagliolo di carta per rimuovere tutti i residui solidi e asciugare all'aria per 3 min.
- Pulire gli elettrodi ITO e FTO con acqua deionizzata. Metterli in un bagno ad ultrasuoni (320 W, 37 kHz) in acetone per 15 min e poi in isopropanolo per il prossimo 15 min.
- Bruciare gli elettrodi di platino, cavi e bobine utilizzando un cannello a gas ad alta temperatura (> 1000 ° C) per 1 min. Fate attenzione! Utilizzare una pinzetta per tenere gli elettrodi. Attendere 5 min dopo la masterizzazione e prima di usare gli elettrodi.
- Pulire le celle elettrochimiche e Spettroelettrochimica (Figura 1) con acqua e poi con acetone. Utilizzare un tovagliolo di carta per rimuovere tutti i residui solidi. Pulire tutti gli altri elementi (ca. parti PTFE) con acetone e asciugare all'aria prima dell'uso.
2. CV analisi
- Accendere il potenziostato e il computer.
- Riempire una cella elettrochimica con 1,5 mL di soluzione elettrolitica per essere analizzati.
- Mettere tutti i tre elettrodi (WE, AE e RE) in una cella (tappo della cella è dotato di pinza porta elettrodo PTFE, quindi mettere gli elettrodi attraverso i fori in questo supporto) e collegarlo a un potenziostato. Mantenere la WE e RE come vicino a vicenda come possibile (Figura 1). Seguire le informazioni riguardanti la connessione di ogni filo del potenziostato al suo rispettivo elettrodo.
- Se necessario (ad esempio durante l'analisi di riduzione), mettere il tubo di argon (o azoto) (attraverso un ulteriore buco nel portaelettrodo) e iniziare a gorgogliare la soluzione per almeno 5 min. Dopo questo, spostare il tubo di argon (o azoto) sopra il livello della soluzione e mantenere il flusso di gas in esecuzione per la misurazione.
Nota: La cella sembra chiusa, ma non può essere completamente a prova di perdite; di conseguenza, un metodo per rimuovere il ridondante quantità di gas dalla cella deve essere disponibile (ad es., di un ulteriore piccolo buco nel portaelettrodo). La pressione dipende la pipe utilizzata; impostato la pressione alta, come è necessario impostare il flusso di gas lento. Se un solvente altamente volatile viene utilizzato nell'esperimento (ad es., diclorometano) o l'esperimento sta prendendo molto tempo (più di 30 min), quindi utilizzare la bottiglia Drechsel per saturare il gas argon (o azoto) con il solvente utilizzato nella misurazione. - Eseguire il software potenziostato, sceglie la procedura di CV e utilizzare le seguenti impostazioni: avviare il potenziale di 0.00 V, il vertice superiore potenziale di 2.0 V, potenziale di vertice inferiore di 0.00 V (se studiando il processo di riduzione, quindi un più basso potenziale di vertice di −2.5 V), smettere di potenziale di 0.00 V, numero dell'incrocio di fermata di 6 e la scansione velocità di 0,05 V/s.
- Nella sezione esportazione ASCII dati, definire un nome file e scegliere una cartella per salvare i dati in e spingere il pulsante START (diminuire o aumentare il potenziale di vertice inferiore e superiore per adattare la finestra elettrochimica o il campione attività elettrochimica)
- Se eventuali picchi sono visibili nell'intervallo positivo del potenziale, quindi ripetere la procedura di pulizia. Se c'è un picco nell'intervallo negativo potenziale, quindi mettere il tubo di argon (o azoto) nella soluzione e iniziare bubbling per altri 5 minuti.
- Aggiungere una goccia di 1 mM ferrocene (nel solvente utilizzato per preparare l'elettrolita) per la soluzione elettrolitica utilizzando una siringa.
- Impostare il potenziale di inizio a 0.00 V, il vertice superiore potenziale a 0,85 V, il vertice inferiore potenziale a 0.00 V, il potenziale di arresto a 0.00 V, il numero di fermata incrocio a 10 e la velocità di scansione a 0,05 V/s. Nella sezione esportazione ASCII dati, modificare il nome del file e premere il pulsante START .
- Dopo la misurazione, finitura ripetendo le operazioni di pulizia come indicato ai punti 1.2 e 1.3.
- Preparare 4 mL di 1 mM test composto nell'elettrolita preparato (punto 1.1).
- Riempire la cella elettrochimica con la soluzione di test composto (1,5 mL); mettere tutti e tre gli elettrodi in una cella e connettersi il potenziostato (punto 2.4). Per studiare il processo di riduzione, rimuovere l'ossigeno (punto 2.5).
- Impostare l'inizio potenziale a 0.00 V, il vertice superiore potenziale a 0,50 V (o 0.00 V in caso di indagine del processo di riduzione), il vertice inferiore potenziale a 0.00 V (−0.50 V in caso di indagine del processo di riduzione), il potenziale di arresto a 0.00 V , il numero di fermata incrocio a 10 e la velocità di scansione a 0,05 V. Nella sezione esportazione ASCII dati, modificare il nome del file e premere il pulsante START .
- Ripetere il passaggio 2.13 aumentando i potenziali vertice superiore (o facendo diminuire il potenziale di vertice inferiore) di 0.1 V fino a registrazione di picco (Figura 2a). Se la scansione successiva è spostata nel potenziale (Figura 2b), quindi pulire il RE e lasciarlo in soluzione elettrolitica. Quindi, ripetere la misurazione.
- Misurare i processi di ossidazione e di riduzione sul CV stesso per la determinazione di IP ed EA: impostare il potenziale di inizio a 0.00 V, il potenziale di vertice superiore a 1,00 V, il potenziale di vertice inferiore di −2.70 V e il potenziale di arresto a 0.00 V. Scegli il vert superiore e inferiore ex potenziale in un modo che registra la completa riduzione e ossidazione picchi ed evita ulteriori passi di redox (se ne esistono) (Figura 2a).
