

Summary
Neste artigo, descrevemos eletroquímica, ressonância paramagnética electrónica e ultravioleta, visível e infravermelho próximo spectroelectrochemical métodos para analisar compostos orgânicos para aplicação em eletrônica orgânica.
Abstract
Voltametria cíclica (CV) é uma técnica usada na análise de compostos orgânicos. Quando esta técnica é combinada com a ressonância paramagnética eletrônica (EPR) ou espectroscopia de ultravioleta-visível e infravermelho próximo (UV-Vis-NIR), obtemos informações úteis tais como a afinidade eletrônica, potencial de ionização, energias de banda-lacuna, o tipo de as transportadoras de carga e informações de degradação que podem ser usadas para sintetizar dispositivos eletrônicos orgânicos estáveis. Neste estudo, apresentamos eletroquímico e spectroelectrochemical métodos para analisar os processos que ocorrem nas camadas ativas de um dispositivo orgânico, bem como os portadores de carga gerado.
Introduction
Em todo o mundo, pesquisadores estão continuamente à procura de novos materiais orgânicos que podem ser usados em eletrônica orgânica com desempenho desejável ou estabilidade, que deixa cair devido ao uso prolongado. No caso de dispositivos orgânicos, é importante compreender o comportamento da transportadora carga para conhecer plenamente as regras o comportamento do dispositivo de condução. Análise do efeito da molecular estrutura sobre a geração da transportadora de carga e a dinâmica e a manutenção do equilíbrio injetado de portadores de cargas, ambos positivos (buracos) e negativa (elétrons), é crucial para melhorar a eficiência e a estabilidade dos dispositivos orgânicos. Isto garante a recombinação eficaz destas acusações individuais e consequentemente significativamente melhora a eficiência de fotoluminescência da luz orgânica emitindo diodos (OLEDs)1,2. Para sistemas fotovoltaicos orgânicos (OPVs)3,4 , bem como orgânico campo efeito transistores (OFETs)5,6, é necessário ter materiais com mobilidade de portador de carga elevada. A análise de portadores de cargas, além de vários parâmetros importantes de materiais orgânicos eletroativos ajudam na previsão de onde o material pode ser usado: potencial de ionização (IP), níveis de energia do elétron afinidade (EA) e banda-intervalo entre eles,7 ,8,9,10.
Neste trabalho, apresentamos um método para a medição eficiente de voltametria cíclica (CV) que pode ser usada na análise de todos os tipos de materiais eletroativos. Esta técnica fornece informações sobre propriedades redox, o mecanismo de doping/dedoping, a estabilidade, a conversão e armazenamento de energia, etc. Também permite a estimativa da energia de ionização e afinidade eletrônica dos compostos teste forma um muito mais barato e mais rápido em comparação com outros métodos de altos vácuo. Os parâmetros acima mencionados se correlacionam com os níveis de energia do orbital molecular ocupado mais alto (HOMO) e orbital molecular mais baixo desocupado (LUMO).
O método apresentado neste artigo pode ser usado para analisar todos os tipos de compostos conjugados, tais como aqueles com elétrons π deslocalizados em suas estruturas. Compostos conjugados podem ser moléculas pequenas com grandes cadeias poliméricas. Pequenas moléculas podem também ser monômeros; durante a reação inicial podem se formar polímeros em monômeros (fotoquímicos, eletroquímicos ou químicos). No aplicativo de OLED, os valores de nível de energia são necessários habilitar o uso do host correto para o emissor em um tèrmica ativado atrasado sistema de anfitrião do convidado de fluorescência (TADF) ou decidir com qual compostos a camada de doador-aceitador exciplex poderia ser formado e que camadas adicionais (elétron transporte camada (ETL), buraco transporte camada (HTL), camada de bloqueio de elétron (EBL) e camada de bloqueio de buraco (HBL)) será necessárias para sintetizar estável eficientemente cobrado equilibrado dispositivos OLED11 , 12 , 13 , 14 , 15 , 16 , 17. medições eletroquímicas adicionais permitem a investigação de reações colaterais possíveis durante o processo de degradação da camada ativa e a formação de baixo móvel cobrar as transportadoras (bipolarons)18,19 ,20,21,22.
Acoplamento de eletroquímico e spectroelectrochemical métodos permite a fácil, precisa e confiável de determinação do grau de oxidação ou redução de compostos conjugados e sua degradação potencial, que é crucial para a estabilidade23 , 24 , 25 , 26 , 27 , 28. espectroscopia de ultravioleta-visível e infravermelho próximo (UV-Vis-NIR), juntamente com a eletroquímica pode caracterizar as propriedades cromáticas fundamentais de todos os novos compostos conjugados, tais como a mudança da banda de absorção durante a dopagem 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30.
Em um estudo relacionado ao mecanismo de doping, é importante definir o tipo de portadores de cargas. Neste processo, participam duas classes de quasiparticles carregadas, um com rotação descompensada (polarons) e o segundo sendo diamagnético (bipolarons); espectroscopia de ressonância paramagnética (EPR) elétrons fornece assistência inestimável, o que permite diretamente a observar e controlar as alterações nas populações de polarons paramagnético29,30,31,32 . Em pequenas moléculas, é difícil de forma bipolarons, mas estas moléculas podem ser conjugadas bastante e tem propriedades bipolaron de indução; é importante verificar se e em que potenciais polarons e bipolarons são formados na estrutura. Bipolarons são pelo menos uma ordem menor em mobilidade do que polarons; Portanto, se bipolarons formam-se em dispositivos de trabalho, então isso pode levar a uma relação desequilibrada dos transportadores de carga, que resultaria em alta corrente e sobreaquecimento do dispositivo OLED ou pode muito bem ser os centros de degradação33.
O método de medição proposto neste estudo é mais barato e mais rápido e permite a determinação dos parâmetros operacionais mais valiosos para um grande número de materiais eletroativos sem a necessidade de dispositivos especiais que são baseados na recém sintetizado materiais para verificar seu desempenho. Aplicando a eletroquímica e spectroelectrochemistry, é possível selecionar um material que é verdadeiramente promissor de centenas de novos materiais. Além disso, é possível obter informações detalhadas sobre os processos de doping e seus efeitos sobre a estrutura química do teste conjugados usando eletroquímica de sistemas e métodos de spectroelectrochemical, que permite a construção de mais dispositivos de eletrônica orgânica eficiente.
Protocol
1. preparação do experimento
- Prepare-se 25 mL de solução 0,1 M de eletrólito.
Nota: Dependendo dos compostos sob investigação, usar eletrólitos diferentes como a mistura de solvente e sal orgânico: o mais comum de sais usados na análise de moléculas orgânicas são Hexafluorofosfato de tetrabutilamónio (Bu4NPF6 ) ou tetrafluoroborate de tetrabutilamónio (Bu4NBF4); o solvente mais comum utilizado na análise são diclorometano, acetonitrila ou tetrahidrofurano.- Escolha o solvente de eletrólito baseado a solubilidade dos compostos teste. Isso deve ser uma solução durante a investigação... mas estar em um estado sólido quando o material é usado na fabricação do dispositivo, como um filme fino depositado na superfície do eletrodo de trabalho.