Nota: Stimare IP ed EA misurando l'insorgenza di potenziale. Ci sono un paio di modi per calcolare questi parametri da CV, ma il più utile è quello di calcolare usando l'insorgenza di potenziale. A volte, non è possibile osservare la coppia redox reversibile, soprattutto per n e p il doping. Miscelazione di due diverse tecniche anche è indesiderabile in quanto può fornire conclusioni e risultati errati.- Per misurare l'insorgenza di potenziale, segna la linea tangente al picco di CV (Figura 3). Calcolare i valori desiderati dal potenziale insorgenza della riduzione o il processo di ossidazione utilizzando equazioni (equazioni 1 e 2)10,18,19,36,37 , 38.
 (1)
(1)
dove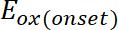 è l'inizio dell'ossidazione potenziale calibrato con ferrocene*
è l'inizio dell'ossidazione potenziale calibrato con ferrocene*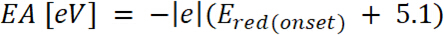 (2)
(2)
dove è l'inizio della riduzione potenziale calibrata con ferrocene*
è l'inizio della riduzione potenziale calibrata con ferrocene*
* Potenziale calibrato con ferrocene significa che il valore del potenziale dalla misurazione è diminuito il potenziale di ossidazione del ferrocene.
- Per misurare l'insorgenza di potenziale, segna la linea tangente al picco di CV (Figura 3). Calcolare i valori desiderati dal potenziale insorgenza della riduzione o il processo di ossidazione utilizzando equazioni (equazioni 1 e 2)10,18,19,36,37 , 38.
- Estrarre gli elettrodi e versare la soluzione nel contenitore per rifiuti adeguato.
- Ripetere la procedura di pulizia (punti 1.2 e 1.3).
- Ripetere i passaggi da 2.9 – 2.12 con 0.25, 0,10 e 0,20 frequenze di scansione V/s.
- Aggiungere una goccia di 1 mM ferrocene (nel solvente utilizzato per preparare la soluzione elettrolitica) alla soluzione della prova utilizzando una siringa. Impostare il potenziale di inizio a 0.00 V, il vertice superiore potenziale a 0.60 V, il potenziale di vertice inferiore a 0.00, il potenziale di arresto a 0.00 V, il numero di fermata incrocio a 6 e la velocità di scansione a 0,05 V. Push il tasto START . Una volta completata la misurazione, è possibile cambiare il vertice superiore potenziale su un valore che registra la coppia ferrocene con il primo picco di ossidazione della sostanza in esame.
- Estrarre gli elettrodi e versare la soluzione nel contenitore per rifiuti adeguato.
- Se l'elettropolimerizzazione si verifica durante l'ossidazione elettrochimica, lavare accuratamente la WE con la soluzione elettrolitica utilizzando la siringa (3 ´ 0,5 mL). Pulire il RE, AE e cella elettrochimica utilizzando la procedura standard (passaggi 1.2-1.3).
- Per studiare prodotti elettrochimici depositati su noi, procedere al punto 2.23. Per studiare una soluzione, procedere al passaggio 1.2.1 e iniziare nuovamente dal punto 2.3.
- Riempire la cella elettrochimica con una soluzione elettrolitica. Mettere tutti i tre elettrodi in una cella, quindi collegarli a un potenziostato (punto 2.4). Per studiare il processo di riduzione, disossidare la soluzione (punto 2.5).
- Ripetere i passaggi da 2.10 – 2.12 e poi passo 2.16.
 (1)
(1)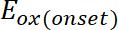 è l'inizio dell'ossidazione potenziale calibrato con ferrocene*
è l'inizio dell'ossidazione potenziale calibrato con ferrocene*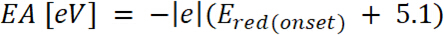 (2)
(2) è l'inizio della riduzione potenziale calibrata con ferrocene*
è l'inizio della riduzione potenziale calibrata con ferrocene*3. UV-Vis-NIR Spettroelettrochimica analisi
- Allestimento fiere
- Attivare lo spettrometro e potenziostato.
- Riempire la cella Spettroelettrochimica con 0,5 mL di soluzione elettrolitica (punto 1.1).
- Mettere la guarnizione PTFE su un lato della cella; poi mettere l'elettrodo ITO alla cella in questo modo di avere un lato conduttivo dell'elettrodo toccare la guarnizione PTFE. Mettere il resto delle parti PTFE come mostrato in Figura 1. Mettere il RE e AE attraverso il supporto di elettrodo PTFE e coprire la parte superiore dell'elettrodo ITO con lamina di rame come mostrato nella Figura 1 per migliore conducibilità.
- Mettere la cella montata nel supporto dello spettrometro e collegare tutti gli elettrodi di un potenziostato. Seguire le informazioni riguardanti la connessione di ogni filo del potenziostato al suo rispettivo elettrodo.
- Eseguire il software potenziostato e spettrometro.
- Nel software spettrometro, scegliere File | Nuovo | Assorbanza misura.
- Scegli uno dei rivelatori elencati.
- Assicurarsi che il pulsante sui rivelatori è in posizione "aperta". Quindi, fare clic sulla lampadina incandescente; chiudere il rivelatore (spostare il pulsante in posizione chiusa) e quindi fare clic sulla lampadina scura.
- Selezionare il secondo rilevatore e ripetere il punto 3.1.7.
- Ricollegare gli elettrodi dal potenziostato ed estrarre gli elettrodi e altri elementi della cella Spettroelettrochimica.
- Ripetere le procedure di pulizia (punti 1.2 e 1.3).
- Riempire la cella Spettroelettrochimica con un diluito (1´10− 5 M) soluzione della sostanza in soluzione elettrolitica se prova la piccola molecola in esame. Riempire la cella Spettroelettrochimica con l'elettrolita se testando lo strato polimerico (oligomerico) depositato sull'elettrodo ITO.
- Montare la cella Spettroelettrochimica (punto 3.1.3).
- Fare clic su Salva.
- Selezionare uno dei rivelatori elencati.
- Definire il salvataggio posizione e nome del file (includono il rivelatore selezionato).
- Selezionare il secondo rilevatore e ripetere il passaggio 3.1.15.
- Analisi potenziostatica18.
- Applicare un potenziale neutro (cioè, 0.00 V). Questo spettro sarà lo spettro di partenza. Aumentare il potenziale di 0,1 V ed attendere ca. 10 s, il processo si stabilizza e registra gli spettri di assorbimento (misure 3.1.13–3.1.16). Continuare la misurazione al primo cambiamento negli spettri UV-Vis-NIR; attendere per 10 s e quindi salvare gli spettri (passi 3.1.13–3.1.16).
- Aumentare la potenzialità di 0,05 V; attendere per 10 s e salvare (passi 3.1.13–3.1.16).
- Ripetere 3.2.2 fino a realizzare il potenziale del potenziale di ossidazione di primo o secondo (questi potenziali sono noti dalla misura di CV); il potenziale di invertire, quindi torna alla potenziale iniziale.
- Quando le misurazioni sono completate, misurare il CV della soluzione da analizzare. Aggiungere 1 mM ferrocene (10 μL) e misurare nuovamente il CV. Il valore del ferrocene aiuterà a stimare il potenziale del processo e unificare i dati.
- Questa analisi fornisce informazioni riguardanti la band-gap ottica, la massima lunghezza d'onda degli stati drogati e drogati e punti isosbestico del processo elettrochimico. Calcolare la banda ottiche-lacune da valori di insorgenza delle bande di π-π* sugli spettri UV-Vis e usando l'equazione 318,19,23,24.
 (3)
(3)
dove h è la costante di Planck, λinsorgenza è composto assorbimento insorgenza e c è la velocità della luce nel vuoto. - Analisi di Potentistatic Time-Resolved19,32,33
- Preparare la misura simile al metodo descritto in 3.1.1–3.1.16 e mettere il tubo di argon (o azoto) (attraverso un ulteriore buco nel portaelettrodo) e spumeggiante la soluzione almeno 5 min prima della misurazione. Dopo questo, spostare il tubo di argon sopra il livello della soluzione e mantenere il flusso di gas in esecuzione per la misurazione.
- Eseguire il software potenziostato, scegliere la procedura cronoamperometria con le seguenti impostazioni: potenziale inizio potenziale a 0.00 V, potenziale superiore a 1.0 V, potenziale inferiore a −1.00 V, durata 5 s, intervallo di tempo come 0.010 s, numero di ripetizioni a 10 e ritardo tempo di 10 s. Nella sezione esportazione ASCII dati, definire un nome file e scegliere una cartella per salvare i dati.
- Nel software spettrometro, scegliere File | Nuovo | Acquisizione ad alta velocità. Verrà visualizzata una nuova finestra; impostare i seguenti parametri: tempo di integrazione — 10 ms, scansioni alla media — 1, Wirth Boxcar — 0, numero di scansioni — 10000. Non fare clic sul pulsante Vai .
- Nel potenziostato software, premere il pulsante START . Quando il conto alla rovescia scende a 1 s, premere il pulsante Vai nel software spettrometro.
- Si noti che non è possibile vedere risultati di assorbimento fino al termine del processo.
- Analizzare i dati per fornire i parametri. Utilizzare le informazioni salvate nel file di tale trasmissione, la corrente, la tensione e la densità di corrente per fornire i parametri di funzionamento:
- Calcolare la densità ottica (ΔOD), l'assorbanza della sostanza durante il doping (dedoping) con 4 equazionein esame:
 (4)
(4)
dove Tb è trasmittanza in forma drogato, uno stato di "candeggina" [%] e Tc è trasmittanza in forma drogata, uno stato "colorato" [%]. - Calcolare l'efficienza di colorazione (CE, η), la quantità di colore elettrocromico formata dalla quantità usata di carica e come la connessione tra la carica iniettato/espulso in funzione dell'area dell'elettrodo (Q,d) e il cambiamento della densità ottica (ΔOD) valore di una specifica lunghezza d'onda (λmax) con equazione 5. Suo valore dipende dalla lunghezza d'onda scelta per lo studio, in modo che il valore è misurato a λmax della banda di assorbimento ottico dello stato colorato.
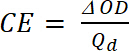 (5)
(5)
dove ΔOD è la densità ottica [-] e Qd è la densità di carica [C/cm2]. - Calcolare il rapporto di contrasto (CR), l'intensità misurata del cambiamento di colore durante il doping elettrochimica e generalmente caratterizzata da λmax della forma colorata con equazione 6:
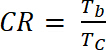 (6)
(6)
dove Tb è trasmittanza in forma drogato, stato "candeggina" [%] e Tc è trasmittanza in forma drogata, "colorato" stato [%].
- Calcolare la densità ottica (ΔOD), l'assorbanza della sostanza durante il doping (dedoping) con 4 equazionein esame:
 (3)
(3) (4)
(4)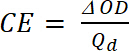 (5)
(5)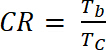 (6)
(6)4. EPR Spettroelettrochimica analisi
- Allestimento fiere
- Accendere lo spettrometro e il potenziostato.
- Riempire la cella Spettroelettrochimica con elettrolito (punto 1.1).
- Insieme la potenza di 1 mW e sintonizzare il risonatore per ottenere un segnale forte, centrato in Q-dip (usare l'opzione autotuned).
- Impostate il marcatore di manganese (manganese standard) di 600 e chiudere la finestra di Q-dip. Se non c'è alcun marcatore di Mn, poi saltare questo passaggio e andare al punto 4.8.
- Impostare "sweep larghezza ±" 2.5´ 10 mT, "mod larghezza ±" a 1.0´0.1 mT, "ampiezza" a 5´100, la "costante di tempo" a 0,03 s e "centro campo" valore di ca. 338 mT e avviare la misurazione.
- Se non registra sei linee spettrali del marcatore di Mn, quindi modificare il valore di "centro campo".
- Durante la registrazione di sei linee spettrali del marcatore Mn (Figura 4), impostare il valore di "centro campo" tra la terza e la quarta linea e diminuire il valore di "sweep larghezza ±" su un valore che copre solo queste due righe. Riprendere la misurazione.
- Andare in opzione Q-Dip, impostate il marcatore di Mn di 0 e chiudere la finestra di Q-dip.
- Misurare l'elettrolito puro come sfondo e controllare se ci sono eventuali segnali. Se i segnali sono visibili, il segnale potrebbe essere dalla contaminazione, composti potenziali bassa ossidazione, o dal vetro.
- Pulire la cella Spettroelettrochimica (punto 1.3).
- Indagine di piccole molecole (soluzioni).
- Riempire la cella Spettroelettrochimica con un diluito (1´10− 5 M) soluzione del test composto nell'elettrolita; mettere tutti i tre elettrodi attraverso il supporto di elettrodo alla cella in questo modo affinché il WE e RE sono all'interno della spirale AE (Figura 1).