- Limpe os eletrodos para ser usado no experimento29.
- Para medições de eletroquímicas, use um eletrodo de disco de platina de diâmetro de 1 mm como o eletrodo de trabalho (nós), uma espiral de platina ou fio como o eletrodo auxiliar (AE) e um eletrodo de prata/cloreto de prata (Ag/AgCl) como o eletrodo de referência (RE).
- Para medições de spectroelectrochemical EPR, use fio de platina como a nós, uma espiral de platina ou fio como o AE e Ag/AgCl, como o RE.
- Para medições de spectroelectrochemical de UV-Vis-NIR, use um óxido da lata do índio quartzo (ITO) ou óxido de estanho dopado com flúor (FTO) como a nós, um fio de platina como o AE e um eletrodo de Ag/AgCl como o RE.
- Polir o eletrodo de disco de platina, esfregando a desvantagem do eletrodo (área de trabalho de eletrodo de platina) sobre uma almofada de lustro coberta com 1 µm chorume de alumina por 3 min.
- Lave o eletrodo com água deionizada para remover todos as lamas do elétrodo e limpa em um banho de ultra-som (320 W, 37 kHz) com água desionizada para 15 min.
- Enxague o eletrodo utilizando uma seringa de 1 mL com isopropanol (3 x 1 mL) e, em seguida, com acetona (3 x 1 mL).
- Use uma toalha de papel para remover todos os resíduos sólidos e secar ao ar por 3 min.
- Limpe os eléctrodos ITO e FTO com água desionizada. Coloque-os em um banho de ultra-som (320 W, 37 kHz) em acetona por 15 min e depois em isopropanol para o próximos 15 min.
- Queimar os eléctrodos de platina, fios e bobinas usando uma tocha de gás de alta temperatura (> 1000 ° C) por 1 min. cuidado! Use uma pinça para segurar os eléctrodos. Espere por 5 min após a queima e antes de usar os eletrodos.
- Limpe as células eletroquímicas e spectroelectrochemical (Figura 1) com água e depois com acetona. Use uma toalha de papel para remover todos os resíduos sólidos. Limpe todos os outros elementos (ca. peças PTFE) com acetona e secar ao ar antes da utilização.
2. análise de CV
- Liga o potentiostat e o computador.
- Preencha uma célula eletroquímica com 1,5 mL de solução eletrolítica para ser analisado.
- Colocar todos os três eléctrodos (WE, AE e RE) em uma célula (tampa da célula é equipada com o eléctrodo PTFE, então colocar eletrodos através dos furos neste suporte) e conectá-lo a um potentiostat. Manter o WE e RE como próximos uns dos outros quanto possível (Figura 1). Siga as informações sobre a conexão de cada fio do potentiostat para seus respectivos eletrodos.
- Se for necessário (por exemplo durante a análise de redução), coloque o tubo de argônio (ou azoto) (através de um orifício adicional no porta-eletrodo) e começar a borbulhar a solução pelo menos 5 min. Depois disso, mover o tubo de argônio (ou nitrogênio) acima do nível da solução e manter o fluxo de gás, correndo para a medição.
Nota: A célula parece fechada, mas não pode ser completamente estanques; Portanto, um método para remover a quantidade redundante de gás da célula deve ser disponível (ex., por um pequeno buraco adicional no porta-eletrodo). A pressão depende da tubulação usada; como a pressão alta conforme ele é necessário para definir o fluxo de gás lento. Se um solvente volátil é usado no experimento (por exemplo, diclorometano) ou o experimento vai levar muito tempo (mais de 30 min), use a garrafa de Drechsel para saturar o gás argônio (ou nitrogênio) com o solvente utilizado na medição. - Executar o software potentiostat, escolher o procedimento de CV e use as seguintes configurações: começar o potencial de 0.00 V, potencial de vértice superior de 2,0 V, menor potencial de vértice de 0.00 V (se a investigar o processo de redução e, em seguida, um potencial de vértice inferior de −2.5 V), parar o potencial de 0.00 V, número de cruzamento de paragem de 6 e digitalizar taxa de 0,05 V/s.
- Na seção dados exportação ASCII, definir um nome de arquivo e escolher uma pasta para salvar os dados em e pressione o botão Iniciar (diminuir ou aumentar o potencial de vértice superior e inferior para encaixar a janela eletroquímica ou a amostra atividade eletroquímica)
- Se qualquer picos são visíveis no intervalo de potencial positivo e, em seguida, repita o procedimento de limpeza. Se há um pico no intervalo negativo potencial, então coloque o tubo de argônio (ou nitrogênio) na solução e iniciar a subida por um adicional 5 min.
- Adicione uma gota do ferroceno 1 mM (no solvente usado para preparar o eletrólito) para a solução eletrolítica, usando uma seringa.
- Defina o potencial de iniciar a 0.00 V o vértice superior potencial de 0,85 V, o vértice de menor potencial de 0.00 V, o potencial de paragem de 0.00 V, o número de cruzamento de paragem para 10 e a taxa de varredura para 0.05 V/s. Na seção dados exportação ASCII, mudar o nome do arquivo e pressione o botão Iniciar .
- Após a medição, termine, repetindo os procedimentos de limpeza conforme mencionado nas etapas 1.2 e 1.3.
- Prepare-se 4 mL de 1 mM teste composto em preparado eletrólito (etapa 1.1).
- Preencher a célula eletroquímica com a solução de teste composto (1,5 mL); colocar todos os três eletrodos em uma cela e ligue para o potentiostat (passo 2.4). Para investigar o processo de redução, remova o oxigênio (etapa 2.5).
- Definir o início potencial para 0.00 V, o vértice superior potencial de 0,50 V (ou 0.00 V em caso de investigação do processo de redução), o vértice de menor potencial de 0.00 V (−0.50 V em caso de investigação do processo de redução), o potencial de paragem de 0.00 V , o número de cruzamento de paragem para 10 e a taxa de varredura para 0.05 V. Na seção dados exportação ASCII, mudar o nome do arquivo e pressione o botão Iniciar .
- Repita a etapa 2.13, aumentando o potencial do vértice superior (ou diminuindo o potencial de vértice inferior) por 0.1 V até registro de pico (Figura 2a). Se o scan sucessivo é deslocado em potencial (Figura 2b), então limpe o RE e deixá-lo na solução eletrolítica. Em seguida, repita a medição.
- Medir os processos de oxidação e a redução no mesmo CV para a determinação do IP e EA: definir o potencial de iniciar a 0.00 V, o potencial de vértice superior a 1.00 V, o potencial de vértice inferior de −2.70 V e o potencial de paragem para 0.00 V. escolher o vert superior e inferior ex de potencial de uma forma que registra redução completa e oxidação picos e evita novas medidas de redox (se alguma existe) (Figura 2a).