- Mettere la WE vicino al fondo della cella e il RE sul lato superiore del principio attivo (metallo non rivestito di PTFE) parte della WE (Figura 1). Posizionare il cellulare dotato di un elettrodo all'interno nella cavità campione dello spettrometro e collegare gli elettrodi con un potenziostato (seguire le informazioni riguardanti la connessione di ogni filo del potenziostato al suo rispettivo elettrodo).
- Applicare il potenziale corrispondente alla comparsa del primo picco redox (valore noto dalla misura della CV). Se viene visualizzata la finestra di un segnale, l'apparecchio è impostato correttamente.
- Andare in opzione Q-Dip, impostate il marcatore di Mn di 600, chiudere la finestra di Q-tuffo e avviare la misurazione. La registrazione del segnale con il segnale del marcatore Mn permette il calcolo di g-fattore del segnale ricevuto.
- Andare in opzione Q-Dip, impostate il marcatore di Mn di 0, chiudere la finestra di Q-tuffo e aspettare ca. 5 min.
- Impostare il valore di "centro campo" al centro del segnale, diminuire il valore di "sweep larghezza ±" su un valore che copre il segnale, ma non tagliare l'inizio e la fine il segnale (quando "sweep larghezza ±" è cambiato, intero segnale di controllo) e diminuire il valore di "m od larghezza ± "per ottenere spettri ben risolti.
- Se il segnale è piccolo, quindi aumentare il valore di "ampiezza" e "time" (tempo di acquisizione) della misura.
- Per diminuire il rapporto segnale-rumore, impostare l'acquisizione di 4, 9 o 16 a seconda di quanto rumoroso il segnale è. Se il segnale è molto rumoroso, selezionare 16.
- Riempire la cella Spettroelettrochimica con un diluito (1´10− 5 M) soluzione del test composto nell'elettrolita; mettere tutti i tre elettrodi attraverso il supporto di elettrodo alla cella in questo modo affinché il WE e RE sono all'interno della spirale AE (Figura 1).
- Indagini dello strato polimerico depositato sulla superficie della WE.
- Riempire la cella Spettroelettrochimica con una soluzione elettrolitica (punto 1.1).
- Mettere gli elettrodi nella cella. Fare attenzione a non distruggere lo strato polimerico sulla WE. Collegare l'elettrodo con il potenziostato (punto 4.2.1).
- Applicare il potenziale neutro (cioè 0.00 V) per garantire che il composto è in uno stato neutrale; Questo spettro saranno gli spettri EPR partenza.
- Aumentare il potenziale di 0.10 V. attendere ca. 10 s fino a quando il processo si stabilizza e registrare gli spettri EPR.
- Ripetere il passaggio 4.3.4 quando viene visualizzato il segnale EPR.
- Aumentare il potenziale di 0,05 V. attendere ca. 10 s fino a quando il processo si stabilizza e registrare gli spettri EPR.
- Ripetere il passo 4.3.6 fino al primo o secondo potenziale di ossidazione è raggiunto e così via (i valori di questi potenziali sono noti da misura di CV); quindi, il potenziale di invertire e tornare a partire potenziali.
- Applicare il potenziale a cui l'EPR segnale apparve nel ciclo precedente (4.3.5), andare in opzione Q-Dip, impostate il marcatore di Mn di 600, chiudere la finestra di Q-tuffo e avviare la misurazione. Registrare il segnale con la terza e la quarta riga spettrale del marcatore Mn (4.1.7).
- Quando la misurazione è completata, è possibile analizzare i dati.
- Alcune delle analisi come un numero di giri può essere eseguito solo per i composti depositati (polimeri insolubili) sulla WE. Per calcolare il numero di giri, doppio-integrare il segnale EPR, seguito da un confronto del valore ottenuto con calibrata standard interno di Mn. Calcolare il numero di unità che formano il polimero sull'elettrodo usando equazione 724,32,33ripetute:
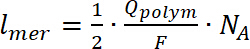 (7)
(7)
dove lmer – numero di unità nel polimero ripetitive, Qpolym è la carica di polimerizzazione, F è la costante di Faraday e NA è il numero di Avogadro. - Per calcolare la carica anti-doping, utilizzare il CV del polimero depositato, registrato in una cella di Spettroelettrochimica prima della misurazione di EPR, utilizzando l'equazione 8:
 (8)
(8)
dove Qd è la carica anti-doping, i è la corrente, E è l'insieme di potenziale, E0 è il potenziale iniziale e v è la velocità di scansione e è la carica elementare. - Utilizzare carica anti-doping calcolato per calcolare il livello di anti-doping, utilizzando l'equazione 9:
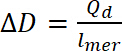 (9)
(9)
dove ∆D- livello, Qd il doping è la carica anti-doping e lmer – numero di unità nel polimero ripetute - Per calcolare il numero di bipolaroni, assumere un iniziale doping livello pari a zero. Utilizzare il numero di polaroni unitario dalla concentrazione di giri, quindi il numero di bipolaroni dovrebbe essere calcolato utilizzando equazione 10. Nei calcoli, si supponga che la quantità iniziale di bipolaroni è zero e che la corrente faradica è molto superiore a quello della non-faradica corrente.
 (10)
(10)
dove zio è la carica elementare di portacarica, niè il numero di portatori di carica per unità di polimero, (p) è il polaroni e (b) è i bipolaroni. - Estrarre il g-fattore valore dagli spettri EPR. Determinare tramite Mn come standard interno (passaggi 4.2.3 e 4.3.8), sapendo che la quarta riga di manganese in esso ha una g-fattore di 2.03324. Calcolare lo spessore della linea di segnale EPR come la distanza in metri tra minimo e massimo del segnale EPR. Questo valore è importante come può dire dove (su quali atom) si formano i radicali.
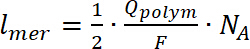 (7)
(7) (8)
(8)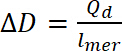 (9)
(9) (10)
(10)Representative Results
L'applicazione più comune di analisi CV è la stima dell'IP ed EA. Anche se ci sono un paio di modi per ottenere dati di CV, si consiglia a calcolarle basato sull'insorgenza di picchi di redox (Figura 3). Questo approccio consente di unificare la procedura di calcolo. Non tutti i materiali testati subiscono processi di ossidazione/riduzione reversibile (Figura 3); in tali situazioni, non è possibile calcolare in media base potenziali (media dai potenziali corrispondono ad un massimo di riduzione catodica e ossidazione anodica picchi). Tuttavia, è quasi sempre possibile eseguire la tangente ad un picco, come mostrato nella Figura 3. Con l'intersezione con la linea di fondo e l'uso delle equazioni 1 e 2, i valori IP ed EA sono stimati come 5,35 eV e −2.90 eV, rispettivamente. Ci sono anche parecchie scale differenti utilizzate per valutare IP ed EA basato su misurazioni di CV. Il più comunemente usato bilance per materiali organici sono −4.8 e −5.1 eV come equivalente a 0.00 V rispetto al normale idrogeno elettrodo (NHE). Tuttavia, tutte le scale sono solo un'approssimazione; ricordare questo quando si confrontano i risultati diversi. La cosa fondamentale è quello di indicare quali parametri sono stati considerati per il calcolo. In questo caso, il valore −5.1 eV è stato scelto come deve corrispondere al potenziale formale della coppia redox ferrocene; della scala di Fermi, è 0,40 V contro elettrodo di calomelano saturo (SCE) in acetonitrile, che è in accordo con la misura precedente.
Ci sono molti articoli pubblicati riguardanti l'analisi dei CV nel presente documento, ci mostra quando il processo non sta per essere come ci si aspetta. L'analisi si basa su derivato tiofene: NtVTh (struttura illustrata nella Figura 4), che subisce degradazione all'ossidazione (Figura 5).
NtVTh ha due potenziali di ossidazione: il primo a 0.70 V e il secondo a 0,84 V (Figura 5). Durante il primo ciclo, il picco di riduzione non è osservato, che indica un processo irreversibile. Caratteristiche elettrochimiche di NtVTh mostrano che la polimerizzazione non si verifica e dopo il primo potenziale di ossidazione, alcuni layer electro-inattivi dei prodotti di reazione si deposita sulla superficie dell'elettrodo, ostacolando in tal modo il processo di polimerizzazione. Ciò che è visibile è la reazione con il catione radicale dell'obbligazione di vinile, dove la molecola sta perdendo la sua coniugazione e forma dimeri sull'elettrodo.
Mentre è difficile trarre informazioni sui portatori di carica dal CV, è possibile distinguere tra polaroni e bipolaroni quando supportato da uno spettrometro UV-Vis-NIR. Il poly neutro (OiPrThEE) è stato caratterizzato da due ampie assorbimento π | π * transizioni bande con maxima picchi presso λmax1 = 363 nm e λmax2 = 488 nm, relazionati alla forma aromatica del derivato spossi politiofene. Durante il doping ossidativo, vengono generate nuove band polaronici e bipolaronic. Lo spettro UV-Vis ottenuto durante l'ossidazione di poli (OiPrThEE) ha rivelato la diminuzione la banda di assorbimento del polimero neutro (300-550 nm) (Figura 6) insieme con la formazione di una nuova banda di assorbimento (550-950 nm) dei cationi radicali di bithiophene e p- phenylenevinylene con maxima presso 692 nm. Il punto isosbestico del processo di ossidazione si trovava a 604 nm. La band bipolaronic è apparsa tra i 950 nm e 1700 nm con un massimo situato a 1438 nm.
Spettroscopia EPR è la tecnica che rileva i materiali con un elettrone spaiato, questo include radicali organici39. Ci sono diversi parametri che potrebbero essere estratti dagli spettri EPR, ma uno dei più interessanti è quello di stimare dove sono localizzati i radicali. Gli elettroni, simili ai protoni, possiedono spin. Inserendo un elettrone in un campo magnetico esterno, questo spin può essere suddiviso in due modi: parallelo ed antiparallelo al campo magnetico, dando due livelli di energia. Questo fenomeno è noto come l'effetto di Zeeman40,41. In caso di radicali organici, l'elettrone non accoppiato interagisce non solo con il campo magnetico esterno, ma anche con i nuclei magnetici (nuclei che hanno una spin diverso da zero; I≠0). Un numero di livelli di energia degenerati è uguale a 2I + 1, dove I è il numero di quantum di rotazione del nucleo con la quale l'elettrone spaiato interagisce42. L'interazione dell'elettrone non accoppiato con un maggior numero di nuclei magnetici conduce ad ulteriore spaccare i livelli di energia e alla struttura iperfine dell'EPR spettri registrazione43 (Figura 7).
Per molecole dove l'elettrone spaiato interagisce con un numero ancora maggiore di nuclei, potrebbe sovrapporsi la singola linea spettrale, che si traduce nella registrazione di un segnale singolo, ampia44,45,46 (Figura 8). questo è tipico per polimeri coniugati, dove lo ione radicale generato durante un processo redox è delocalizzata47,48.
La combinazione della spettroscopia EPR con metodi elettrochimici permette la caratterizzazione degli elementi portanti della carica (ione radicale) generati durante il processo redox, come pure la determinazione del meccanismo di questi processi49,50 . Se ben risolto (picchi sono separati; non in forma di un vasto picco) spettri sono registrati, come nel caso di riduzione elettrochimica di s -tetrazine derivato (Figura 7), poi l'analisi della struttura iperfine degli spettri porta a conclusioni circa la localizzazione di un elettrone spaiato. Per analizzare questo tipo di spettri è di condurre la simulazione con software speciale e per adattarsi simulati spettri con uno sperimentale51. Questo è particolarmente utile quando la struttura iperfine è complessa a causa dell'interazione dell'elettrone non accoppiato con un gran numero di protoni. In caso il s- tetrazine derivato illustrato nella Figura 7, la simulazione degli spettri EPR (linea rossa) indica l'interazione dell'elettrone spaiato con quattro atomi di azoto di s- tetrazine.

Figura 1: elettrochimica e Spettroelettrochimica celle utilizzate per misurazioni. La figura presenta il programma di installazione di schema delle cellule elettrochimiche/Spettroelettrochimica mediante voltammetria ciclica, ultravioletto-visibile e vicino infrarosso (UV-Vis-NIR) ed elettrone misure di Spettroelettrochimica di risonanza paramagnetica elettronica (EPR). Clicca qui per visualizzare una versione più grande di questa figura.