Nota: Estimar IP e EA medindo o potencial de aparecimento. Existem algumas maneiras de calcular estes parâmetros de CV, mas é o mais útil para calcular usando o potencial de aparecimento. Às vezes, não é possível observar o par redox reversível, especialmente para n e p de doping. Mistura de duas técnicas diferentes também é indesejável, como pode fornecer conclusões e resultados errados.- Para medir o potencial de aparecimento, marca a linha tangente ao pico de CV (Figura 3). Calcular os valores desejados do potencial de aparecimento do redução ou oxidação processo usando equações (equações 1 e 2)10,18,19,36,37 , 38.
 (1)
(1)
onde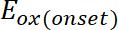 é o aparecimento de oxidação potencial calibrado com ferroceno*
é o aparecimento de oxidação potencial calibrado com ferroceno*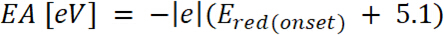 (2)
(2)
onde é o início da redução potencial calibrado com ferroceno*
é o início da redução potencial calibrado com ferroceno*
* Potencial calibrado com ferroceno significa que o valor do potencial da medida é diminuído o potencial de oxidação do ferroceno.
- Para medir o potencial de aparecimento, marca a linha tangente ao pico de CV (Figura 3). Calcular os valores desejados do potencial de aparecimento do redução ou oxidação processo usando equações (equações 1 e 2)10,18,19,36,37 , 38.
- Retirar os eletrodos e deite fora a solução de teste dentro do recipiente de lixo adequado.
- Repita o procedimento de limpeza (etapas 1.2 e 1.3).
- Repita as etapas de 2.9 – 2.12 com 0,25, 0,10 e 0,20 V/s taxas de varredura.
- Colocar uma gota do ferroceno 1 mM (no solvente usado para preparar a solução eletrolítica) para a solução de teste usando uma seringa. Definir o potencial de iniciar a 0.00 V o vértice superior potencial de 0,60 V, o potencial de vértice inferior de 0,00, o potencial de paragem de 0.00 V, o número de cruzamento de paragem para 6 e a taxa de varredura de 0,05 V. Push botão Iniciar . Depois de concluída a medição, altere o vértice superior potencial para um valor que registra o casal do ferroceno com o primeiro pico de oxidação de compostos no teste.
- Retirar os eletrodos e deite fora a solução de teste dentro do recipiente de lixo adequado.
- Se o electropolymerization ocorre durante a oxidação eletroquímica, lave o WE cuidadosamente com a solução eletrolítica, utilizando a seringa (3 ´ 0,5 mL). Limpe o RE, AE e célula eletroquímica usando o procedimento padrão (etapas 1.2-1.3).
- Para investigar produtos eletroquímicos depositados em nós, vá para a etapa 2.23. Para investigar uma solução, prossiga para o passo 1.2.1 e começar de novo da etapa 2.3.
- Preencha a célula eletroquímica com uma solução eletrolítica. Em seguida, colocar todos os três eletrodos em uma cela e conectá-los a um potentiostat (passo 2.4). Para investigar o processo de redução, deoxidate a solução (etapa 2.5).
- Repita as etapas de 2.10 – 2.12 e depois passo 2.16.
 (1)
(1)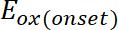 é o aparecimento de oxidação potencial calibrado com ferroceno*
é o aparecimento de oxidação potencial calibrado com ferroceno*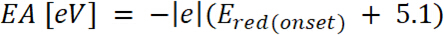 (2)
(2) é o início da redução potencial calibrado com ferroceno*
é o início da redução potencial calibrado com ferroceno*3. UV-Vis-NIR Spectroelectrochemical análise
- Preparação de medição
- Ligue o espectrômetro e potentiostat.
- Preencha a célula spectroelectrochemical com 0,5 mL de solução eletrolítica (etapa 1.1).
- Colocar a vedação PTFE, de um lado da célula; Então coloque o eletrodo de ITO à célula desta forma ter um lado condutor do eléctrodo tocando o selo PTFE. Colocar o resto das peças PTFE como mostrado na Figura 1. Coloque o RE e AE através do eléctrodo PTFE e cobrir a parte superior do eletrodo ITO com folha de cobre, como mostrado na Figura 1 para melhor condutividade.
- Colocar a célula montada no suporte do aparelho e conectar todos os eletrodos de um potentiostat. Siga as informações sobre a conexão de cada fio do potentiostat para seus respectivos eletrodos.
- Execute o software potentiostat e o espectrômetro.
- No software do espectrômetro, escolher arquivo | Novo | Absorvância medição.
- Escolha um dos detetores listados.
- Certifique-se que o botão dos detectores de está na posição "aberta". Em seguida, clique na lâmpada de incandescência; Feche o detector (Mova o botão para a posição fechada) e em seguida, clique na lâmpada luz negra.
- Selecione o segundo detector e repita a etapa 3.1.7.
- Reconecte-se eletrodos da potentiostat e retirar os eletrodos e outros elementos da célula spectroelectrochemical.
- Repita os procedimentos de limpeza (etapas 1.2 e 1.3).
- Preencher a célula de spectroelectrochemical com um diluído (1´105 M) solução do teste composto na solução eletrolítica se testando a pequena molécula. Preencha a célula de spectroelectrochemical com o eletrólito se testando camada polimérica (oligoméricas) depositada sobre o eléctrodo de ITO.
- Monte a célula spectroelectrochemical (passo 3.1.3).
- Clique em salvar.
- Selecione um dos detectores de listados.
- Definir a salvar localização e nome do arquivo (incluem o detector selecionado).
- Selecione o segundo detector e repita o passo 3.1.15.
- Análise de Potentiostatic18.
- Aplica um potencial neutro (ou seja, 0,00 V). Este espectro será espectros de partida. Aumentar o potencial de 0,1 V e esperar ca. 10 s até que o processo se estabiliza e registra os espectros de absorção (passos 3.1.13–3.1.16). Continuar a medição para a primeira mudança nos espectros de UV-Vis-NIR; aguarde 10 s e salve os espectros (passos 3.1.13–3.1.16).
- Potenciais de aumento por 0.05 V; esperar 10 s e salvar (passos 3.1.13–3.1.16).
- Repita 3.2.2 até atingir o potencial do potencial de oxidação de primeira ou segunda (estas potencialidades são conhecidas desde a medição de CV); em seguida, reverter o potencial e voltar para o potencial de partida.
- Quando as medições são concluídas, medir o CV da solução para análise. Adicionar ferroceno 1 mM (10 μL) e medir o CV mais uma vez. O valor do ferroceno ajudarão a estimar o potencial do processo e unificar os dados.
- Esta análise fornece informações sobre a banda óptica-lacuna, comprimento de onda máximo de Estados undoped e dopados e pontos de isosbestic do processo eletroquímico. Calcule as banda ópticas-lacunas de valores início das bandas de π-π* sobre os espectros de UV-Vis e utilizando a equação 318,19,23,24.
 (3)
(3)
onde h é a constante de Planck, λinício é o aparecimento de compostos de absorção e c é a velocidade da luz no vácuo. - Análise de Potentistatic de tempo-resolvido19,32,33
- Preparar a medição semelhante ao método descrito em 3.1.1–3.1.16 e coloque o tubo de argônio (ou azoto) (através de um orifício adicional no porta-eletrodo) e borbulhando a solução pelo menos 5 min antes da medição. Depois disso, mova o tubo de argônio acima do nível da solução e manter o fluxo de gás, correndo para a medição.
- Executar o software potentiostat, escolher o procedimento de chronoamperometry com as seguintes configurações: potencial iniciar potencial de 0.00 V potencial superior a 1.0 V, menor potencial para −1.00 V, duração como 5 s, o intervalo de tempo como 0.010 s, número de repetições de 10 e o atraso tempo de 10 s. Na seção dados exportação ASCII, definir um nome de arquivo e escolha uma pasta para salvar os dados.
- No software do espectrômetro, escolher arquivo | Novo | Aquisição de alta velocidade. Uma nova janela irá aparecer; configurar os seguintes parâmetros: tempo de integração — 10 ms, Scans de média — 1, Boxcar Wirth — 0, número de varreduras — 10000. Não clique no botão ir .
- No software potentiostat, aperte o botão Iniciar . Quando a contagem cai para 1 s, pressione o botão ir no software do espectrômetro.
- Note que não é possível ver os resultados de absorção, até que o processo seja concluído.
- Analise os dados para fornecer parâmetros. Use as informações salvas no arquivo tal transmissão, corrente, tensão e densidade de corrente para fornecer parâmetros de trabalho:
- Calcule a densidade óptica (ΔOD), a absorbância do teste composto durante a dopagem (dedoping) com a equação 4:
 (4)
(4)
onde Tb transmitância forma dopada, um estado de "bleach" [%] e Tc está no transmitância em forma undoped, um estado "de cor" [%]. - Calcular a eficiência de coloração (CE, η), a quantidade de eletrocrômico cor formada pela quantidade utilizada de forma gratuita e como a conexão entre a carga injetado/ejetado em função da área do eletrodo (Qd) e a variação da densidade óptica (ΔOD) valor em um comprimento de onda específico (λmáx) com a equação 5. Seu valor depende do comprimento de onda escolhido para estudo, então o valor é medido nos λmáx da banda de absorção óptica do estado colorido.
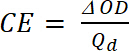 (5)
(5)
onde ΔOD é a densidade óptica [-] e Qd é a densidade de carga [C/cm2]. - Calcular o rácio de contraste (CR), a intensidade medida da mudança de cor durante a dopagem eletroquímica e geralmente caracterizada por λmáx do formulário colorido com equação 6:
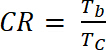 (6)
(6)
onde Tb transmitância forma dopada, "bleach" estado [%] e Tc está no transmitância em forma undoped, estado "de cor" [%].
- Calcule a densidade óptica (ΔOD), a absorbância do teste composto durante a dopagem (dedoping) com a equação 4:
 (3)
(3) (4)
(4)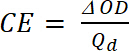 (5)
(5)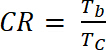 (6)
(6)4. EPR Spectroelectrochemical análise
- Preparação de medição
- Ligue o espectrômetro e o potentiostat.
- Preencha a célula spectroelectrochemical com electrólito (etapa 1.1).
- Regule a potência de 1 mW e tune o ressonador para conseguir um sinal forte, centrado na Q-mergulho (use a opção autotuned).
- Definir o marcador de Mn (manganês padrão) para 600 e fechar a janela de Q-mergulho. Se não há nenhum marcador de Mn, Então pule esta etapa e vá para a etapa 4.8.
- Defina "varrer largura ±" para 2.5´ 10 mT, "mod largura ±" 1.0´0.1 mT, "amplitude" para 5´100, "constante de tempo" para 0,03 s e "centro do campo" valor de ca. 338 mT e iniciar a medição.
- Se não registrar seis linhas espectrais de marcador de Mn, altere o valor de "centro do campo".
- Ao registrar seis linhas espectrais de marcador de Mn (Figura 4), defina o valor de "centro de campo" entre a terceira e quarta linha e diminuir o valor de "varredura largura ±" para um valor que cobre apenas essas duas linhas. Inicie a medição novamente.
- Vá para a opção de Q-Dip, como o marcador de Mn 0 e fechar a janela de Q-mergulho.
- Medir o eletrólito puro como plano de fundo e verificar se há algum sinal. Se os sinais são visíveis, então o sinal pode ser da contaminação, compostos de potencial de oxidação baixa, ou de vidro.
- Limpe a célula spectroelectrochemical (etapa 1.3).
- Investigação de pequenas moléculas (soluções).
- Preencher a célula de spectroelectrochemical com um diluído (1´105 M) solução do teste composto no eletrólito; colocar todos os três eletrodos através do eléctrodo à célula dessa maneira para que os nós e RE estão dentro da espiral AE (Figura 1).
- Coloque o WE perto do fundo da célula e o RE no lado superior do ativo (metal não abrangido pelo PTFE) parte a nós (Figura 1). Coloque o celular equipado com um eletrodo dentro na cavidade do espectrómetro de amostra e conectar os eletrodos com um potentiostat (siga as informações sobre a conexão de cada fio do potentiostat para seus respectivos eletrodos).
- Aplica o potencial correspondente ao aparecimento do primeiro pico redox (valor conhecido de medição CV). Se aparecer um sinal, então o equipamento está configurado corretamente.
- Vá para a opção de Q-Dip, defina o marcador de Mn para 600, feche a janela de Q-mergulho e iniciar a medição. O registo do sinal com o sinal de marcador de Mn permite o cálculo do g-factor do sinal recebido.
- Vá para a opção de Q-Dip, como o marcador de Mn 0, feche a janela de Q-mergulho e esperar ca. 5 min.
- Definir o valor do "centro de campo" no centro do sinal, diminua o valor de "varredura largura ±" para um valor que cobre o sinal, mas não corta o início e o fim do sinal (quando "varrer largura ±" é alterada, todo sinal de seleção) e diminuir o valor de "m uma overdose de largura ± "para obter espectros bem resolvidos.
- Se o sinal for pequeno, então aumente o valor de "amplitude" e "tempo" (aquisição) da medição.
- Para diminuir o sinal à relação de ruído, defina a aquisição de 4, 9 ou 16 dependendo barulhento como o sinal é. Se o sinal é muito barulhento, selecione 16.
- Preencher a célula de spectroelectrochemical com um diluído (1´105 M) solução do teste composto no eletrólito; colocar todos os três eletrodos através do eléctrodo à célula dessa maneira para que os nós e RE estão dentro da espiral AE (Figura 1).
- Investigações da camada polimérica depositado na superfície da nós.
- Preencha a célula de spectroelectrochemical com uma solução eletrolítica (etapa 1.1).
- Colocar eletrodos dentro da célula. Tenha cuidado para não destruir a camada polimérica sobre a nós. Conecte o eletrodo com o potentiostat (passo 4.2.1).