Figura 2: voltammetria ciclica (CV) di composti correttamente misurato COPO1 (a) e il CV con un instabile di riferimento potenziale dibenzotiofene-S, S-diossido con ferrocene (b)52. La figura mostra due voltammograms ciclico. (a) presenta correttamente registrato CV e (b) Mostra un voltammogram registrato utilizzando un elettrodo di riferimento con nessun potenziale stabile. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 3: voltammetria ciclica (CV) di composti COPO1 in una vasta gamma di potenziali. Stima dei potenziali di inizio per il calcolo di EA e IP della COPO1 composti52. EA = −2.90 eV e IP = 5,35 eV. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 4. Linea spettrale EPR sei di manganese standard. Il segnale paramagnetico di manganese usato per la taratura dello spostamento segnale. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 5: voltammetria ciclica (CV) del composto NtVTh. Voltammograms ciclico di 15 mM NtVTh in 0.1 M Bu4NBF4/CH3CN e relativo ferrocene standard che presenta il processo di degradazione coinvolto su elettrodo di lavoro. Velocità di scansione 0.05 V/s: (un) è stata scattata nella gamma −1.4 V a 0,8 V e (b) in −1.4 la gamma V a 0,9 V. per favore clicca qui per visualizzare una versione più grande di questa figura.

Figura 6: ultravioletto-visibile e nel vicino infrarosso (UV-Vis-NIR) spectroelectrochemistry del derivato di poli (OiPrThEE). Spettri UV-Vis-NIR che presenta l'evoluzione della banda di assorbimento attraverso la generazione di elementi portanti della carica sulla struttura del polimero. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 7: analisi Spettroelettrochimica EPR di tetrazine derivato. (a) struttura del s- tetrazine derivato; (b) gli spettri EPR registrato durante la riduzione elettrochimica di s- tetrazine derivato (spettro simulato nero linea rossa e linea-sperimentale). Clicca qui per visualizzare una versione più grande di questa figura.

Figura 8: segnale EPR Spettroelettrochimica polarone del polimero coniugato. Gli spettri di risonanza paramagnetica elettronica (EPR) dell'elettrone registrata durante la prima fase di ossidazione dei polimeri coniugati (spettri EPR di polarone specie). Clicca qui per visualizzare una versione più grande di questa figura.

Figura 9: voltammetria ciclica (CV) di ferrocene e decamethylferrocene. Confronto di due elettrochimici standard come puro e come miscela mostrando lo spostamento del potenziale. Clicca qui per visualizzare una versione più grande di questa figura.
Discussion
Tecniche di elettrochimica e Spettroelettrochimica non hanno limitazioni; si può analizzare sia allo stato solido e liquidi soluzioni in una vasta gamma di temperatura e altre condizioni con queste tecniche. La cosa importante in tutti questi casi è che i composti/materiali vengono analizzati sotto il potenziale applicato, replicando le condizioni del mondo reale per l'utilizzo di dispositivi di elettronica organica. L'unica differenza è che in elettrochimica, la formazione di portatori di carica, è osservata.
I metodi presentati qui illustrano l'utilità dell'analisi di portatori generati nei composti organici che correlano con la loro applicabilità in elettronica organica. Inoltre, le tecniche di elettrochimica e Spettroelettrochimica sono meno costosi e meno esigenti di quello dei tipici metodi utilizzati nell'analisi di vettore di carica, ma ci sono alcuni passaggi critici e modifiche del protocollo che sono necessari a seconda del risultati ottenuti.
Durante la caratterizzazione elettrochimica, iniziare sempre con una particolare concentrazione. Se un insieme dei composti è rispetto, tutti i materiali hanno bisogno di avere la stessa concentrazione molare. Il migliore è di iniziare con concentrazione di 1 mM e 50 mV/s velocità di scansione come indicato nel protocollo in questo studio, ma è bene sapere che la concentrazione del campione sul comportamento elettrochimico osservato. Sempre cercare di misurare almeno tre scansioni. Le prime due scansioni sono solitamente differenti perché le condizioni di partenza (equilibrio) sono diverse. Il secondo e il terza scansioni dovrebbero essere lo stesso. Se le scansioni seconda e terza sono uguali, quindi non ci sono probabilmente nessun reazioni collaterali osservati in questo sistema (Figura 2a). In un processo di ossidazione, un nuovo picco a un potenziale inferiore appare mostrando che il materiale conduttivo è stato depositato il WE18,19,24,25,29,30 , 31 , 32. se l'altezza del picco inferiore aumenta nelle successive scansioni, quindi probabilmente il polimero coniugato è stato depositato18,19,24,25,29 , 30 , 31 , 32. se tutte le correnti diminuiscono nelle successive scansioni, quindi il prodotto non conduttivo di degradazione è stato depositato sull'elettrodo. Se un molto piccolo picco si osserva prima il principale ossidazione o riduzione peak (soprattutto per i polimeri), allora questo è probabilmente carica-intrappolamento processo19,23,31,34. Se si osserva un picco di dedoping acuto di ossidazione o riduzione, quindi questo è probabilmente causato dalla decomposizione delle strutture cristalline su un elettrodo formata attraverso il processo di electrocrystallization durante l'ossidazione35.
Controllare sempre il comportamento della sostanza prima, durante e dopo i picchi di redox in esame. Vuol dire che devono essere registrate almeno tre scansioni CV: con tomaia (nel caso ossidazione) o più basso potenziale di vertice inferiore o superiore, rispettivamente, quindi il potenziale di massimo picco, con un potenziale di vertice superiore o inferiore impostata esattamente il massimo del picco e con Vertex potenzialità superiore (ossidazione) e inferiore (riduzione) che il potenziale del massimo del picco. Il processo osservato può variare e talvolta due processi possono essere osservati sotto un unico picco teoricamente. Sempre confrontare i raccolti voltammograms ciclico dell'elettrolito (punto 2.6), il ferrocene (punto 2.9), il composto (passo 2.13) e il ferrocene con composto (passo 2.19); Ci sono diversi problemi da tenere in considerazione.