- Aplicar o potencial neutro (ou seja, 0,00 V) para garantir que o composto é um estado neutro; Este espectro será os espectros EPR a partida.
- Aumentar o potencial por 0,10 V. Wait ca. 10 s até que o processo se estabiliza e gravar os espectros EPR.
- Repita o passo 4.3.4 quando aparece o sinal EPR.
- Aumentar o potencial por 0.05 V. Wait ca. 10 s até que o processo se estabiliza e gravar os espectros EPR.
- Repita a etapa 4.3.6 até o primeiro ou segundo potencial de oxidação é alcançado e assim por diante (valores desses potenciais são conhecidos de medição CV); em seguida, reverter o potencial e voltar a começar potenciais.
- Aplica o potencial em que o EPR sinal apareceu no ciclo anterior (4.3.5), vá para a opção de Q-Dip, defina o marcador de Mn para 600, feche a janela de Q-mergulho e iniciar a medição. Registre o sinal com a terceira e quarta linha espectral de marcador de Mn (4.1.7).
- Quando a medição estiver concluída, analise os dados.
- Algumas das análises tais como um número de rotações pode ser realizado apenas para os compostos depositados (polímeros insolúveis) sobre os nós. Para calcular o número de rotações, double-integre o sinal EPR, seguido por uma comparação do valor obtido com calibrado padrão interno de Mn. Calcular o número de repetir unidades formando o polímero sobre o eletrodo usando equação 724,32,33:
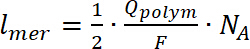 (7)
(7)
onde lmer – número de repetir unidades em polímero, Qpolym é a acusação de polimerização, F é a constante de Faraday e NA é o número de Avogadro. - Para calcular a acusação de doping, use o CV de polímero depositado, gravado em uma célula de spectroelectrochemical antes da medição do EPR, usando a equação 8:
 (8)
(8)
onde Qd é a acusação de doping, i é a corrente, E é o conjunto de potencial, E0 é o potencial inicial, v é a velocidade de varredura e e é a carga elementar. - Use a acusação de doping calculada para calcular o nível de dopagem, usando a equação 9:
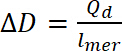 (9)
(9)
onde ∆D- nível, Qd de doping é a acusação de doping ou lmer – número de repetir unidades em polímero - Para calcular o número de bipolarons, assumir um doping inicial nível igual a zero. Use o número de polarons por unidade de concentração de rodadas, então o número de bipolarons deve ser calculado usando equação 10. Os cálculos, suponha que a quantidade inicial de bipolarons é zero e corrente faradaic é muito maior do que a corrente não-faradaic.
 (10)
(10)
onde z, é a carga elementar do transportador de carga, nié o número de portadores de cargas por unidade de polímero, (p) é a polarons e (b) é o bipolarons. - Extraia o g-fator de valor a partir dos espectros EPR. Determinar usando Mn como um padrão interno (etapas 4.2.3 e 4.3.8), sabendo que a quarta linha de manganês nele tem um g-factor de 2.03324. Calcule a largura da linha de sinal EPR como a distância em mT entre mínimo e máximo do sinal do EPR. Este valor é importante porque pode dizer onde (no qual átomo) são formados os radicais.
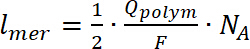 (7)
(7) (8)
(8)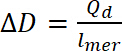 (9)
(9) (10)
(10)Representative Results
A aplicação mais comum de análise de CV é a estimativa de IP e EA. Embora existam algumas maneiras de obter dados de CV, é altamente recomendável para calculá-las com base no início dos picos de redox (Figura 3). Esta abordagem permite unificar o procedimento de cálculo. Nem todos os materiais testados passam por processos de oxidação/redução reversível (Figura 3); em tais situações, não é possível calcular com base em média potenciais (média de potenciais correspondem a um máximo de picos catódica de redução e oxidação anódica). No entanto, quase sempre é possível executar a tangente de um pico, como mostrado na Figura 3. Com a intersecção com a linha de fundo e a utilização das equações 1 e 2, os valores IP e EA são estimados como 5,35 eV e −2.90 eV, respectivamente. Existem também várias escalas diferentes, utilizadas para avaliar o IP e EA com base em medições de CV. O mais comumente usado escalas para materiais orgânicos são −4.8 e −5.1 eV como equivalente a 0.00 V contra o eletrodo de hidrogênio Normal (NHE). No entanto, todas as escalas são apenas uma aproximação; Lembre-se isso ao comparar resultados diferentes. O ponto crucial é a declarar parâmetros que foram considerados para os cálculos. Neste caso, o valor −5.1 eV foi escolhido como ele deve corresponder ao potencial formal do casal redox ferroceno; na escala de Fermi, é 0,40 V contra eletrodo de calomelano saturado (SCE), em acetonitrila, que está de acordo com a medição anterior.
Há muitos artigos publicados sobre a análise de CV. aqui, nós mostramos quando o processo não vai ser como é esperado. A análise baseia-se derivado de tiofeno: NtVTh (estrutura mostrada na Figura 4), que sofre degradação após oxidação (Figura 5).
NtVTh tem dois potenciais de oxidação: o primeiro em 0,70 V e a segunda em 0,84 V (Figura 5). Durante o primeiro ciclo, o pico de redução não é observado, indicando um processo irreversível. Características eletroquímicas de NtVTh mostram que a polimerização não ocorrerá e após a primeira potencial de oxidação, depósitos de alguma camada electro-inativos dos produtos de reação na superfície do elétrodo, dificultando assim o processo de polimerização. O que é visível é a reação com o cátion radical sobre o vínculo de vinil, onde a molécula está perdendo suas conjugação e forma dímeros sobre o eletrodo.
Enquanto é difícil extrair informações sobre as transportadoras de carga do CV, é possível distinguir entre polarons e bipolarons, quando apoiado por um espectrômetro UV-Vis-NIR. O poli neutro (OiPrThEE) foi caracterizado por dois grande absorção π | π * transições de bandas com maxima de picos em λmax1 = 363 nm e λmax2 = 488 nm, relacionado ao formulário do derivado polythiophene undoped aromático. Durante o doping oxidativa, são geradas novas bandas polaronic e bipolaronic. O espectro de UV-Vis, obtido durante a oxidação de poli (OiPrThEE) revelou que a diminuição da banda de absorção do polímero neutro (300-550 nm) (Figura 6) juntamente com a formação de uma nova banda de absorção (550-950 nm) dos cátions radicais de bithiophene e p- phenylenevinylene com maxima em 692 nm. O ponto de isosbestic do processo de oxidação foi localizado a 604 nm. A banda de bipolaronic apareceu entre a 950 nm e 1700 nm com um máximo localizado em 1438 nm.