Sempre confrontare i segnali CV dell'elettrolito e il test composto, se alcun segnale dall'elettrolito è visibile sul voltammogram ciclico del composto misurato, poi l'elettrolito deve essere cambiato perché la finestra elettrochimica è troppo bassa, o il dell'elettrolito è contaminato. Se il segnale (coppia redox) del ferrocene (punto 2.9) e ferrocene con composto (passo 2.19) sono nella stessa posizione, quindi tutto viene eseguito correttamente. Se i picchi sono spostati tra l'altro, quindi controllare il RE e ripetere la misurazione. Se il segnale (ossidazione, riduzione o potenziale di coppia redox) del test composto con aggiunto ferrocene (passo 2.19) è a un potenziale più elevato di quello del puro compound (passo 2.13), quindi prendere in considerazione i valori (ossidazione, riduzione o redox coppia potenziali) da il voltammogram ciclico del composto puro. Lo spostamento è causato dalla maggiore quantità di ferrocene nella soluzione. Quando si osservano due processi di ossidazione, il primo processo (ossidazione o riduzione) che è sempre su WE può interessare la superficie attiva; Questo può causare un aumento del potenziale di ossidazione del secondo processo (Figura 9).
Disclosures
Gli autori non hanno nulla a rivelare.
Acknowledgments
Gli autori riconoscono con gratitudine il sostegno finanziario del progetto "Excilight" "Donatore-accettore Light Emitting Tespesio come materiali per facile-a-tailor Lightning OLED ultra-efficiente" (H2020-MSCA-ITN-2015/674990) finanziato da Marie Skłodowska-Curie Azioni nell'ambito del programma quadro per la ricerca e innovazioni "Horizon 2020".
Materials
| Name | Company | Catalog Number | Comments |
| Potentiostat | Metrohm | Autolab PGSTAT100 | |
| EPR | JEOL | JES-FA200 | |
| UV-Vis detector | Oceanoptics | QE6500 | |
| NIR detector | Oceanoptics | NIRQuest | |
| Dichloromethane (DCM) | Sigma-Aldrich | 106048 | |
| Tetrabutylammonium tetrafluoroborate (Bu4NBF4) | Sigma-Aldrich | 86896 | |
| 2-propanol, 99.9% | Sigma-Aldrich | 675431 | |
| Acetone, 99.9% | Sigma-Aldrich | 439126 | |
| Ultrasonic Bath | Elma | S30H | |
| Tetrahydrofuran >99.9% | Sigma-Aldrich | 401757 | |
| ferrocene >98% | Sigma-Aldrich | F408 | |
| decamethylferrocene >97% | Sigma-Aldrich | 378542 |
References
- O'Brien, D. F., Baldo, M. A., Thompson, M. E., Forrest, S. R. Improved energy transfer in electrophosphorescent devices. Applied Physics Letters. 74, 442-444 (1999).
- Chin, B. D., et al. Effects of interlayers on phosphorescent blue organic light-emitting diodes. Applied Physics Letters. 86, 133505-133507 (2005).
- Cardon, C. M., Li, W., Kaifer, A. E., Stockdale, D., Bazan, G. C. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bang, A. -M., et al. A novel random terpolymer for high-efficiency bulk-heterojunction polymer solar cells. RSC Advances. 7, 1975-1980 (2017).
- Tybrandt, K., Kollipara, S. B., Berggren, M. Organic electrochemical transistors for signal amplification in fast scan cyclic voltammetry. Sensors and Actuators B: Chemical. 195, 651-656 (2014).
- Schaur, S., Stadler, P., Meana-Esteban, B., Neugebauer, H., Sariciftci, N. S. Electrochemical doping for lowering contact barriers in organic field effect transistors. Organic Electronics. 13, 1296-1301 (2012).
- Mullen, K., Scherf, U. Organic Light Emitting Devices: Synthesis, Properties and Applications. , Wiley-VCH. Weinheim. (2006).
- Monk, P. M. S., Mortimer, R. J., Rosseinsky, D. R. Electrochromism and Electrochromic Devices. , Cambridge University Press. Cambridge. (2007).
- Skotheim, T. A., Elsenbaumer, R. L., Reynolds, J. R. Handbook of Conducting Polymers. , Marcel Dekker. New York. (1998).
- Data, P., et al. Kesterite Inorganic-Organic Heterojunction for Solution Processable Solar Cells. Electrochimica Acta. 201, 78-85 (2016).
- Data, P., Pander, P., Okazaki, M., Takeda, Y., Minakata, S., Monkman, A. P. Dibenzo[a,j]phenazine-Cored Donor-Acceptor-Donor Compounds as Green-to-Red/NIR Thermally Activated Delayed Fluorescence Organic Light Emitters. Angewandte Chemie International Edition. 55 (19), 5739-5744 (2016).
- Jankus, V., et al. Highly Efficient TADF OLEDs: How the Emitter-Host Interaction Controls Both the Excited State Species and Electrical Properties of the Devices to Achieve Near 100% Triplet Harvesting and High Efficiency. Advanced Functional Materials. 24 (39), 6178-6186 (2014).
- Data, P., et al. Exciplex Enhancement as a Tool to Increase OLED Device Efficiency. Journal of Physical Chemistry C. 120 (4), 2070-2078 (2016).
- Dias, F. B., et al. The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Advanced Science. , (2016).
- Okazaki, M., et al. Thermally Activated Delayed Fluorescent Phenothiazine-Dibenzo[a,j]phenazine-Phenothiazine Triads Exhibiting Tricolor-Changing Mechanochromic Luminescence. Chemical Science. 8, 2677-2686 (2017).
- Data, P., et al. Efficient p-phenylene based OLEDs with mixed interfacial exciplex emission. Electrochimica Acta. 182, 524-528 (2015).
- Goushi, K., Yoshida, K., Sato, K., Adachi, C. Organic light-emitting diodes employing efficient reverse intersystem crossing for triplet-to-singlet state conversion. Nature Photonics. 6, 253-258 (2012).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of heteroaryl group on electrochemical and spectroscopic properties of conjugated polymers. Electrochimica Acta. 83, 271-282 (2012).
- Data, P., Motyka, M., Lapkowski, M., Suwinski, J., Monkman, A. Spectroelectrochemical Analysis of Charge Carries as a Way of Improving Poly(p-phenylene) Based Electrochromic Windows. Journal of Physical Chemistry C. 119, 20188-20200 (2015).