Espectroscopia EPR é a técnica que detecta materiais com um elétron não pareado, isto inclui os radicais orgânicos39. Existem vários parâmetros que podem ser extraídos de espectros EPR, mas um dos mais interessantes é estimar onde os radicais são localizados. Elétrons, prótons, semelhantes possuem rotação. Colocando um elétron em um campo magnético externo, esta rotação pode ser dividida duas maneiras: paralelas e antiparalelas ao campo magnético, dando dois níveis de energia. Este fenómeno é conhecido como o efeito Zeeman40,41. No caso de radicais orgânicos, o elétron não pareado interage não só com o campo magnético externo, mas também com núcleos magnéticos (núcleos que tem uma rotação diferente de zero; I≠0). Um número de níveis de energia degenerados é igual a 2I + 1, onde I é o número quântico de spin do núcleo com o qual o elétron não pareado interage42. A interação de um elétron não pareado com um maior número de núcleos magnéticos conduz a dividir ainda mais os níveis de energia e a estrutura hiperfina das de registo de espectros EPR43 (Figura 7).
Para moléculas onde o elétron não pareado interage com um número ainda maior de núcleos, a linha espectral individual poderia sobrepor-se, que resulta em registro de um único, amplo sinal44,45,46 (Figura 8). isso é típico de polímeros conjugados, onde o íon radical gerado durante o processo de redox é deslocalizados47,,48.
A combinação de espectroscopia EPR com métodos eletroquímicos permite a caracterização de portadores de cargas (íon radical) gerados durante o processo de redox, bem como a determinação do mecanismo desses processos49,50 . Se bem resolvido (picos são separados; não sob a forma de um pico de amplo) espectros são registrados, como no caso de redução eletroquímica de s -derivado de tetrazine (Figura 7) e, em seguida, a análise da estrutura hiperfina dos espectros leva a conclusões sobre a localização do elétron não pareado. Uma maneira de analisar esse tipo de espectros é para realizar a simulação com software especial e caber espectros simulados com o experimental uma51. Isso é especialmente útil quando a estrutura hiperfina é complexa devido a interação de um elétron não pareado com um grande número de prótons. No caso do s- tetrazine derivado mostrado na Figura 7, simulação de espectros EPR (linha vermelha) indica a interação de um elétron não pareado com quatro átomos de nitrogênio de s- tetrazine.

Figura 1: eletroquímica e células de spectroelectrochemical utilizados para medições. A figura apresenta a configuração do esquema de células eletroquímicas/spectroelectrochemical utilizando voltametria cíclica, ultravioleta-visível e infravermelho-próximo (UV-Vis-NIR) e elétron medições de spectroelectrochemical de ressonância paramagnética (EPR). Clique aqui para ver uma versão maior desta figura.

Figura 2: referência a voltametria cíclica (CV) de COPO1 composto corretamente medido (a) e o CV com um instável potenciais dibenzothiophene-S, S-dióxido com ferroceno (b)52. A figura mostra dois voltammograms cíclicas. (a) apresenta corretamente registrado CV e (b) mostra um voltammogram registrado usando um eletrodo de referência com potencial não estável. Clique aqui para ver uma versão maior desta figura.

Figura 3: voltametria cíclica (CV) do composto COPO1 em uma ampla gama de potenciais. Estimativa dos potenciais início para calcular EA e IP, a COPO1 compostos52. EA = −2.90 eV e IP = 5,35 eV. Clique aqui para ver uma versão maior desta figura.

Figura 4. EPR seis linha espectral de manganês padrão. O sinal paramagnético de manganês usado para calibração da mudança de sinal. Clique aqui para ver uma versão maior desta figura.

Figura 5: voltametria cíclica (CV) do composto NtVTh. Voltammograms cíclico de 15mm NtVTh em 0,1 M Bu4NBF4/CH3CN e em relação ao padrão de ferroceno, apresentando o processo de degradação envolvido no eletrodo de trabalho. Taxa de varredura 0.05 V/s: (um) foi tirada no intervalo −1.4 V para 0,8 V e (b) no intervalo −1.4 V a 0,9 V. , por favor clique aqui para ver uma versão maior desta figura.

Figura 6: ultravioleta, visível e infravermelho próximo (UV-Vis-NIR) spectroelectrochemistry de derivado de poli (OiPrThEE). Espectros de UV-Vis-NIR apresentando a evolução da banda de absorção através da geração de portadores de cargas na estrutura do polímero. Clique aqui para ver uma versão maior desta figura.

Figura 7: análise de spectroelectrochemical EPR de derivado de tetrazine. (a) estrutura de s- tetrazine derivado; (b) espectros EPR registrado durante a redução eletroquímica de s- tetrazine derivado (espectro simulado de linha preta de linha-experimental e vermelho). Clique aqui para ver uma versão maior desta figura.

Figura 8: sinal de polaron EPR spectroelectrochemical do polímero conjugado. Espectros de ressonância paramagnética (EPR) elétron registrado durante a primeira etapa de oxidação de polímeros conjugados (espectros EPR de polaron espécies). Clique aqui para ver uma versão maior desta figura.

Figura 9: voltametria cíclica (CV) do ferroceno e decamethylferrocene. Comparação dos dois padrões de eletroquímicas como puras e como mistura mostrando a mudança do potencial. Clique aqui para ver uma versão maior desta figura.
Discussion
Técnicas eletroquímicas e spectroelectrochemical não têm limites; um pode analisar tanto o estado sólido e líquidas soluções em uma ampla gama de temperatura e outras condições com estas técnicas. O importante em todos estes casos é que os compostos/materiais são analisados sob o potencial aplicado, replicando as condições do mundo real para trabalhar com dispositivos de eletrônica orgânica. A única diferença é que em eletroquímica, a formação de portadores de cargas, é observado.
Os métodos aqui apresentados mostram a utilidade da análise das transportadoras carregadas geradas em compostos orgânicos que se correlacionam com a sua aplicabilidade em eletrônica orgânica. Além disso, as técnicas eletroquímicas e spectroelectrochemical são mais baratos e menos exigentes do que a do típicos métodos utilizados na análise de transportadora de carga, mas existem alguns passos críticos e modificações no protocolo que são necessários dependendo do resultados obtidos.
Durante a caracterização eletroquímica, sempre começam com uma determinada concentração. Se um conjunto de compostos é comparado, então todos os materiais precisam ter a mesma concentração molar. O melhor é começar com concentração de 1 mM e 50 mV/s taxa de varredura conforme indicado no protocolo neste estudo, mas é bom saber que a concentração da amostra sobre o comportamento eletroquímico observado. Sempre tente medir pelo menos três exames. Os dois primeiros exames são geralmente diferentes, porque as condições de partida (equilíbrio) são diferentes. O segundo e os terceiros exames devem ser o mesmo. Se os exames de segundo e terceiros são as mesmas, então há provavelmente sem reações colaterais observadas neste sistema (Figura 2a). Em um processo de oxidação, um novo pico em um potencial inferior aparece mostrando que o material condutor foi depositado sobre o WE18,19,24,25,29,30 , 31 , 32. se a altura do pico mais baixo aumenta em varreduras sucessivas, então provavelmente o polímero conjugado foi depositado18,19,24,25,29 , 30 , 31 , 32. se todas as correntes diminuem em varreduras sucessivas e, em seguida, o produto não condutora de degradação foi depositado sobre o eletrodo. Se um pico muito pequeno é observado antes da oxidação principal ou o pico de redução (especialmente para polímeros), então este é provavelmente retêm carga processo19,23,31,34. Se for observado um pico de dedoping muito afiado de oxidação ou redução, então isso provavelmente é causado pela decomposição de estruturas cristalinas em um eletrodo formado através do processo de electrocrystallization durante a oxidação35.
Verifique sempre o comportamento do teste composto antes, durante e depois de picos de redox. Significa que pelo menos três exames de CV devem ser registradas: com superior (no caso oxidação) ou menor potencial de vértice inferior ou superior, respectivamente, então o potencial máximo do pico, com potencial de vértice inferior ou superior definido como exatamente no pico máximo e com vértice potenciais mais elevados (oxidação) e inferior (redução) do que o potencial máximo do pico. O processo observado pode variar e por vezes dois processos podem ser observados sob um pico teoricamente. Sempre comparar o voltammograms cíclico coletados do eletrólito (passo 2.6), o ferroceno (passo 2.9), o composto (etapa 2.13) e do ferroceno com composto (etapa 2.19); Há várias questões a ter em conta.
Sempre comparar os sinais de CV do eletrólito e o ensaio composto, se quaisquer sinais de eletrólito é visível sobre o voltammogram cíclico do composto medido, então o eletrólito deve ser mudado porque sua janela eletroquímica é muito baixa, ou o eletrólito é contaminado. Se o sinal (par redox) de ferroceno (passo 2.9) e ferroceno com composto (etapa 2.19) estão na mesma posição, então tudo é executado corretamente. Se os picos são deslocados entre si, então verifique a RE e repita a medição. Se o sinal (oxidação, redução ou par redox potencial) do teste composto de ferroceno adicionado (etapa 2.19) está em um potencial mais elevado do que o puro composto (etapa 2.13), em seguida, considere os valores (oxidação, redução ou redox casal potenciais) de o voltammogram cíclico do composto puro. A mudança é causada pela maior quantidade de ferroceno na solução. Quando são observados dois processos de oxidação, o primeiro processo (oxidação ou redução), que está sempre ligado a nós pode afetar a superfície ativa; Isto pode causar um aumento no potencial de oxidação do segundo processo (Figura 9).
Disclosures
Os autores não têm nada para divulgar.
Acknowledgments
Os autores reconhecem com gratidão o apoio financeiro do projeto "Excilight", "Doador-aceitador Light Emitting Exciplexes como materiais para fácil-para-alfaiate ultra-eficiente OLED relâmpago" (H2020-ACEM-ITN-2015/674990) financiado pela Marie Skłodowska-Curie Ações no âmbito do programa quadro de investigação e de inovações "Horizonte 2020".
Materials
| Name | Company | Catalog Number | Comments |
| Potentiostat | Metrohm | Autolab PGSTAT100 | |
| EPR | JEOL | JES-FA200 | |
| UV-Vis detector | Oceanoptics | QE6500 | |
| NIR detector | Oceanoptics | NIRQuest | |
| Dichloromethane (DCM) | Sigma-Aldrich | 106048 | |
| Tetrabutylammonium tetrafluoroborate (Bu4NBF4) | Sigma-Aldrich | 86896 | |
| 2-propanol, 99.9% | Sigma-Aldrich | 675431 | |
| Acetone, 99.9% | Sigma-Aldrich | 439126 | |
| Ultrasonic Bath | Elma | S30H | |
| Tetrahydrofuran >99.9% | Sigma-Aldrich | 401757 | |
| ferrocene >98% | Sigma-Aldrich | F408 | |
| decamethylferrocene >97% | Sigma-Aldrich | 378542 |
References
- O'Brien, D. F., Baldo, M. A., Thompson, M. E., Forrest, S. R. Improved energy transfer in electrophosphorescent devices. Applied Physics Letters. 74, 442-444 (1999).
- Chin, B. D., et al. Effects of interlayers on phosphorescent blue organic light-emitting diodes. Applied Physics Letters. 86, 133505-133507 (2005).
- Cardon, C. M., Li, W., Kaifer, A. E., Stockdale, D., Bazan, G. C. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bang, A. -M., et al. A novel random terpolymer for high-efficiency bulk-heterojunction polymer solar cells. RSC Advances. 7, 1975-1980 (2017).
- Tybrandt, K., Kollipara, S. B., Berggren, M. Organic electrochemical transistors for signal amplification in fast scan cyclic voltammetry. Sensors and Actuators B: Chemical. 195, 651-656 (2014).
- Schaur, S., Stadler, P., Meana-Esteban, B., Neugebauer, H., Sariciftci, N. S. Electrochemical doping for lowering contact barriers in organic field effect transistors. Organic Electronics. 13, 1296-1301 (2012).
- Mullen, K., Scherf, U. Organic Light Emitting Devices: Synthesis, Properties and Applications. , Wiley-VCH. Weinheim. (2006).
- Monk, P. M. S., Mortimer, R. J., Rosseinsky, D. R. Electrochromism and Electrochromic Devices. , Cambridge University Press. Cambridge. (2007).
- Skotheim, T. A., Elsenbaumer, R. L., Reynolds, J. R. Handbook of Conducting Polymers. , Marcel Dekker. New York. (1998).
- Data, P., et al. Kesterite Inorganic-Organic Heterojunction for Solution Processable Solar Cells. Electrochimica Acta. 201, 78-85 (2016).
- Data, P., Pander, P., Okazaki, M., Takeda, Y., Minakata, S., Monkman, A. P. Dibenzo[a,j]phenazine-Cored Donor-Acceptor-Donor Compounds as Green-to-Red/NIR Thermally Activated Delayed Fluorescence Organic Light Emitters. Angewandte Chemie International Edition. 55 (19), 5739-5744 (2016).
- Jankus, V., et al. Highly Efficient TADF OLEDs: How the Emitter-Host Interaction Controls Both the Excited State Species and Electrical Properties of the Devices to Achieve Near 100% Triplet Harvesting and High Efficiency. Advanced Functional Materials. 24 (39), 6178-6186 (2014).
- Data, P., et al. Exciplex Enhancement as a Tool to Increase OLED Device Efficiency. Journal of Physical Chemistry C. 120 (4), 2070-2078 (2016).
- Dias, F. B., et al. The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Advanced Science. , (2016).
- Okazaki, M., et al. Thermally Activated Delayed Fluorescent Phenothiazine-Dibenzo[a,j]phenazine-Phenothiazine Triads Exhibiting Tricolor-Changing Mechanochromic Luminescence. Chemical Science. 8, 2677-2686 (2017).
- Data, P., et al. Efficient p-phenylene based OLEDs with mixed interfacial exciplex emission. Electrochimica Acta. 182, 524-528 (2015).