- Enengl, C., et al. Explaining the Cyclic Voltammetry of a Poly(1,4-phenylene-ethynylene)-alt-poly(1,4-phenylene-vinylene) Copolymer upon Oxidation by using Spectroscopic Techniques. ChemPhysChem. 18, 93-100 (2017).
- Gudeika, D., et al. Hydrazone containing electron-accepting and electron-donaiting moieties. Dyes Pigments. 91, 13-19 (2011).
- Swist, A., Cabaj, J., Soloducho, J., Data, P., Lapkowski, M. Novel acridone-based branched blocks as highly fluorescent materials. Synthetic Metals. 180, 1-8 (2013).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of alkyl chain on electrochemical and spectroscopic properties of polyselenophenes. Electrochimica Acta. 87, 438-449 (2013).
- Laba, K., et al. Electrochemically induced synthesis of poly(2,6-carbazole). Macromolecular Rapid Communications. 36, 1749-1755 (2015).
- Pluczyk, S., et al. Unusual Electrochemical Properties of the Electropolymerized Thin Layer Based on a s-Tetrazine-Triphenylamine Monomer. Journal of Physical Chemistry C. 120, 4382-4391 (2016).
- Blacha-Grzechnik, A., Turczyn, R., Burek, M., Zak, J. In situ Raman spectroscopic studies on potential-induced structural changes in polyaniline thin films synthesized via surface-initiated electropolymerization on covalently modified gold surface. Vibrational Spectroscopy. 71, 30-36 (2014).
- Laba, K., et al. Diquinoline derivatives as materials for potential optoelectronic applications. Journal of Physical Chemistry C. 119, 13129-13137 (2015).
- Enengl, S., et al. Spectroscopic characterization of charge carriers of the organic semiconductor quinacridone compared with pentacene during redox reactions. Journal of Materials Chemistry C. 4, 10265-10278 (2016).
- Data, P., et al. Electrochemically Induced Synthesis of Triphenylamine-based Polyhydrazones. Electrochimica Acta. 230, 10-21 (2017).
- Brzeczek, A., et al. Synthesis and properties of 1,3,5-tricarbazolylbenzenes with star-shaped architecture. Dyes Pigments. 113, 640-648 (2015).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Electrochemistry and spectroelectrochemistry of a novel selenophene-based monomer. Electrochimica Acta. 59, 567-572 (2012).
- Data, P., et al. Electrochemical and spectroelectrochemical comparison of alternated monomers and their copolymers based on carbazole and thiophene derivatives. Electrochimica Acta. 122, 118-129 (2014).
- Data, P., et al. Evidence for Solid State Electrochemical Degradation Within a Small Molecule OLED. Electrochimica Acta. 184, 86-93 (2015).
- Data, P., et al. Unusual properties of electropolymerized 2,7- and 3,6- carbazole derivatives. Electrochimica Acta. 1282, 430-438 (2014).
- Etherington, M. K., et al. Regio- and conformational isomerization critical to design of efficient thermally-activated delayed fluorescence emitters. Nature Communications. 8, 14987 (2017).
- Trasatti, S. The absolute electrode potential: an explanatory note. Pure and Applied Chemistry. 58, 955-966 (1986).
- Cardona, C. M., et al. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bredas, J. -L. Mind the gap! Mater Horiz. 1, 17-19 (2014).
- Gerson, F., Huber, W. Electron Spin Resonance Spectroscopy of Organic Radicals. , (2003).
- Brustolon, M., Giamello, E. Electron Paramagnetic Resonance. A Practitioner's Toolkit. , John Wiley & Sons Inc. New Jersey. (2009).
- Weil, J., Bolton, J. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. , John Wiley & Sons, Inc. (2007).
- Rieger, P. H. Electron Spin Resonance. Analysis and Interpretation. , The Royal Society of Chemistry. (2007).
- Roznyatovskiy, V. V., Gardner, D. M., Eaton, S. W., Wasielewski, M. R. Radical Anions of Trifluoromethylated Perylene and Naphthalene Imide and Diimide Electron Acceptors. Organic Letters. 16, 16-19 (2013).
- Pluczyk, S., Kuznik, W., Lapkowski, M., Reghu, R. R., Grazulevicius, J. V. The Effect of the Linking Topology on the Electrochemical and Spectroelectrochemical Properties of Carbazolyl Substituted Perylene Bisimides. Electrochimica Acta. 135, 487-494 (2014).
- Ledwon, P., et al. A Novel Donor-acceptor Carbazole and Benzothiadiazole Material for Deep Red and Infrared Emitting Applications. Journal of Materials Chemistry C. 4, 2219-2227 (2016).
- Heinze, J., Frontana-Uribe, B. A., Ludwigs, S. Electrochemistry of Conducting Polymers-Persistent Models and New Concepts. Chemical Reviews. 110, 4724-4771 (2010).
- Kurowska, A., Kostyuchenko, A. S., Zassowski, P., Skorka, L., Yurpalov, V. L., Fisyuk, A. S., Pron, A., Domagala, W. Symmetrically Disubstituted Bithiophene Derivatives of Properties. Journal of Physical Chemistry C. 118, 25176-25189 (2014).
- Zotti, G., Schiavon, G., Zecchin, S., Morin, J. F., Leclerc, M. Electrochemical, Conductive, and Magnetic Properties of 2,7-Carbazole-Based Conjugated Polymers. Macromolecules. 35, 2122-2128 (2002).
- Kaim, W., Fiedler, J. Spectroelectrochemistry: The Best of Two Worlds. Chemical Society Reviews. 38, 3373-3382 (2009).
- Duling, D. R. Simulation of Multiple Isotropic Spin-Trap EPR Spectra. Journal of Magnetic Resonance, Series B. 104, 105-110 (1994).
- Benson, C. R., et al. Multiplying the Electron Storage Capacity of a Bis-Tetrazine Pincer Ligand. Dalton Transactions. 43, 6513-6524 (2014).
- Nobuyasu, R. S., et al. Rational Design of TADF Polymers Using a Donor-Acceptor Monomer with Enhanced TADF Effi ciency Induced by the Energy Alignment of Charge Transfer and Local Triplet Excited States. Advanced Optical Materials. 4, 597-607 (2016).