- Goushi, K., Yoshida, K., Sato, K., Adachi, C. Organic light-emitting diodes employing efficient reverse intersystem crossing for triplet-to-singlet state conversion. Nature Photonics. 6, 253-258 (2012).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of heteroaryl group on electrochemical and spectroscopic properties of conjugated polymers. Electrochimica Acta. 83, 271-282 (2012).
- Data, P., Motyka, M., Lapkowski, M., Suwinski, J., Monkman, A. Spectroelectrochemical Analysis of Charge Carries as a Way of Improving Poly(p-phenylene) Based Electrochromic Windows. Journal of Physical Chemistry C. 119, 20188-20200 (2015).
- Enengl, C., et al. Explaining the Cyclic Voltammetry of a Poly(1,4-phenylene-ethynylene)-alt-poly(1,4-phenylene-vinylene) Copolymer upon Oxidation by using Spectroscopic Techniques. ChemPhysChem. 18, 93-100 (2017).
- Gudeika, D., et al. Hydrazone containing electron-accepting and electron-donaiting moieties. Dyes Pigments. 91, 13-19 (2011).
- Swist, A., Cabaj, J., Soloducho, J., Data, P., Lapkowski, M. Novel acridone-based branched blocks as highly fluorescent materials. Synthetic Metals. 180, 1-8 (2013).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Influence of alkyl chain on electrochemical and spectroscopic properties of polyselenophenes. Electrochimica Acta. 87, 438-449 (2013).
- Laba, K., et al. Electrochemically induced synthesis of poly(2,6-carbazole). Macromolecular Rapid Communications. 36, 1749-1755 (2015).
- Pluczyk, S., et al. Unusual Electrochemical Properties of the Electropolymerized Thin Layer Based on a s-Tetrazine-Triphenylamine Monomer. Journal of Physical Chemistry C. 120, 4382-4391 (2016).
- Blacha-Grzechnik, A., Turczyn, R., Burek, M., Zak, J. In situ Raman spectroscopic studies on potential-induced structural changes in polyaniline thin films synthesized via surface-initiated electropolymerization on covalently modified gold surface. Vibrational Spectroscopy. 71, 30-36 (2014).
- Laba, K., et al. Diquinoline derivatives as materials for potential optoelectronic applications. Journal of Physical Chemistry C. 119, 13129-13137 (2015).
- Enengl, S., et al. Spectroscopic characterization of charge carriers of the organic semiconductor quinacridone compared with pentacene during redox reactions. Journal of Materials Chemistry C. 4, 10265-10278 (2016).
- Data, P., et al. Electrochemically Induced Synthesis of Triphenylamine-based Polyhydrazones. Electrochimica Acta. 230, 10-21 (2017).
- Brzeczek, A., et al. Synthesis and properties of 1,3,5-tricarbazolylbenzenes with star-shaped architecture. Dyes Pigments. 113, 640-648 (2015).
- Data, P., Lapkowski, M., Motyka, M., Suwinski, J. Electrochemistry and spectroelectrochemistry of a novel selenophene-based monomer. Electrochimica Acta. 59, 567-572 (2012).
- Data, P., et al. Electrochemical and spectroelectrochemical comparison of alternated monomers and their copolymers based on carbazole and thiophene derivatives. Electrochimica Acta. 122, 118-129 (2014).
- Data, P., et al. Evidence for Solid State Electrochemical Degradation Within a Small Molecule OLED. Electrochimica Acta. 184, 86-93 (2015).
- Data, P., et al. Unusual properties of electropolymerized 2,7- and 3,6- carbazole derivatives. Electrochimica Acta. 1282, 430-438 (2014).
- Etherington, M. K., et al. Regio- and conformational isomerization critical to design of efficient thermally-activated delayed fluorescence emitters. Nature Communications. 8, 14987 (2017).
- Trasatti, S. The absolute electrode potential: an explanatory note. Pure and Applied Chemistry. 58, 955-966 (1986).
- Cardona, C. M., et al. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Advanced Materials. 23, 2367-2371 (2011).
- Bredas, J. -L. Mind the gap! Mater Horiz. 1, 17-19 (2014).
- Gerson, F., Huber, W. Electron Spin Resonance Spectroscopy of Organic Radicals. , (2003).
- Brustolon, M., Giamello, E. Electron Paramagnetic Resonance. A Practitioner's Toolkit. , John Wiley & Sons Inc. New Jersey. (2009).
- Weil, J., Bolton, J. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. , John Wiley & Sons, Inc. (2007).
- Rieger, P. H. Electron Spin Resonance. Analysis and Interpretation. , The Royal Society of Chemistry. (2007).
- Roznyatovskiy, V. V., Gardner, D. M., Eaton, S. W., Wasielewski, M. R. Radical Anions of Trifluoromethylated Perylene and Naphthalene Imide and Diimide Electron Acceptors. Organic Letters. 16, 16-19 (2013).
- Pluczyk, S., Kuznik, W., Lapkowski, M., Reghu, R. R., Grazulevicius, J. V. The Effect of the Linking Topology on the Electrochemical and Spectroelectrochemical Properties of Carbazolyl Substituted Perylene Bisimides. Electrochimica Acta. 135, 487-494 (2014).
- Ledwon, P., et al. A Novel Donor-acceptor Carbazole and Benzothiadiazole Material for Deep Red and Infrared Emitting Applications. Journal of Materials Chemistry C. 4, 2219-2227 (2016).
- Heinze, J., Frontana-Uribe, B. A., Ludwigs, S. Electrochemistry of Conducting Polymers-Persistent Models and New Concepts. Chemical Reviews. 110, 4724-4771 (2010).
- Kurowska, A., Kostyuchenko, A. S., Zassowski, P., Skorka, L., Yurpalov, V. L., Fisyuk, A. S., Pron, A., Domagala, W. Symmetrically Disubstituted Bithiophene Derivatives of Properties. Journal of Physical Chemistry C. 118, 25176-25189 (2014).
- Zotti, G., Schiavon, G., Zecchin, S., Morin, J. F., Leclerc, M. Electrochemical, Conductive, and Magnetic Properties of 2,7-Carbazole-Based Conjugated Polymers. Macromolecules. 35, 2122-2128 (2002).
- Kaim, W., Fiedler, J. Spectroelectrochemistry: The Best of Two Worlds. Chemical Society Reviews. 38, 3373-3382 (2009).
- Duling, D. R. Simulation of Multiple Isotropic Spin-Trap EPR Spectra. Journal of Magnetic Resonance, Series B. 104, 105-110 (1994).
- Benson, C. R., et al. Multiplying the Electron Storage Capacity of a Bis-Tetrazine Pincer Ligand. Dalton Transactions. 43, 6513-6524 (2014).
- Nobuyasu, R. S., et al. Rational Design of TADF Polymers Using a Donor-Acceptor Monomer with Enhanced TADF Effi ciency Induced by the Energy Alignment of Charge Transfer and Local Triplet Excited States. Advanced Optical Materials. 4, 597-607 (2016).
